

Abstract
Subtle alterations in the coupling of drug binding to nucleotide hydrolysis were observed following mutation of all seven endogenous cysteine residues to serines in the human multidrug resistance transporter, P-glycoprotein. Wild-type (wt) and the mutant (cys-less) forms of P-gp were expressed in Trichoplusia ni (High Five) cells and purified by metal affinity chromatography in order to undertake functional studies.
No significant differences were observed in substrate ([3H]-azidopine) binding to wt or cys-less P-gp. Furthermore, neither the transported substrate vinblastine, nor the modulator nicardipine, differed in their respective potencies to displace [3H]-azidopine from the wt or cys-less P-gp. These results suggest that respective binding sites for these drugs were unaffected by the introduced cysteine to serine substitutions.
The Michaelis-Menten characteristics of basal ATP hydrolysis of the two isoforms of P-gp were identical. The maximal ATPase activity in the presence of vinblastine was marginally reduced whilst the Km was unchanged in cys-less P-gp compared to control. However, cys-less P-gp displayed lower overall maximal ATPase activity (62%), a decreased Km and a lower degree of stimulation (76%) in the presence of the modulator nicardipine.
Therefore, the serine to cysteine mutations in P-gp may suggest that vinblastine and nicardipine transduce their effects on ATP hydrolysis through distinct conformational pathways. The wt and cys-less P-gp isoforms display similarity in their fundamental kinetic properties thereby validating the use of cys-less P-gp as a template for future cysteine-directed structure/function analysis.
Keywords: ABC transporter, cysteine scanning mutagenesis, P-glycoprotein, purification and reconstitution, recombinant expression, Trichoplusia ni (High Five) cells, drug binding, ATP hydrolysis
Introduction
Multidrug resistance (MDR), a major obstacle to effective chemotherapy, is often associated with overexpression of the 180 kDa P-glycoprotein (wt P-gp), a drug efflux pump that couples ATP hydrolysis with active extrusion of a wide variety of structurally dissimilar and unrelated drugs. A major focus of research has been to screen established pharmacological reagents for their ability to inhibit P-gp mediated drug extrusion and so chemosensitize MDR cells. However, clinical chemotherapeutic regimens incorporating inhibitors of wt P-gp were only moderately successful and hindered by lack of drug specificity (Mickisch et al., 1991; Motzer et al., 1995; Raderer & Scheithauer, 1993). The need to design specific inhibitors that lack other pharmacological effects would be facilitated by mechanistic knowledge of the P-gp-mediated drug transport process.
P-gp is a member of the ABC transporter family and thus shares the common architecture of two cytoplasmic nucleotide binding and two membrane bound domains (Higgins, 1992). Each of the membrane bound domains contains six membrane-spanning segments. To elucidate which transmembrane segment of the protein contributes to the drug binding sites is of particular importance. Attempts to localize drug-binding sites on P-gp have been numerous, however the precise identity remains elusive. Chemical and photoaffinity labelling approaches have been used predominantly to address this issue and have implicated TM segments 4, 6, 10, 11 and 12 in contributing to the drug binding sites (Bruggemann et al., 1992; Greenberger, 1993; Isenberg et al., 2001; Loo & Clarke, 2001; Safa et al., 1990; Tamai & Safa, 1991). However, these approaches suffer from inherent mobility of the photolabelled compounds, which frequently results in labelling of residues distinct from the ‘true' binding site (Glossmann et al., 1987). Another key issue in elucidating the transport mechanism of P-gp is how the binding of drug is coupled to energy production in the nucleotide binding domains. It has been well established that drug binding elicits conformational changes that alter the binding or hydrolysis of ATP at the NBDs (Mechetner & Roninson, 1992; Sonveaux et al., 1999; Wang et al., 1998). Similarly, binding of nucleotides elicits conformational changes that manifest in perturbation of drug binding (Martin et al., 2000b). The first intracellular loop (Muller et al., 1996) and the linker region between the homologous N-terminal and C-terminal halves of the protein (Hrycyna et al., 1998) have been proposed to mediate transduction in P-gp, although the mechanism is unclear. Further investigation is required to delineate other regions of P-gp involved in NBD-TMD domain interactions and characterize how inhibitors of MDR may alter the ability of these regions to couple drug binding with ATP hydrolysis, thereby leading to transport.
The introduction of a unique cysteine residue into a protein (cysteine scanning mutagenesis; CSM) provides a reactive sulphydryl group that may be used to elucidate membrane protein structure, to map regions of the protein involved in substrate binding and to correlate conformational changes associated with drug efflux and ATP hydrolysis (Frillingos et al., 1998). CSM requires that prior mutation of endogenous cysteines does not affect protein structure and function. Previously, a mutant of P-gp, where all cysteines were mutated to alanine, was reported (Loo & Clarke, 1995b). The authors demonstrated that cys-less P-gp retained overall function, since the levels of cellular resistance to cytotoxic drugs was 70 – 80% of that in cells expressing wild-type protein. What is the basis for the 20 – 30% drop in the ability to confer resistance and how does it relate to the ability to recognize and respond to drugs at a molecular level? A subsequent investigation by the authors using cys-less P-gp in native plasma membrane preparations only reported a minor alteration in the potency of one drug, verapamil, to stimulate ATP hydrolysis (Loo & Clarke, 1995a). Similarly, the observations in native membrane preparations are difficult to translate into molecular mechanisms due to the presence of many contaminating ATPases. Therefore, we have no detailed information on the precise effects on (i) drug binding and (ii) the efficacy of this event on P-gp function for this important mutant isoform. In the present study, we have characterized in detail the pharmacological properties of P-gp in which all seven endogenous cysteines were replaced with serines. We found that the fundamental properties were not significantly altered; hence, the cys-less P-gp is able to provide a template for subsequent CSM studies. There were however, subtle alterations in the ability of nicardipine, but not vinblastine, to stimulate ATP hydrolysis indicating different routes of communication from their respective binding sites to the NBDs.
Methods
Materials
Nicardipine and vinblastine were obtained from Sigma-Aldrich (U.K.). [3H]-azidopine (47 Ci mmol−1) [3H]-phosphatidylcholine (84 Ci mmol−1) and AmplifyTM were from Amersham-Pharmacia Biotech (U.K.). [14C]-octyl-glucopyranoside (340 mCi mmol−1) was obtained from Isotopchim (France). The anthranilic acid derivative, XR9576, was synthesized and provided by Xenova Ltd (U.K.). SM2-Biobeads and the detergent compatible DC-Brad protein assay were purchased from BioRad (Hemel Hempstead, U.K.) Octyl-glucoside was obtained from Calbiochem (Nottingham, U.K.) and nickel-NTA chromatographic resin from Novagen (Nottingham, U.K.). All other reagents were of analytical grade or better.
Generation of recombinant baculovirus encoding (His6)-tagged wild-type and cys-less P-gp
Plasmid pKSmdr (a gift from Dr D. Gill, Oxford University) is derived from pBluescriptKS (Stratagene) and encodes the MDR1 cDNA (genbank accession: M14758) from the translation initiation codon at position 425 (see below, underlined) to the translation termination codon at position 4265. The sequence 5′ to the cDNA is 5′- GGATCC GCCGGGTTAAccacc ATG gAT-3′ and includes a BamHI restriction endonuclease site (italics) and a consensus Kozak sequence (lower case). To the 3′ of the MDR1 coding sequence there is a short untranslated cDNA sequence and a SalI restriction endonuclease site. The cDNA was subcloned into pAlterTM (Promega) and mutated as described by Promega (Altered sitesR), to introduce the coding sequence for a C-terminal hexa-histidine tag and, 3′ to the translational stop codon, a NotI restriction site (see Table 1 for oligonucleotide sequences). To make MDR-cys−, all seven cysteine codons were replaced with codons for serine and several silent unique restriction sites were introduced. Fidelity of mutated DNA was confirmed by sequencing. The tagged wt and MDR-cys− were sub-cloned into the baculovirus transfer vector pBacPAK9 (Clontech) via the BamHI and NotI restriction sites to generate pBP9-MDR and pBP9-MDR-cys−, respectively.
Table 1.
Oligonucleotide sequences used to generate cDNA for P-gp devoid of cysteine residues

Cell lines and viruses
Spodoptera frugiperda (Sf9) and Trichoplusia ni (High Five) insect cells were maintained at 27°C in serum-free SF-900 II medium (Life Technologies, U.K.) and serum-free Ex-cell 405 medium (JRH Biosciences, U.S.A.) respectively, supplemented with antibiotics (100 U ml−1 and 0.1 mg ml−1 streptomycin sulphate). Recombinant baculovirus was produced by lipofectin-mediated cotransfection of Sf9 monolayers with pBacPAK6-viral DNA and the pBP9-MDR and pBP-MDR-cys− plasmid vectors. The recombinant virus was isolated and purified by two rounds of plaque picks, according to standard procedures (Page & Rodgers, 1995). Expression of P-gp was confirmed in Western immunoblots of cell lysates following infection using the isolated recombinant baculovirus.
Isolation of cell membranes
High Five cell suspensions were infected at a density of 1.5×106 cells ml−1 with high titre recombinant virus at a multiplicity of infection of five. Cells were harvested 3 days post infection and centrifuged at 2000×g for 10 min at 4°C. The pellet was washed once with ice-cold PBS and resuspended in 5 volumes of ice-cold 10 mM Tris, 250 mM sucrose, 0.2 mM CaCl2, 20 μM leupeptin, 1 mM benzamidine, 1 μM pepstatin, pH 7.4. Cells were lysed by three rounds of nitrogen cavitation (1200 p.s.i., 15 min, 4°C) and cell debris removed by low speed centrifugation (2000×g, 15 min, 4°C). Membranes, isolated by ultracentrifugation (1000,000×g, 45 min, 4°C), were resuspended to 40 mg protein ml−1 in the same buffer but without CaCl2 and stored at −80°C.
Purification and reconstitution of (His6)-tagged wt and cys-less P-gp
Insect cell membranes were pelleted (100,000×g, 20 min, 4°C), then solubilized at 4°C with 2% (w v−1) octyl β-D-glucoside at a concentration of 5 mg protein ml−1 in Buffer A (150 mM NaCl, 20 mM Tris, 1.5 mM MgCl2, 20% glycerol, 20 μM leupeptin, 1 mM benzamidine, 1 μM pepstatin, pH 6.8). To maintain P-gp function, buffer A was supplemented with a 0.4% (w v−1) lipid mixture comprising crude Escherichia coli (E. Coli) phospholipids (Avanti, U.S.A.) and cholesterol at a ratio of 80 : 20 (w w−1) respectively. Following incubation on ice for 40 min, with periodic extrusion through a 22 G needle, insoluble material was removed by ultracentrifugation (100,000×g, 20 min, 4°C).
The soluble protein fraction was added to Ni-NTA resin at a volumetric ratio of 1 : 20 (packed resin : solubilized protein). To reduce non-specific interactions between proteins and the resin, imidazole was added to a final concentration of 10 mM. The protein and resin were incubated with agitation for 1 h at 4°C to ensure total binding of (His6)-tagged P-gp. The slurry was applied to an 18 ml Econo-column (Bio-Rad, U.K.). The resin was washed twice with 20 bed volumes of ice-cold 10 mM imidazole, then once with 20 bed volumes each of 20 mM imidazole and 30 mM imidazole, all in Buffer B (as buffer A but at pH 8) containing 1.25% (w v−1) octyl β-D-glucoside and 0.1% (w v−1) lipid mixture (as above). The resin was further washed with 20 bed volumes of ice-cold Buffer A containing 0.1% (w v−1) lipid mixture (as above), 1.25% (w v−1) octyl β-D-glucoside and 2 mM imidazole. Protein was eluted in ice-cold Buffer A containing 0.1% (w v−1) lipid mixture (as above), 1.25% (w v−1) octyl β-D-glucoside and 120 mM imidazole and two bed volume fractions collected. To monitor purification, proteins were separated by 7.5% SDS – PAGE (Laemmli, 1970) and visualized by silver staining (ICN, Thame, U.K.). Dilute protein samples were concentrated by trichloroacetic acid precipitation, as described elsewhere (Bensadoun & Weinstein, 1976), prior to electrophoresis.
Wt P-gp or cys-less P-gp containing fractions were pooled an routinely spiked with [3H]-phosphatidylcholine. Trace amounts of [14C]-octyl glucoside were added to monitor detergent removal. Reconstitution was achieved following removal of octyl glucoside by incubation with 225 mg ml−1 of washed Biobeads SM-2 for 2 h at 20°C. To assess reconstitution efficiency, 200 μl of the proteoliposomes were added to an equal volume of 60% (w v−1) sucrose in Buffer A containing 0.05% (v v−1) Triton X-100 and overlaid with 400 μl of 20, 10, 5 and 0% sucrose solutions in the same buffer, as previously described (Rigaud et al., 1988). Sample was centrifuged at 150,000×g at 4°C for 12 h to reach equilibrium. The distribution of proteins was determined by 7.5% SDS – PAGE of 200 μl aliquots from the gradient and detected with silver staining. Lipid distribution was determined by liquid scintillation counting 20 μl fractions from each aliquot.
Measurement of ATPase activity
Proteoliposomes (0.3 μg) were incubated at 37°C for 20 min in buffer containing (mM): NH4Cl 150, TrisHCl 50, MgSO4 5, 0.02% (w v−1) NaN3, pH 7.4 Activity was measured by either (i) varying Na2ATP concentration (0 – 2.5 mM) at a 50 μM drug concentration, or (ii) varying drug concentrations (0 – 100 μM) at 2 mM Na2ATP. In experiments to examine inhibition of ATP hydrolysis by XR9576 or vanadate, a 50 μM concentration of the modulator nicardipine was used to stimulate the ATPase activity of P-gp. These proteoliposomes were incubated with 2 mM Na2ATP and 0 – 100 μM inhibitor. Total assay volume was 50 μl and due to their hydrophobicity, all drugs were added from concentrated stocks of DMSO. The final DMSO concentration did not exceed 1% (v v−1) and we have previously demonstrated that neither ATP hydrolysis or drug binding are sensitive to DMSO levels up to 1% (v v−1) (Martin et al., 1997). This was also confirmed in the present investigation. ATP hydrolysis was stopped by addition of 40 μl of a 12% (w v−1) SDS solution and released inorganic phosphate was determined colorimetrically, as previously described (Chifflet et al., 1988).
For experiments conducted in the presence of a range of ATP concentrations, ATPase activity (μmol Pi min−1 mg protein−1), was plotted as a function of increasing ATP concentration and fitted, using non-linear regression, by the Michaelis-Menten equation:
where Vmax is the maximal activity, [A] is ATP concentration (M) and Km is the Michaelis-Menten affinity constant (M). Alternatively, ATPase activity (μmol Pi min−1 mg protein−1) was plotted as a function of increasing drug concentration and fitted, using non-linear regression, by the general dose-response equation (Delean et al., 1978):
where Vmax is the maximal activity, Vmin is the minimum activity, [B] is drug concentration (M), EC50 is the concentration of drug required to elicit 50% response.
Photoaffinity labelling
Reconstituted wt P-gp or cys-less P-gp was photoaffinity labelled with [3H]-azidopine. Proteoliposomes (0.5 – 1.5 μg) were incubated (2 h, 20°C) in the dark with either (i) increasing concentrations of [3H]-azidopine (0 – 1.5 μM), or (ii) 400 nM [3H]-azidopine and increasing concentrations (0 – 1 mM) of nicardipine or vinblastine. Total incubation volume was 45 μl and drugs were added from 50× concentrated stocks in DMSO. [3H]-azidopine was cross-linked to protein by irradiating with a UV source (100 W; samples located 5 cm from source) on ice for 5 min and free [3H]-azidopine removed by 7.5% SDS – PAGE. Gels were fixed, treated with AmplifyTM according to manufacturers instructions prior to drying and exposed to Kodak Biomax MR-1 film (Sigma-Aldrich, U.K.). The degree of labelling was quantified from digitally scanned autoradiograms using NIH 2.0 Image software and normalized to the maximal observed signal.
Where [3H]-azidopine labelling was plotted as a function of increasing total [3H]-azidopine concentrations the saturating value of binding was designated a value of one, and all other concentrations normalized to this. The normalized binding was fitted, using non-linear regression analysis, by equation 2.The values of Vmax and Vmin correspond to maximal and minimal binding capacities respectively and the EC50 corresponds to KD, a measure of radioligand binding affinity. For experiments with competing drugs, the photolabelling observed in the absence of drug was assigned a value of one and values in the presence of competing drug were normalized against this. The relationship was also fitted by equation 2and EC50 is the concentration of drug required to displace 50% of bound [3H]-azidopine.
Protein concentration
Protein content of membranes was determined by the DC Protein Assay (Bio-Rad, U.K.). P-gp concentration in proteoliposomes could not be determined using any commercial kit due to interference from imidazole and/or lipids. Therefore, protein concentration was estimated by quantitative densitometric analysis of Coomassie stained SDS – PAGE using NIH Image software, with bovine serum albumin as a standard.
Data analysis
Curve-fitting was performed using GraphPad Prism 3.0 (GraphPad software, U.S.A.). Statistical comparisons between mean values of two parameters were performed using the Student's t-test. ANOVA followed by Newman-Keul's post-hoc test was used to compare multiple parameters. In both methods of analysis P<0.05 was considered to be a statistically significant difference. All reported functional parameters are expressed as mean±s.e.m.
Results
Purification and reconstitution of wt and cys-less P-gp
In this study we have characterized the pharmacology of both wt and cys-less P-gp with respect to drug binding and ATP hydrolysis. To enable measurement of these parameters the two forms of P-gp were expressed in High Five insect cells and purified by Ni-NTA affinity chromatography, making use of a C-terminal (His)6 tag. Membrane protein solubilization and chromatography in the presence of asolectin, crude egg PC various portions of PC : PE or crude E. coli phospholipids produced P-gp devoid of ATPase activity (data not shown). The 80 : 20 (w w−1) mixture of E. coli bulk lipid and cholesterol was the only lipid mixture able to retain significant P-gp function.
A one step purification by immobilized nickel chromatography yielded pure wt P-gp (Figure 1a). Starting with 50 mg of total membrane protein, we were able to purify 160±23 μg and 80±19 μg of wt and cys-less P-gp respectively. Since approximately 50% of the P-gp was solubilized by octyl β-D-glucoside (data not shown) therefore these protein yields (assuming no loss during chromatography) indicate that P-gp comprised 0.6 – 1% of total membrane protein. The apparent molecular mass of both wt and cys-less P-gp was 140 – 150 kDa, compared to 180 kDa observed for wt P-gp in mammalian cells. This difference in molecular mass has been attributed to core glycosylation in insect cells without the further elaboration found in mammalian systems (Muller et al., 1996). Sucrose density gradient centrifugation demonstrated co-migration of greater than 90% protein with approximately 80% of the total phospholipid (Figure 1b), indicating efficient incorporation of wt protein into liposomes. Similar results were obtained for the reconstitution of cys-less P-gp (not shown).
Figure 1.
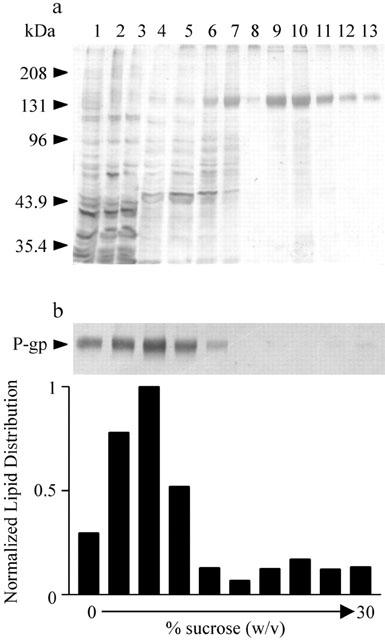
Purification and reconstitution of wt P-gp. (a) Silver stained SDS – PAGE samples (2 μg of protein/lane) from crude High Five membranes infected with pBacPAK9-wt-P-gp (lane 1), solubilized protein (lane 2), and protein not bound to Ni-NTA resin (lane 3), washing steps with 10 mM imidazole, pH 8 (lanes 4, 5); 20 mM imidazole, pH 8 (lane 6); 30 mM imidazole, pH 8 (lane 7); and 2 mM imidazole, pH 6.8 (lane 8). Proteins eluted with 120 mM imidazole, pH 6.8 (lanes 9 – 13). 4% (v v−1) of each elution fraction was loaded on each lane. (b) Following reconstitution, co-migration of both lipid and protein on a sucrose gradient confirmed successful incorporation of wt P-gp into liposomes. Wt P-gp content of fractions from the sucrose gradient were subjected to SDS – PAGE and visualized by silver staining, lipid content was determined by liquid scintillation counting.
Drug binding and displacement
Wild-type P-gp interacts with a wide variety of structurally disimilar and apparently unrelated drugs. For cys-less P-gp to be a constructive reporter of wt P-gp function, this characteristic must be preserved. Therefore, the ability of purified and reconstituted wt and cys-less P-gp to interact with [3H]-azidopine, a photoactive transported 1,4-dihydropyridine substrate (Tamai & Safa, 1991), was examined. Representative autoradiograms and corresponding [3H]-azidopine binding isotherms are shown in Figure 2 for both wt and cys-less P-gp. Non-linear regression analysis of the binding data enabled KD, a measure of radioligand binding affinity, to be determined. There was no significant difference in the affinity of [3H]-azidopine to photolabel either wt or cys-less P-gp as demonstrated from the mean data obtained from several independent preparations (Table 2). This indicates that the binding site for azidopine was unaffected by the removal of endogenous cysteine residues in P-gp.
Figure 2.
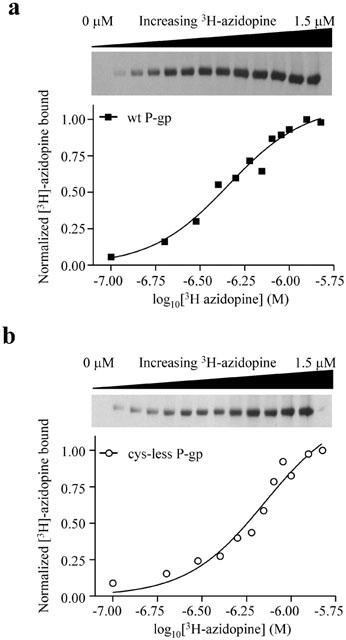
Photoaffinity labelling of purified and reconstituted wild-type (a) and cys-less P-gp (b) with increasing concentrations of [3H]-azidopine. Following removal of free [3H]-azidopine by 7.5% SDS – PAGE, bound material was visualized by autoradiography (upper panels). The amount of [3H]-azidopine bound was quantified using NIH 2.0 Image software and normalized to maximal observed binding (lower panels). KD values were determined from non-linear regression analysis of the general dose-response equation.
Table 2.
Interaction of selected drugs with purified and reconstituted wild-type and cys-less P-gp
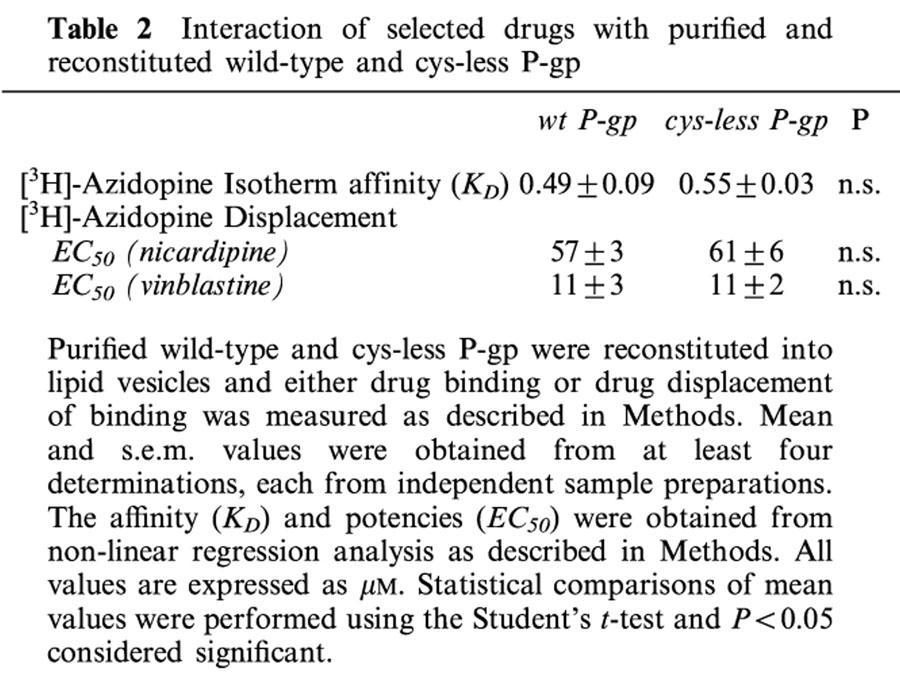
Previously, it has been shown that [3H]-azidopine binding to wt-P-gp in membrane preparations from resistant cells could be displaced by a range of drugs (Tamai & Safa, 1991). Demonstrating a similar displacement of [3H]-azidopine binding by drugs that do not interact at the same site in the reconstituted system would reveal the fidelity of communication between multiple drug binding sites on P-gp. We tested two drugs for their ability to displace [3H]-azidopine; namely nicardipine, a dihydropyridine modulator of P-gp function, and vinblastine, a vinca alkaloid that is transported by P-gp. As demonstrated by the representative data in Figure 3, both nicardipine and vinblastine caused similar dose-dependent reductions in the labelling of P-gp by [3H]-azidopine. The potency of nicardipine to completely displace [3H]-azidopine binding was indistinguishable between wt (EC50=57±3 μM n=4) and the cys-less P-gp isoform (EC50=61±6 μM n=4). Vinblastine displaced binding of [3H]-azidopine with an approximately 5 fold higher potency and like nicardipine, was unable to discriminate between wt and cys-less P-gp (Table 2).
Figure 3.
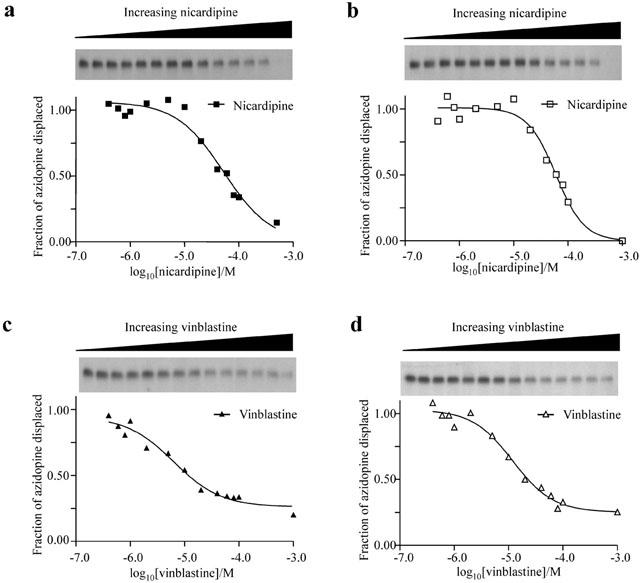
Photoaffinity labelling of purified and reconstituted wild-type (a,c) and cys-less P-gp (b,d) with [3H]-azidopine (400 nM) in the presence of increasing concentrations of either nicardipine or vinblastine. Sigmoidal curve fitting was performed, as described in Figure 2 legend, to determine EC50 values. Upper panels are autoradiograms of [3H]-azidopine bound to P-gp. Lower panels are dose response curves from the autoradiograms.
ATPase activity
The saturation isotherm and drug displacement studies demonstrated that the initial drug interaction with P-gp were unaffected by replacement of cysteine residues with serines. However, this is insufficient evidence to demonstrate that the wt and cys-less forms of P-gp are functionally identical. Consequently, the basal and drug stimulated ATP activities of the two proteins were compared.
As mentioned earlier, the functional integrity of purified P-gp had a specific lipid requirement that was met by an 80 : 20 weight ratio of crude E. coli lipid to cholesterol. Furthermore, measurable basal or drug stimulated ATPase activity was only observed following reconstitution, indicating that either (i) P-gp function is dependent on a lamellar lipid contact and/or (ii) octyl-glucoside causes a reversible inactivation of P-gp. As shown by the representative data in Figure 4, the basal ATPase activity of wt and cys-less P-gp followed Michaelis-Menten kinetics. Under basal conditions, the two proteins displayed similar maximal activities. Although the affinity constant, Km, was 30% lower in the cys-less mutant the difference was not statistically significant (Table 3). Addition of vinblastine (50 μM) to either wt or cys-less P-gp did not alter the Michaelis affinity constant for ATP hydrolysis from their respective basal values (Table 3), however the maximal velocity was 26% lower in cys-less P-gp (P<0.05). In addition, the maximal specific activity for vinblastine-stimulated ATP hydrolysis of both wt and cys-less P-gp were similar. Nicardipine caused more profound effects on the ATP activity of cys-less compared to wt P-gp. For example, 50 μM nicardipine reduced the Km for ATP by 28% (P<0.05) in the cys-less P-gp but did not affect the wt protein (P<0.05, Table 3). The Vmax for ATPase activity in the presence of 50 μM nicardipine was 38% lower in the cys-less P-gp compared to the wild type (P<0.05, Table 3).
Figure 4.
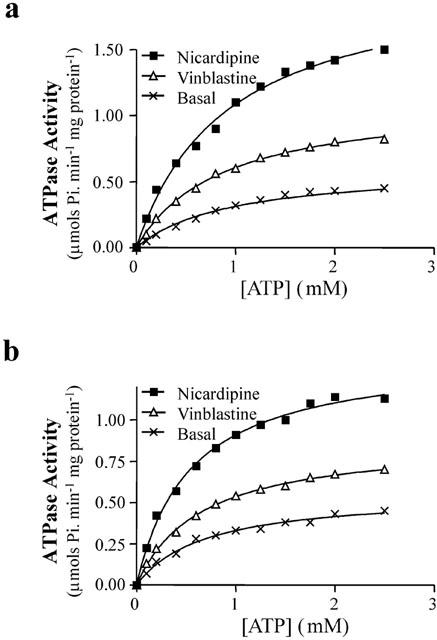
ATPase activity of purified and reconstituted wt P-gp (a) and cys-less P-gp (b) was determined at varying ATP concentrations by measuring the release of inorganic phosphate, as described in Methods. ATPase activity was measured either under basal conditions, or following addition of either 50 μM nicardipine or 50 μM vinblastine. Data was fitted, using non-linear regression, by the Michaelis-Menten equation to determine Km, the Michaelis constant, and Vmax, the maximal activity.
Table 3.
ATPase activity of purified and reconstituted wild-type and cys-less P-gp

Complete dose-response relationships provided further insight into the nature of the altered nicardipine stimulation of ATPase activity by cys-less P-gp. Vinblastine produced a similar stimulation of ATPase activity (Sxt) at similar potency (EC50) in both wt and cys-less P-gp (Table 3). Nicardipine also stimulated the ATPase activity of cys-less P-gp with higher potency than that observed in wt protein (Table 3). The reduction in extent of stimulation produced in the cys-less P-gp by nicardipine (Table 3) was in close agreement with the reduction in Vmax reported above. Therefore, it appears that mutations of the cysteines to serines has caused a subtle change in the relationship between P-gp and ATP that were apparent following stimulation by nicardipine.
Nicardipine stimulation affects nucleotide hydrolysis via long-range communication from the TM regions to the NBDs of P-gp. It is unclear which stage of hydrolysis is affected. Hence, the ability of nicardipine to alter trapping to P-gp in a transition complex (Pgp.MgADP.Vi) that mimics the post-hydrolytic state was examined. In the presence of nicardipine, vanadate was able to trap this intermediate state in both wt and cys-less P-gp to a similar extent and with similar potency (Table 3). The ability of the high affinity modulator XR9576 to potently inhibit the ATPase activity of P-gp (Martin et al., 1999) is also likely to involve intra-protein conformational changes. In the presence of a fixed concentration of nicardipine a dose-response analysis of XR9576 on ATPase activity was measured. The potency of XR9576 to inhibit ATPase activity was significantly reduced (P<0.001) in the cys-less (EC50=0.43±0.03 μM, n=9) compared to the wt (EC50=0.25±0.02 μM, n=9) protein (Table 3). This indicates that in the cys-less protein, communication between the XR9576 binding site and the NBDs has been perturbed.
Discussion
The purpose of this study was to determine whether substitution of all seven endogenous cysteine residues witth serines compromises function of the human multidrug resistant protein, P-gp. If the cys-less form of P-gp retains function, it may be used in CSM as a template for introduction of novel cysteine residues at desired sites. Specific chemical labelling of these cysteine residues may then be used to elucidate specific protein structure-function relationships pertaining to the transportcycle of P-gp. A mutant P-gp, where the cysteines were mutated to alanines, was shown to confer similar levels of drug resistance in whole cells to the wild-type protein (Loo & Clarke, 1995b). However there were no direct details presented on the ability of this specific P-gp isoform to bind drugs or how the binding of drugs affects the rate of ATP hydrolysis. The characterization in the present study has uncovered subtle differences in the ATPase activity of cys-less P-gp when compared to wild-type protein. We have presented evidence to indicate that these differences were not caused by altered binding of the two drugs to P-gp. Given that nicardipine and vinblastine bind at distinct sites on the protein (Martin et al., 2000a), it suggests different routes of communication to the NBDs.
To examine the effects of mutating all seven endogenous cysteines to serines, it was essential to establish expression, purification and reconstitution conditions that preserved the functional integrity of the protein. Maintenance of functional integrity following purification and reconstitution was assessed by comparing values obtained for drug binding and ATP hydrolysis of wt P-gp with published values. We found that the Km (ATP) describing basal ATP hydrolysis, which was observed to follow Michaelis-Menten kinetics, agreed with previously reported values of 0.5 – 1.4 mM (Al-Shawi & Senior, 1993; Muller et al., 1996; Ramachandra et al., 1998; Shapiro & Ling, 1994). In concurrence with others, the Km for ATP was unchanged following binding of vinblastine (Ramachandra et al., 1998); and the extent to which nicardipine and vinblastine stimulated ATPase activity agreed with values previously reported (Ramachandra et al., 1998). Finally, the KD for [3H]-azidopine binding, and its displacement by vinblastine, was within the reported range (Safa et al., 1987; Tamai & Safa, 1991). These comparisons strongly suggest that the functional integrity of wt P-gp was maintained following expression, purification and reconstitution. Furthermore, addition of the C-terminal (His)6 tag and underglycosylation of the protein in insect cells did not compromise function. Previous studies have shown that wt P-gp function was unaltered by tagging the extreme C-terminus (Chen & Simon, 2000; Loo & Clarke, 1995; Loo & Clarke, 1995), and that glycosylation is unimportant for the drug transport activity (Gribar et al., 2000). We can therefore be confident that differences between wt and cys-less P-gp function result from cysteine mutations.
Interaction of drugs with the TM domain of P-gp appears not to be compromised by cysteine substitutions. This was evidenced by similar KD values for [3H]-azidopine binding, and the EC50 values for both nicardipine and vinblastine displacement of [3H]-azidopine are close to those found in wt P-gp. The similarity in the structures of the 1,4-dihydropyridines azidopine and nicardipine suggests that the two drugs compete for the same binding site. Taken together, this indicates that the drug binding site for nicardipine and azidopine is able to provide a similar initial interaction with these drugs in cys-less P-gp compared to wt. The partial displacement of [3H]-azidopine binding by the vinca alkaloid vinblastine agrees with a previous report demonstrating a non-competitive interaction between these drugs on P-gp (Tamai & Safa, 1991). Consequently, the allosteric coupling that occurs between distinct drug-binding sites in P-gp is unaffected by the cysteine substitutions (Martin et al., 2000b).
The cysteine mutations in P-gp did not significantly affect the Michaelis-Menten characteristics of ATP hydrolysis. However, the ability of drugs, particularly nicardipine, to affect the catalytic cycle of P-gp was modulated by the cysteine substitutions. With cys-less P-gp, in the presence of nicardipine, the maximal ATPase activity and affinity for ATP was reduced compared to the wt P-gp. Given that these alterations cannot be explained by the initial binding event for nicardipine on P-gp, it appears that communication between the drug binding site and the NBDs is affected. Examination of the catalytic cycle of P-gp (Al-Shawi et al., 1994; Urbatsch et al., 1995) indicates that the overall Km for the reaction will depend on (i) ATP binding, (ii) hydrolysis of the γPi moiety, (iii) dissociation of Pi and (iv) ADP release. Stage (i) may be ruled out since a saturating concentration of ATP was employed in this investigation. Similarly, stage (iii) may be discounted since the dissociation of phosphate is rapid, produces a large free energy change and is therefore unlikely to be a rate-limiting step (Urbatsch et al., 1995). The equilibrium position between Pgp.MgATP⇔Pgp.MgADP.Pi has not yet been elucidated and it remains unclear whether drug binding affects this step (stage ii). The similar potency of vanadate to trap nicardipine stimulated wt compared to cys-less P-gp may be complicated by the altered drug binding observed at this stage of the catalytic cycle (Martin et al., 2000b). The other possibility for nicardipine to alter the overall Km for ATP hydrolysis was by affecting MgADP release at stage (iv), which has been suggested to be a rate-limiting step in the catalytic cycle for P-gp (Kerr et al., 2001; Urbatsch et al., 1995). It is not yet possible to assign the specific mutation involved in this communication route from the nicardipine-binding site to the NBDs since seven endogenous cysteine residues were substituted. It has previously been shown that vinblastine and nicardipine bind to different sites on P-gp (Ferry et al., 1992; Martin et al., 2000a) and data from the present manuscript may suggest that they communicate with the NBDs by distinct routes.
A characterization of the ATPase activity of purified reconstituted P-gp containing cysteine to alanine mutations has recently been reported (Urbatsch et al., 2001). We chose to mutate the cysteines to serine due to the greater similarity of these two amino acids compared with alanine. The largely identical rates of maximal ATP hydrolysis and affinity of ATP indicates that both cysteine→serine and cysteine→alanine mutations are tolerated by P-gp. Similar to our observations with nicardipine, it was reported that the P-gp modulator verapamil displayed a lower degree of stimulation of ATPase activity and a reduced affinity to do so. We have shown that cysteine→serine mutations do not affect nicardipine binding, however this does not preclude an effect of the cysteine→alanine mutation on verapamil binding to protein. Furthermore, it is not clear whether nicardipine and verapamil bind at identical or distinct sites on P-gp; nor can we exclude the possibility that they may bind at distinct sites, which may transduce their effects on ATP hydrolysis through a common conformational pathway.
Is cys-less P-gp able to be used as a background for introduction of novel cysteine residues and subsequent cysteine-directed chemical labelling analyses? This study demonstrates that endogenous cysteine residues are not critical for the overall function of P-gp. As described above, changing cysteines to serines was, however, not without effect in P-gp. The maximal rates of transport by the cysteine-less forms of lactose permease, the subunit of the yeast vacuolar proton translocating ATPase, the NHaA H+/Na+ antiporter from E. coli, and of the yeast mitochondrial citrate transporter all displayed 50 – 80% of wild-type activity (Leng et al., 1999; Olami et al., 1997; van iwaarden et al., 1991; Xu et al., 2000). This magnitude of change has not precluded cysteine-less proteins being used as effective reporters of wild-type structure – function relationships. The alterations we have observed are considerably milder and therefore, cysteine-less P-gp may be used as a template for further CSM studies. Indeed, we have also previously demonstrated that the cys-less P-gp is transport competent with similar properties to wild-type protein (Blott et al., 1999).
In summary, this study demonstrates that endogenous cysteines are not critical for (i) drug binding; (ii) ATP hydrolysis; or (iii) instigating modulation of ATP hydrolysis following drug binding. However, substitution of cysteines to serines reduced basal Km (ATP) and altered the ability of nicardipine to stimulate ATP hydrolysis. We speculate that these subtle changes in properties highlighted distinct and drug-specific routes of communication between TMDs and NBDs on P-gp.
Acknowledgments
This work was supported by a Cancer Research Campaign program grant (ARG/CS SP1861/0301). We wish to thank Xenova Ltd. for providing the XR9576 used in these investigations.
Abbreviations
- ATP
adenosine triphosphate
- CSM
cysteine scanning mutagenesis
- E. coli
Escherichia coli
- MDR
multidrug resistance
- NBD
nucleotide binding domain
- octyl glucoside
octyl β-D-glucopyranoside
- PAGE
polyacrylamide gel electrophoresis
- wt P-gp
wild-type P-glycoprotein
- cys-less P-gp
P-glycoprotein where all endogenous cysteines have been replaced by serines
- Sf9
Spodoptera frugiperda cell line
- TM
transmembrane
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- Ni-NTA
nickel nitrilotriacetic acid
References
- AL-SHAWI M.K., SENIOR A.E. Characterization of the adenosine triphosphatase activity of Chinese hamster P-glycoprotein. J. Biol. Chem. 1993;268:4197–4206. [PubMed] [Google Scholar]
- AL-SHAWI M.K., URBATSCH I.L., SENIOR A.E. Covalent inhibitors of P-glycoprotein ATPase activity. J. Biol. Chem. 1994;269:8986–8992. [PubMed] [Google Scholar]
- BENSADOUN A., WEINSTEIN D. Assay of proteins in the presence of interfering materials. Anal. Biochem. 1976;70:241–250. doi: 10.1016/s0003-2697(76)80064-4. [DOI] [PubMed] [Google Scholar]
- BLOTT E.J., HIGGINS C.F., LINTON K.J. Cysteine-scanning mutagenesis provides no evidence for the extracellular accessibility of the nucleotide-binding domains of the multidrug resistance transporter P-glycoprotein. EMBO J. 1999;18:6800–6808. doi: 10.1093/emboj/18.23.6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRUGGEMANN E.P., CURRIER S.J., GOTTESMAN M.M., PASTAN I. Characterization of the azidopine and vinblastine binding site of P-glycoprotein. J. Biol. Chem. 1992;267:21020–21026. [PubMed] [Google Scholar]
- CHEN Y., SIMON S.M. In situ biochemical demonstration that P-glycoprotein is a drug efflux pump with broad specificity. J. Cell. Biol. 2000;148:863–870. doi: 10.1083/jcb.148.5.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHIFFLET S., TORRIGLIA A., CHIESA R., TOLOSA S. A method for the determination of inorganic phosphate in the presence of labile organic phosphate and high concentrations of protein: application to lens ATPases. Anal. Biochem. 1988;168:1–4. doi: 10.1016/0003-2697(88)90002-4. [DOI] [PubMed] [Google Scholar]
- DELEAN A., MUNSON P.J., RODBARD D. Simultaneous analysis of families of sigmoidal curves: application to bioassay, radioligand assay, and physiological dose-response curves. Am. J. Physiol. 1978;235:E97–E102. doi: 10.1152/ajpendo.1978.235.2.E97. [DOI] [PubMed] [Google Scholar]
- FERRY D.R., RUSSELL M.A., CULLEN M.H. P-glycoprotein possesses a 1,4-dihydropyridine-selective drug acceptor site which is allosterically coupled to a vinca-alkaloid-selective binding site. Biochem. Biophys. Res. Commun. 1992;188:440–445. doi: 10.1016/0006-291x(92)92404-l. [DOI] [PubMed] [Google Scholar]
- FRILLINGOS S., SAHIN-TOTH M., WU J., KABACK H.R. Cys-scanning mutagenesis: a novel approach to structure function relationships in polytopic membrane proteins. FASEB J. 1998;12:1281–1299. doi: 10.1096/fasebj.12.13.1281. [DOI] [PubMed] [Google Scholar]
- GLOSSMANN H., FERRY D.R., STREISSNIG J., GOLL A., MOOSBURGER K. Resolving the structure of the Ca2+ channel by photoaffinity labeling. Trends Biochem. Sci. 1987;8:95–100. [Google Scholar]
- GREENBERGER L.M. Major photoaffinity drug labeling sites for iodaryl azidoprazosin in P-glycoprotein are within, or immediately C-terminal to, transmembrane domains 6 and 12. J. Biol. Chem. 1993;268:11417–11425. [PubMed] [Google Scholar]
- GRIBAR J.J., RAMACHANDRA M., HRYCYNA C.A., DEY S., AMBUDKAR S.V. Functional characterization of glycosylation-deficient human P-glycoprotein using a vaccinia virus expression system. J. Membr. Biol. 2000;173:203–214. doi: 10.1007/s002320001020. [DOI] [PubMed] [Google Scholar]
- HIGGINS C.F. ABC Transporters: from microorganisms to man. Annu. Rev. Cell Biol. 1992;8:67–113. doi: 10.1146/annurev.cb.08.110192.000435. [DOI] [PubMed] [Google Scholar]
- HRYCYNA C.A., AIRAN L.E., GERMANN U.A., AMBUDKAR S.V., PASTAN I., GOTTESMAN M.M. Structural flexibility of the linker region of human P-glycoprotein permits ATP hydrolysis and drug transport. Biochemistry. 1998;37:13660–13673. doi: 10.1021/bi9808823. [DOI] [PubMed] [Google Scholar]
- ISENBERG B., THOLE H., TUMMLER B., DEMMER A. Identification and localization of three photobinding sites of iodoarylazidoprazosin in hamster P-glycoprotein. Eur. J. Biochem. 2001;268:2629–2634. doi: 10.1046/j.1432-1327.2001.02155.x. [DOI] [PubMed] [Google Scholar]
- KERR K.M., SAUNA Z.E., AMBUDKAR S.V. Correlation between steady-state ATP hydrolysis and vanadate-induced ADP trapping in Human P-glycoprotein. Evidence for ADP release as the rate-limiting step in the catalytic cycle and its modulation by substrates. J. Biol. Chem. 2001;276:8657–8664. doi: 10.1074/jbc.M010044200. [DOI] [PubMed] [Google Scholar]
- LAEMMLI U.K. Cleavage of structural proteins during the assembly of the head of bactgeriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- LENG X.H., NISHI T., FORGAC M. Transmembrane topography of the 100-kDa a subunit (Vph1p) of the yeast vacuolar proton-translocating ATPase. J. Biol. Chem. 1999;274:14655–14661. doi: 10.1074/jbc.274.21.14655. [DOI] [PubMed] [Google Scholar]
- LOO T.W., CLARKE D.M. Covalent modification of human P-glycoprotein mutants containing a single cysteine in either nucleotide-binding fold abolishes drug-stimulated ATPase activity. J. Biol. Chem. 1995a;270:22957–22961. doi: 10.1074/jbc.270.39.22957. [DOI] [PubMed] [Google Scholar]
- LOO T.W., CLARKE D.M. Membrane topology of a cysteine-less mutant of human P-glycoprotein. J. Biol. Chem. 1995b;270:843–848. doi: 10.1074/jbc.270.2.843. [DOI] [PubMed] [Google Scholar]
- LOO T.W., CLARKE D.M. Defining the drug-binding site in the human multidrug resistance P-glycoprotein using MTS-verapamil. J. Biol. Chem. 2001;14:14. doi: 10.1074/jbc.M100407200. [DOI] [PubMed] [Google Scholar]
- MARTIN C., BERRIDGE G., HIGGINS C.F., CALLAGHAN R. The multi-drug resistance reversal agent SR33557 and modulation of vinca alkaloid binding to P-glycoprotein by an allosteric interaction. Br. J. Pharmacol. 1997;122:765–771. doi: 10.1038/sj.bjp.0701429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTIN C., BERRIDGE G., MISTRY P., HIGGINS C., CHARLTON P., CALLAGHAN R. The molecular interaction of the high affinity reversal agent XR9576 with P-glycoprotein. Br. J. Pharmacol. 1999;128:403–411. doi: 10.1038/sj.bjp.0702807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTIN C., BERRIDGE G., HIGGINS C.F., MISTRY P., CHARLTON P., CALLAGHAN R. Communication between multiple drug binding sites on P-glycoprotein. Mol. Pharmacol. 2000a;58:624–632. doi: 10.1124/mol.58.3.624. [DOI] [PubMed] [Google Scholar]
- MARTIN C., BERRIDGE G., MISTRY P., HIGGINS C., CHARLTON P., CALLAGHAN R. Drug binding sites on P-glycoprotein are altered by ATP binding prior to nucleotide hydrolysis. Biochemistry. 2000b;39:11901–11906. doi: 10.1021/bi000559b. [DOI] [PubMed] [Google Scholar]
- MECHETNER E.B., RONINSON I.B. Efficient inhibition of P-glycoprotein-mediated multidrug resistance with a monoclonal antibody. Proc. Natl. Acad. Sci. U.S.A. 1992;89:5824–5828. doi: 10.1073/pnas.89.13.5824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MICKISCH G.H., MERLINO G.T., AIKEN P.M., GOTTESMAN M.M., PASTAN I. New potent verapamil derivatives that reverse multidrug resistance in human renal carcinoma cells and in transgenic mice expressing the human MDR1 gene. J. Urol. 1991;146:447–453. doi: 10.1016/s0022-5347(17)37822-9. [DOI] [PubMed] [Google Scholar]
- MOTZER R.J., LYN P., FISCHER P., LIANES P., NGO R.L., CORDON-CARDO C., O'BRIEN J.P. Phase I/II trial of dexverapamil plus vinblastine for patients with advanced renal cell carcinoma. J. Clin. Oncol. 1995;13:1958–1965. doi: 10.1200/JCO.1995.13.8.1958. [DOI] [PubMed] [Google Scholar]
- MULLER M., BAKOS E., WELKER E., VARADI A., GERMANN U.A., GOTTESMAN M.M., MORSE B.S., RONINSON I.B., SARKADI B. Altered drug-stimulated ATPase activity in mutants of the human multidrug resistance protein. J. Biol. Chem. 1996;271:1877–1883. doi: 10.1074/jbc.271.4.1877. [DOI] [PubMed] [Google Scholar]
- OLAMI Y., RIMON A., GERCHMAN Y., ROTHMAN A., PADAN E. Histidine 225, a residue of the NhaA-Na+/H+ antiporter of Escherichia coli is exposed and faces the cell exterior. J. Biol. Chem. 1997;272:1761–1768. doi: 10.1074/jbc.272.3.1761. [DOI] [PubMed] [Google Scholar]
- PAGE M.J., RODGERS B.C. Selection of recombinant baculoviruses by visual screening. Methods Mol. Biol. 1995;39:107–127. doi: 10.1385/0-89603-272-8:107. [DOI] [PubMed] [Google Scholar]
- RADERER M., SCHEITHAUER W. Clinical trials of agents that reverse multidrug resistance. A literature review. Cancer. 1993;72:3553–3563. doi: 10.1002/1097-0142(19931215)72:12<3553::aid-cncr2820721203>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- RAMACHANDRA M., AMBUDKAR S.V., CHEN D., HRYCYNA C.A., DEY S., GOTTESMAN M.M., PASTAN I. Human P-glycoprotein exhibits reduced affinity for substrates during a catalytic transition state. Biochemistry. 1998;37:5010–5019. doi: 10.1021/bi973045u. [DOI] [PubMed] [Google Scholar]
- RIGAUD J.L., PATERNOSTRE M.T., BLUZAT A. Mechanisms of membrane protein insertion into liposomes during reconstitution procedures involving the use of detergents. 2. Incorporation of the light-driven proton pump bacteriorhodopsin. Biochemistry. 1988;27:2677–2688. doi: 10.1021/bi00408a007. [DOI] [PubMed] [Google Scholar]
- SAFA A.R., AGRESTI M., TAMAI I., MEHTA N.D., VAHABI S. The alpha 1-adrenergic photoaffinity probe [125I]arylazidoprazosin binds to a specific peptide of P-glycoprotein in multidrug-resistant cells. Biochem. Biophys. Res. Commun. 1990;166:259–266. doi: 10.1016/0006-291x(90)91939-p. [DOI] [PubMed] [Google Scholar]
- SAFA A.R., GLOVER C.J., SEWELL J.L., MEYERS M.B., BIEDLER J.L., FELSTED R.L. Identification of the multidrug resistance-related membrane glycoprotein as an acceptor for calcium channel blockers. J. Biol. Chem. 1987;262:7884–7888. [PubMed] [Google Scholar]
- SHAPIRO A.B., LING V. ATPase activity of purified and reconstituted P-glycoprotein from Chinese hamster ovary cells. J. Biol. Chem. 1994;269:3745–3754. [PubMed] [Google Scholar]
- SONVEAUX N., VIGANO C., SHAPIRO A.B., LING V., RUYSSCHAERT J.M. Ligand-mediated tertiary structure changes of reconstituted P-glycoprotein. J. Biol. Chem. 1999;274:17649–17654. doi: 10.1074/jbc.274.25.17649. [DOI] [PubMed] [Google Scholar]
- TAMAI I., SAFA A.R. Azidopine noncompetitively interacts with vinblastine and cyclosporin A binding to P-glycoprotein in multidrug resistant cells. J. Biol. Chem. 1991;266:16796–16800. [PubMed] [Google Scholar]
- URBATSCH I.L., GIMI K., WILKE-MOUNTS S., LERNER-MARMAROSH N., ROUSSEAU M.E., GROS P., SENIOR A.E. Cysteines 431 and 1074 are responsible for inhibitory disulfide cross-linking between the two nucleotide-binding sites in human P-glycoprotein. J. Biol. Chem. 2001;276:26980–26987. doi: 10.1074/jbc.M010829200. [DOI] [PubMed] [Google Scholar]
- URBATSCH I.L., SANKARAN B., WEBER J., SENIOR A.E. P-glycoprotein is stably inhibited by vanadate-induced trapping of nucleotide at a single catalytic site. J. Biol. Chem. 1995;270:19383–19390. doi: 10.1074/jbc.270.33.19383. [DOI] [PubMed] [Google Scholar]
- VAN IWAARDEN P.R., PASTORE J.C., KONINGS W.N., KABACK H.R. Construction of a functional lactose permease devoid of cysteine residues. Biochemistry. 1991;30:9595–9600. doi: 10.1021/bi00104a005. [DOI] [PubMed] [Google Scholar]
- WANG G., PINCHEIRA R., ZHANG J.T. Dissection of drug-binding-induced conformational changes in P-glycoprotein. Eur. J. Biochem. 1998;255:383–390. doi: 10.1046/j.1432-1327.1998.2550383.x. [DOI] [PubMed] [Google Scholar]
- XU Y., KAKHNIASHVILI D.A., GREMSE D.A., WOOD D.O., MAYOR J.A., WALTERS D.E., KAPLAN R.S. The yeast mitochondrial citrate transport protein. Probing the roles of cysteines, Arg(181), and Arg(189) in transporter function. J. Biol. Chem. 2000;275:7117–7124. doi: 10.1074/jbc.275.10.7117. [DOI] [PubMed] [Google Scholar]