

Abstract
Cibenzoline, (±)-2-(2,2-diphenylcyclopropyl-2-imidazoline succinate, has been clinically used as one of the Class I type antiarrhythmic agents and also reported to block ATP-sensitive K+ channels in excised membranes from heart and pancreatic β cells. In the present study, we investigated if this drug inhibited gastric H+,K+-ATPase activity in vitro.
Cibenzoline inhibited H+,K+-ATPase activity of permeabilized leaky hog gastric vesicles in a concentration-dependent manner (IC50: 201 μM), whereas no effect was shown on Na+,K+-ATPase activity of dog kidney (IC50: >1000 μM). Similarly, cibenzoline inhibited H+,K+-ATPase activity of HEK-293 cells (human embryonic kidney cell line) co-transfected with rabbit gastric H+,K+-ATPase α- and β-subunit cDNAs (IC50: 183 μM).
In leaky gastric vesicles, inhibition of H+,K+-ATPase activity by cibenzoline was attenuated by the addition of K+ (0.5 – 5 mM) in a concentration-dependent manner. The Lineweaver-Burk plot of the H+,K+-ATPase activity shows that cibenzoline increases Km value for K+ without affecting Vmax, indicating that this drug inhibits H+,K+-ATPase activity competitively with respect to K+.
The inhibitory effect of H+,K+-ATPase activity by cibenzoline with normal tight gastric vesicles did not significantly differ from that with permeabilized leaky gastric vesicles, indicating that this drug reacted to the ATPase from the cytoplasmic side of the membrane.
These findings suggest that cibenzoline is an inhibitor of gastric H+,K+-ATPase with a novel inhibition mechanism, which inhibits gastric H+,K+-ATPase by binding its K+-recognition site from the cytoplasmic side.
Keywords: Cibenzoline; hog gastric vesicle; H+,K+-ATPase activity; proton uptake; K+-recognition site
Introduction
The gastric H+,K+-ATPase is involved in acid secretion of the stomach (Sachs et al., 1976; Wallmark et al., 1985). This enzyme is a heterodimeric membrane protein which belongs to the family of P-type cation-transporting ATPases, such as Na+,K+-ATPase and Ca2+-ATPase (Sachs et al., 1995), and catalyzes the electroneutral exchange of luminal potassium ions for cytosolic protons mediated by the hydrolysis of ATP (Sachs et al., 1976). Gastric H+,K+-ATPase is irreversibly inhibited by benzimizazole derivatives such as 5-methoxy-2-(((4-methoxy-3,5-dimethyl-2-pyridinyl)methyl)sulphinyl)-1H-benzimidazole (omeprazole) and 2-((4-(3-methoxypropoxy)-3-methylpyridin-2-yl)methyl-sulphinyl)-1H-benzimidazole sodium salt (rabeprazole), which have been clinically used as anti-ulcer drugs (Fryklund et al., 1988; Morii et al., 1990; Richardson et al., 1998). The H+,K+-ATPase is reversibly inhibited by 3-(cyanomethyl)-2-methyl-8-(phenylmethoxy)-imidazo(1,2a)-pyridine (SCH 28080) (Asano et al., 1999; Beil et al., 1986; Briving et al., 1988; Keeling et al., 1988; Scott et al., 1987; Wallmark et al., 1987) and 3 - butyryl - 4 - (2-methylphenylamino)-8-methoxy-quinoline (SK&F96067) (Keeling et al., 1998; 1991). SCH 28080 and SK&F96067 are K+-site inhibitors, which competitively bind to the K+-high affinity site on the luminal side of the membrane (Beil et al., 1986; Briving et al., 1988; Keeling et al., 1988, 1991; Wallmark et al., 1987).
(±)-2-(2,2-diphenylcyclopropyl)-2-imidazoline succinate (cibenzoline, Figure 1), has been clinically used as one of the Class I type antiarrhythmic agents (Harron et al., 1992; Holck & Osterrieder, 1986; Millar & Williams, 1982; Touboul et al., 1986). This drug relieves arrhythmia by restricting fast inward Na+ current (Millar & Williams, 1982) and blocking the slow inward Ca2+ channel in myocytes (Holck & Osterrieder, 1986). In addition, it has been reported that cibenzoline induces sporadic hypoglycemia as an extracardiac side effect (Gachot et al., 1988; Jeandel et al., 1988) and stimulates insulin secretion from pancreatic β cells (Bertrand et al., 1992; Gachot et al., 1988). It was recently shown that micromolar concentrations of cibenzoline blocked ATP-sensitive K+ (KATP) channel in excised membranes from rat heart and pancreatic β cells (Horie et al., 1992; Ishida-Takahashi et al., 1996; Kakei et al., 1993). Molecular biological findings indicate that KATP channel of pancreatic β cells is composed of a sulfonylurea receptor (SUR) 1, a member of the ATP-binding cassette super family, and a K+ channel (Kir6.2), which forms the ion-pore (Inagaki et al., 1995). And, cardiac KATP channel is composed of SUR2A, a subtype of SUR, and Kir6.2 (Chutkow et al., 1996). More recently, Mukai et al. (1998a, b) and Horie et al. (2000) reported that cibenzoline inhibits KATP channels by a novel inhibitory mechanism in which cibenzoline directly binds to the Kir6.2 subunit rather than the SUR1 subunit. This finding recurred our question whether the K+ recognition site of K+ channel structurally resembles with that of K+-transporting pump.
Figure 1.
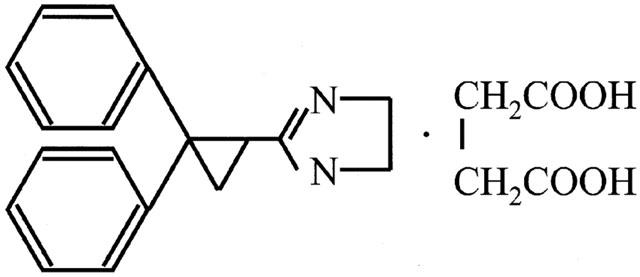
Chemical structure of cibenzoline (molecular mass=380.44).
Methods
Materials
Cibenzoline and N-methylated SCH 28080, 3-(cyanomethyl)-2,3-dimethyl-8-(phenylmethoxy) imidazo(1,2a)-pyridine were a generous gift from Fujisawa Pharmaceutical Co. (Osaka, Japan) and Toyama Chemical Co. (Tokyo, Japan), respectivly. HEK-293 cells (human embryonic kidney cell line) were a kind gift from Prof. Jonathan Lytton (University of Calgary, Calgary, Canada). Dog kidney Na+,K+-ATPase (Product number: A0142) was obtained from Sigma-Aldrich Japan K.K. (Tokyo, Japan), and pcDNA3 vector from Invitrogen Co. (San Diego, CA, U.S.A.). All other reagents were of molecular biology or analytical grade.
Preparation of hog gastric vesicles
Hog gastric vesicles were prepared from mucosa in the fundic region of hog stomachs as described previously (Asano et al., 1989; Takeguchi et al., 1983). Brifely, hog gastric mucosa was homogenized in a buffer solution containing 250 mM sucrose, 1 mM EGTA and 5 mM Tris-HCl (pH 7.4). The homogenate was centrifuged at 20,000×g for 30 min at 4°C, and the supernatant was re-centrifuged at 78,000×g for 30 min at 4°C. The pellet was suspended in the above solution, and centrifuged at 132,000×g for 1 h at 4°C through 250 mM sucrose layered on 250 mM sucrose containing 7% (w v−1) Ficoll. The membrane fraction containing vesicles fractionated at the Ficoll interface was collected and called ‘gastric vesicles'. When indicated, permeabilized leaky vesicles were prepared as following. Vesicles were diluted with 10 volumes of pure water, followed by immediate freezing in liquid nitrogen. Then, the vesicles were lyophilized and resuspended with the original volume of pure water.
Hog gastric vesicles used were originated from intracellular tubulovesicles (tubulocisternal network) and almost all of them have been well established to be ‘inside-out' and tightly sealed by previous studies (Forte et al., 1980; Morii et al., 1984; Ogata & Yamasaki, 2000; Saccomani et al., 1977; Sachs et al., 1976; Wolosin & Forte, 1981). The cytosolic domains of H+,K+-ATPase including the ATP-binding site are present on the external side of the vesicles, and the addition of ATP into the vesicle solution induces H+ uptake into the vesicle interior and K+ release from vesicles (Asano et al., 1992; Forte et al., 1980; Lee et al., 1974; Sachs et al., 1976). Ion permeability in isolated gastric vesicles have been shown to be low by the measurement of solute influx (Rabon et al., 1980; Takeguchi et al., 1983; Wolosin & Forte, 1981). The addition of valinomycin (K+ ionophore) or gramicidin (H+/K+ ionophore) to the gastric vesicle solution increases KCl influx into vesicles, resulting in 4 – 10 fold increase of H+,K+-ATPase activity (Asano et al., 1992; Forte et al., 1980; Saccomani et al., 1977; Sachs et al., 1976), because the intravesicular K+ stimulates the ATPase activity. On the other hand, H+,K+-ATPase in permeabilized leaky gastric vesicles shows high levels of the activity without addition of the ionophores (Briving et al., 1988; Keeling et al., 1988). Furthermore, Briving et al. (1988) and Keeling et al. (1988) showed that N-methylated SCH 28080, which is an analogue of SCH 28080 with a permanent cation, was able to permeate the leaky vesicles but not normal tight vesicles. Thus, lyophilized vesicles are permeable to small molecules with permanent cations as well as ions.
Cell culture, transfection and preparation of HEK-293 membrane fraction
Cell culture of HEK-293 was carried out as described previously (Asano et al., 1996). H+,K+-ATPase α- and β-subunit cDNAs were prepared from rabbit gastric mucosa, and cloned in pcDNA3 vector as described elsewhere (Asano et al., 1996). Transfection was carried out in subconfluent HEK-293 cells with H+,K+-ATPase α- and β-subunit cDNAs (10 μg each) using the calcium phosphate method (Asano et al., 1996). Two days after the transfection, cells were harvested, and membrane fractions of the cells were prepared according to the method of Asano et al. (1996). In brief, cells were washed with phosphate-buffered saline and incubated in low ionic salt buffer (0.5 mM MgCl2 and 10 mM Tris-HCl, pH 7.4) for 10 min on ice. Phenylmethylsulphonyl fluoride (1 mM) and aprotinin (0.09 unit ml−1) were added to the cell suspension. The cells were homogenized with 25 strokes in a Dounce homogenizer, and the homogenate was diluted with equal volume of a solution containing 500 mM sucrose and 10 mM Tris-HCl (pH 7.4). The cell suspension was centrifuged at 800×g for 10 min, and the supernatant was centrifuged at 100,000×g for 90 min, and the pellet (HEK membrane fraction) was suspended in a solution containing 250 mM sucrose and 5 mM Tris-HCl (pH 7.4). The ATPase activity of the membrane fraction was not elevated by the addition of gramicidin, indicating that the membrane was leaky (Asano et al., 1996).
Assay of protein
Protein was measured using the BCA Protein Assay Kit (Pierce, Rockford, IL, U.S.A.) with bovine serum albumin as standard.
Incubation of vesicles and membrane fractions with cibenzoline or N-methylated SCH 28080
Cibenzoline at final concentrations of 1 – 1,000 μM or N-methylated SCH 28080 at final concentrations of 0.01 – 300 μM was dissolved in each reaction buffer. Unless noted otherwise, vesicles or HEK membrane fractions were preincubated with each drug for 10 min at 37°C in the absence of ATP and Mg2+. Then, ATP and Mg2+ were added to start the enzymatic reaction. An equal volume of reaction buffer without each drug was used as the control.
Measurement of H+,K+-ATPase activity
H+,K+-ATPase activity of normal tight or permeabilized leaky vesicles was measured in a 1 ml solution containing 5 μg of each vesicles, 3 mM MgSO4, 3 mM ATP and 40 mM Tris-HCl (pH 6.4 – 8.0) in the presence or absence of KCl (0.5 – 15 mM). With the normal tight vesicles, to increase K+ permeability of the vesicle membrane, 10 μg of valinomycin was added to the reaction solution. When leaky HEK membrane fractions were used, H+,K+-ATPase activity was measured in a 1 ml solution containing 50 μg of the membrane fractions (mM): MgSO4 3, ATP 1, NaN3 5, ouabain 1 and Tris-HCl 40 (pH 7.4) in the presence or absence of 15 mM KCl. After incubation for 10 min (30 min for membrane fractions) at 37°C, the reaction was terminated by the addition of ice-cold stop solution (12% perchloric acid and 3.6% ammonium molybdate). Inorganic phosphate released was measured by the method of Yoda & Hokin (1970). The K+-dependent ATPase activity was calculated as the difference between activities in the presence and absence of KCl, and defined as H+,K+-ATPase activity (Asano et al., 1989; 1996).
Measurement of Na+,K+-ATPase activity
Na+,K+-ATPase activity was measured in a 1 ml of solution containing 0.03 units of dog kidney Na+,K+-ATPase (mM), NaCl 120, KCl 15, MgSO4 3, ATP 3 and Tris-HCl 40 (pH 7.4) in the presence or absence of 100 μM ouabain. After incubation for 10 min at 37°C, the reaction was terminated by the addition of ice-cold stop solution, and inorganic phosphate released was measured as described above. The Na+,K+-ATPase activity was calculated as the difference between the activities in the presence and absence of ouabain.
Proton uptake
Proton uptake into normal gastric vesicles was monitored by measuring the quenching of a fluorescence dye, acridine orange (Takeguchi & Yamazaki, 1986). Gastric vesicles were incubated in a 1 ml solution containing 150 mM KCl, 2 mM MgCl2, 4 μM acridine orange, 10 μg of valinomycin and 1 mM Pipes [piperazine-N,N′-bis(2-ethanesulphonic acid)]-NaOH (pH 7.4) at 25°C. Proton uptake was started by the addition of 0.3 mM ATP. When time-course of inhibition of proton uptake by cibenzoline was evaluated, the drug was added 15 s after the ATP addition. Fluorescence of acridine orange was measured with excitation and emission wavelengths of 493 and 530 nm, respectively. Activity of proton uptake was taken as the initial rate of acridine orange quenching.
Results
Effects of cibenzoline on H+,K+-ATPase activity
Cibenzoline was examined for its ability to inhibit gastric H+,K+-ATPase activity in two different in vitro systems. With permeabilized leaky hog gastric vesicles, the value of H+,K+-ATPase activity in the absence of inhibitors was about 130 μmol Pi (mg protein)−1 h−1. Although low concentrations of cibenzoline (0.1 – 10 μM) did not inhibit the H+,K+-ATPase activity of the vesicles, higher concentrations of the drug markedly inhibited the H+,K+-ATPase activity in a concentration-dependent manner, with the IC50 value being 201 μM (Figure 2). To clarify the effective concentration range of cibenzoline under other in vitro conditions, we investigated effects of cibenzoline on H+,K+-ATPase activities of the membrane fraction obtained from HEK-293 cells expressing rabbit gastric H+,K+-ATPase α- and β-subunits. This HEK membrane was leaky. In this recombinant in vitro system, the value of H+,K+-ATPase activity in the absence of inhibitors was about 1.1 μmol Pi (mg protein)−1 h−1. On the other hand, the enzyme activity of mock-transfected cells was very low (0.06 μmol Pi (mg protein)−1 h−1) (Asano et al., 1996). Cibenzoline at concentrations from 30 to 1,000 μM inhibited the H+,K+-ATPase activity of the HEK membrane fraction in a concentration-dependent manner, with its IC50 value being 183 μM (Figure 2).
Figure 2.
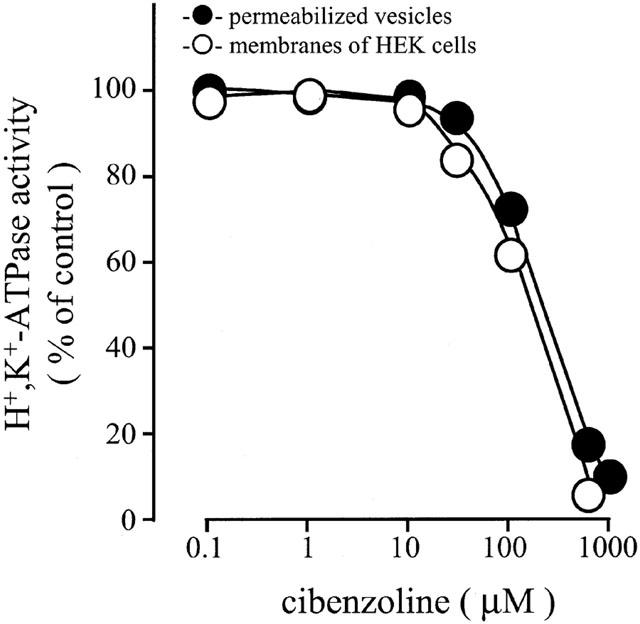
Effects of cibenzoline on the activities of H+,K+-ATPase from permeabilized leaky hog gastric vesicles and membrane fractions of HEK-293 cells co-transfected with rabbit gastric H+,K+-ATPase α- and β-subunit cDNAs. The HEK membrane was leaky. Each enzyme preparation was preincubated with cibenzoline for 10 min, and the K+-dependent ATPase activity was measured. Data show means±s.e.m. of 3 – 4 different experiments.
Effects of cibenzoline on Na+,K+-ATPase activity
H+,K+-ATPase and Na+,K+-ATPase belong to the same family of P-type cation-transporting ATPases. To study the specificity of cibenzoline among the P-type cation-transporting ATPases, effects of this drug on Na+,K+-ATPase activity were studied. In dog kidney membrane preparation, the value of Na+,K+-ATPase activity in the absence of inhibitors was about 180 μmol Pi (mg protein)−1 h−1. Cibenzoline at concentrations from 0.1 – 1,000 μM had no effect on the Na+,K+-ATPase activity, indicating that this drug has specificity to gastric H+,K+-ATPase (Figure 3).
Figure 3.
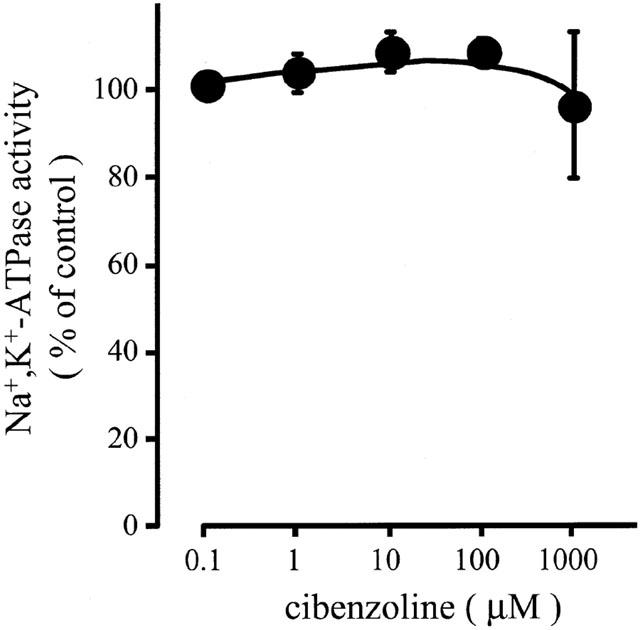
Effects of cibenzoline on the activity of Na+,K+-ATPase from dog kidney. The membrane fraction of dog kidney was preincubated with cibenzoline for 10 min, and the Na+ and K+-dependent ATPase activity was measured. Data show means±s.e.m of four different experiments.
Effects of K+ concentrations and pHs on inhibition of H+,K+-ATPase activity by cibenzoline
To study the inhibition mechanism of H+,K+-ATPase activity of permeabilized leaky gastric vesicles by cibenzoline, we measured H+,K+-ATPase activity as a function of the K+ concentration in the presence of 10, 50 and 100 μM cibenzoline. Inhibition of H+,K+-ATPase activity by cibenzoline was attenuated with the addition of K+ in a concentration-dependent manner. From Lineweaver-Burk plot between K+-dependent ATPase activity and K+ concentration, cibenzoline (0, 10, 50 and 100 μM) was shown to elevate the Km value for K+ (0.20, 0.54, 2.84 and 6.57 mM, respectively) without affecting Vmax (171, 157, 161 and 166 μmol Pi (mg protein)−1 h−1, respectively), and the calculated Ki value for cibenzoline was 807 μM. These data indicate that cibenzoline is a competitive inhibitor with respect to K+ (Figure 4).
Figure 4.
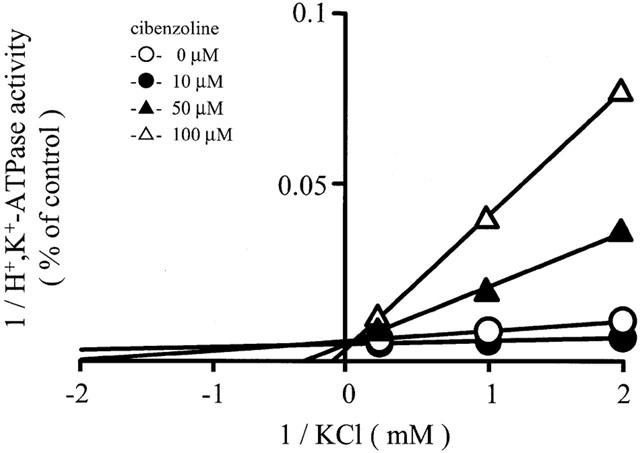
Lineweaver-Burk plots between H+,K+-ATPase activity of permeabilized leaky hog gastric vesicles and KCl concentration (0.5 – 5 mM) in the absence or presence of cibenzoline (10, 50 and 100 μM). Vesicles were preincubated with cibenzoline for 10 min in the presence of KCl, and the K+dependent ATPase activity was measured. Data show means of 4 – 5 different experiments.
Next, effects of medium pH on the inhibition of H+,K+-ATPase activity of leaky gastric vesicles by cibenzoline were studied. H+,K+-ATPase activities in the absence of cibenzoline at pH 6.4, 7.4 and 8.0 were about 90, 130 and 60 μmol Pi (mg protein)−1 h−1, respectively. As shown in Figure 5, the inhibition by cibenzoline did not depend on these pHs of the medium.
Figure 5.
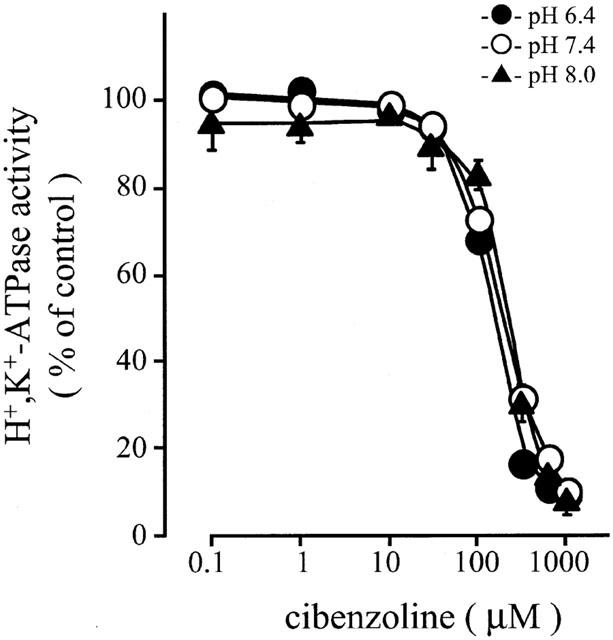
Effects of pH on inhibition of H+,K+-ATPase activity of permeabilized leaky hog gastric vesicles by cibenzoline. Vesicles were preincubated with cibenzoline for 10 min at various pHs (6.4 – 8.0), and the K+-dependent ATPase activity was measured. Data show means±s.e.m. of 3 – 6 different experiments.
Identification of binding site of cibenzoline
To study whether the binding site of cibenzoline is located on the inner or outer surface of gastric vesicles, we used two kinds of vesicle preparations. One is normal tight vesicles. The other is permeabilized leaky vesicles which have high permeability to ion (Briving et al., 1988; Keeling et al., 1988). Since the pKa value for imidazoline ring and distribution coefficient (n-octanol: water−1) of cibenzoline are reported to be 10.6 and 0.126, respectively, the drug exists as a charged form at neutral pH and is difficult to permeate the membrane (Ishida-Takahashi et al., 1996). Here, these vesicles were preincubated in a test solution for 10 min and incubated for another 10 min for the measurement of the H+,K+-ATPase activity. If the binding site of the drug were located on the inner surface of vesicles, the inhibitory effect of the drug in tight vesicles would be smaller than that in leaky vesicles because the drug is difficult to permeate the membrane. On the other hand, if the binding site of cibenzoline were located on the outer surface of vesicles, inhibitory effects of the drug would be similar in both tight and leaky vesicles. The values of H+,K+-ATPase activities in the absence of inhibitors were about 40 and 130 μmol Pi (mg protein)−1 h−1 in the tight and leaky vesicles, respectively. With tight vesicles, cibenzoline inhibited H+,K+-ATPase activity, with its IC50 value being 230 μM. A comparable inhibitory effect of cibenzoline was observed with leaky vesicles (IC50=201 μM) (Figure 6A). These findings indicate that the binding site of cibenzoline is located on the outer surface of vesicles (on the cytoplasmic side).
Figure 6.

Inhibition of H+,K+-ATPase activity by (A) cibenzoline and (B) N-methylated SCH 28080 (SCH 28080-CH3) in permeabilized leaky and normal tight hog gastric vesicles. Vesicles were preincubated with each drug for 10 min, and the K+-dependent ATPase activity was measured. Data show means±s.e.m. of 3 – 4 different experiments.
N-methylated SCH 28080, an analogue of SCH 28080, has a permanent cation and binds to the inner surface of vesicles as in the case of SCH 28080 (Briving et al., 1988; Keeling et al., 1988). Here, we checked effects of N-methylated SCH 28080 on the H+,K+-ATPase activity using the present leaky and tight gastric vesicles. This drug markedly inhibited the H+,K+-ATPase activity of leaky vesicles, with its IC50 value being 0.73 μM. On the other hand, inhibition potency of this drug was dramatically attenuated in the tight vesicles (IC50=40 μM) (Figure 6B). In contrast to cibenzoline which exists as a cationic form at neutral pHs (Figure 6A), these results confirm that binding site of N-methylated SCH 28080 is located on the inner surface of vesicles (on the luminal side).
Effects of cibenzoline on proton uptake
Proton uptake into tight gastric vesicles was monitored by measuring the quenching of acridine orange fluorescence. When ATP was added to the reaction medium, decrease in the fluorescence intensity was observed (data not shown). Pretreatment with cibenzoline (50 – 1000 μM) inhibited the proton uptake into vesicles in a concentration-dependent manner, with its IC50 value being 143 μM (Figure 7). Moreover, time-course of inhibition of proton uptake by the drug was tested. Proton uptake was inhibited immediately after addition of this drug (500 μM) (data not shown).
Figure 7.
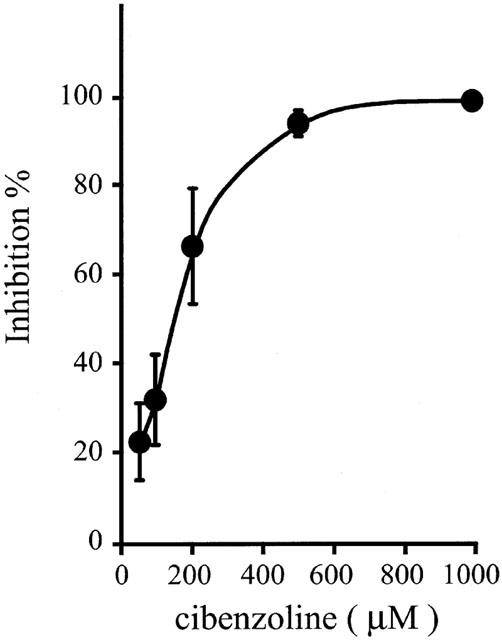
Effects of cibenzoline on proton uptake into normal tight hog gastric vesicles. The vesicles were preincubated with cibenzoline for 10 min, and proton uptake was measured. Data show means±s.e.m. of 3 – 5 different experiments.
Discussion
In the present study, we discovered that cibenzoline inhibits gastric H+,K+-ATPase activity of hog gastric vesicles and the membrane fraction of HEK-293 cells transfected with the H+,K+-ATPase cDNAs. Cibenzoline is clinically used as one of the Class I type antiarrhythmic agents (Harron et al., 1992; Holck & Osterrieder, 1986; Millar & Williams, 1982; Touboul et al., 1986). In whole-cell mode of patch clamp with isolated cardiac myocytes of guinea pig, cibenzoline caused an inhibition of Ca2+ inward current (IC50=14 μM) (Holck & Osterrieder, 1986). In addition, this drug blocked KATP channel in cell-attached mode of patch clamp of rat pancreatic β cells (IC50=1.5 – 5.2 μM) (Horie et al., 1992; Ishida-Takahashi et al., 1996; Kakei et al., 1993). In the present study, IC50 value of cibenzoline for inhibition of gastric H+,K+-ATPase activity and H+ uptake in in vitro systems were approximately 200 μM and 143 μM, respectively. The present difference of IC50 values between H+,K+-ATPase activity and H+ uptake would reflect the different types of observation. Thus, the inhibitory effect of H+,K+-ATPase activity by cibenzoline was 14 and 38 – 133 times smaller than those of Ca2+ inward current and KATP channel, respectively. However, because cibenzoline did not inhibit Na+,K+-ATPase activity (IC50: >1,000 μM), the inhibition of gastric H+,K+-ATPase activity by this drug was specific. In humans, the therapeutic mean maximum plasma concentration was 1.9 μM following the last dose (160 mg t.i.d. for 7 days) (Massarella et al., 1986). It is, therefore, considered that cibenzoline at clinically used doses does not inhibit gastric H+,K+-ATPase. In fact, it has not been reported that such doses of cibenzoline inhibit gastric acid secretion.
Inhibition of H+,K+-ATPase activity by cibenzoline was attenuated with the addition of K+ with permeabilized leaky gastric vesicles, and the Lineweaver-Burk plot of the ATPase activity showed that this drug increased Km value for K+ without affecting Vmax. Therefore, we suggest that cibenzoline is a competitive inhibitor respect to K+ as in the cases of SCH 28080 (Beil et al., 1986; Keeling et al., 1988; Scott et al., 1987; Wallmark et al., 1987) and SK&F96067 (Keeling et al., 1991). The calculated Ki value for cibenzoline was 807 μM, which was much larger than those of SCH 28080 (Ki=0.056 – 0.12 μM) (Scott et al., 1987; Wallmark et al., 1987) and SK&F96067 (Ki=0.39 μM) (Keeling et al., 1991). These hydrophobic compounds of SCH 28080 and SK&F96067 permeate the membrane, accumulate in the acidic compartment as prontonated forms, and bind to the high-affinity K+-binding site on the luminal side of the membrane (Beil et al., 1986; Briving et al., 1988; Keeling et al., 1988; 1991; Wallmark et al., 1987). N-methylated SCH 28080, a permanent cation, was shown to bind to the inner (luminal) surface of vesicles (Briving et al., 1988; Keeling et al., 1988). We found here that the inhibition of H+,K+-ATPase in tight vesicles by N-methylated SCH 28080 (IC50=40 μM) was two orders of magnitude less effective than that in the present leaky vesicles (IC50=0.73 μM), indicating that the inhibition of N-methylated SCH 28080 in tight vesicles was 55 times less effective than that in leaky vesicles. Because cibenzoline exists as a charged form at neutral pH, the drug is difficult to permeate the vesicle membrane (Ishida-Takahashi et al., 1996; Horie et al., 2000). In the present study, the inhibitory effect of H+,K+-ATPase activity by cibenzoline with normal tight gastric vesicles did not significantly differ from that with permeabilized leaky gastric vesicles. Moreover, this drug inhibited proton uptake in tight vesicles soon after addition of the drug. Therefore, it is suggested that the binding site of cibenzoline is on the cytoplasmic side of the membrane (external surface of the vesicles). Ishida-Takahashi et al. (1996) previously showed that the action of cibenzoline to KATP channel in the excised inside-out mode was acute in onset with a small IC50 (0.4 μM) compared with that in the cell-attached mode of patch clamp (IC50=5.2 μM) in rat pancreatic β cells. These results indicate that the binding site of cibenzoline is located on the cytoplasmic side of the cell membrane (Horie et al., 2000).
It has been demonstrated that imidazoline derivatives such as several classical α-adrenoreceptor antagonists, act as stimulators of insulin secretion, and these effects do not result from antagonism of α-adrenoreceptors but from inhibition of KATP channels in the pancreatic β cell (Dunne, 1991; Le Brigand et al., 1999; Proks & Ashcroft, 1997; Plant & Henquin, 1990; Jonas et al., 1992). The KATP channel of pancreatic β cells is a complex of two proteins: a member of the ATP-binding cassette super family, SUR1, and a pore-forming subunit, Kir6.2 (Inagaki et al., 1995). Proks & Ashcroft (1997) indicated that KATP channels blocked by phentolamine, an imidazoline derivative, is mediated by Kir6.2 subunit. Mukai et al. (1998a, 1998b) and Horie et al. (2000) reported that an imidazoline derivative, cibenzoline, inhibits KATP channels by direct binding of this drug to the cytoplasmic side of Kir6.2 subunit. In the present study, we showed that the binding site of cibenzoline is located on the cytoplasmic surface of gastric H+,K+-ATPase. It is interesting that cibenzoline binds to the cytoplasmic surface of both Kir6.2 subunit and H+,K+-ATPase α-subunit. Although the amino acid homology between Kir6.2 (Inagaki et al., 1995) and H+,K+-ATPase α-subunit (Shull & Lingrel, 1986) is extremely low, the three-dimensional structure of the K+-binding site in Kir6.2 subunit is likely to have some extent of similarity with that of the K+ binding site of H+,K+-ATPase α-subunit.
In conclusion, cibenzoline inhibits gastric H+,K+-ATPase by binding to the K+-recognition site of the enzyme from the cytoplasmic side. In our knowledge, this is the first drug that inhibits gastric H+,K+-ATPase with this unique inhibitory action.
Acknowledgments
This work was supported in part by Grants-in-Aid for Scientific Research (to S. Asano and N. Takeguchi) from the Ministry of Education, Science, Sports and Culture of Japan.
Abbreviations
- cibenzoline
(±)-2-(2,2,-diphenylcyclopropyl)-2-imidazoline succinate
- KATP channel
ATP-sensitive K+ channel
- NAIBS
non-adrenergic idazoxan binding site
- Pipes
piperazine-N,N′-bis(2-ethanesulfonic acid)
- SCH 28080
3-(cyanomethyl)-2-methyl-8-(phenylmethoxy)imidazo(1,2a)-pyridine
- N-methylated SCH 28080
3-(cyanomethyl)-2,3-dimethyl-8-(phenylmethoxy) imidazo(1,2a)-pyridine
- SK&F96067
3-butryrl-4-(2-methylphenylamino)-8-methoxyquinoline
- SUR
sulphonylurea receptor
References
- ASANO S., KAMIYA S., TAKEGUCHI N. The energy transduction mechanism is different among P-type ion-transporting ATPases. J. Biol. Chem. 1992;267:6590–6595. [PubMed] [Google Scholar]
- ASANO S., MATSUDA S., HOSHINA S., SAKAMOTO S., TAKEGUCHI N. A chimeric gastric H+,K+-ATPase inhibitable with both ouabain and SCH 28080. J. Biol. Chem. 1999;274:6848–6854. doi: 10.1074/jbc.274.11.6848. [DOI] [PubMed] [Google Scholar]
- ASANO S., TABUCHI Y., TAKEGUCHI N. Monoclonal antibody HK4001 completely inhibits K+-dependent ATP hydrolysis and H+ transport of hog gastric H+,K+-ATPase. J. Biochem. 1989;106:1074–1079. doi: 10.1093/oxfordjournals.jbchem.a122967. [DOI] [PubMed] [Google Scholar]
- ASANO S., TEGA Y., KONISHI K., FUJIOKA M., TAKEGUCHI N. Functional expression of gastric H+,K+-ATPase and site-directed mutagenesis of the putative cation binding site and the catalytic center. J. Biol. Chem. 1996;271:2740–2745. doi: 10.1074/jbc.271.5.2740. [DOI] [PubMed] [Google Scholar]
- BEIL W., HACHBARTH I., SEWING K.F. Mechanism of gastric antisecretory effect of SCH 28080. Br. J. Pharmacol. 1986;88:19–23. doi: 10.1111/j.1476-5381.1986.tb09466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERTRAND G., GROSS R., PETIT P., LOUBATIERES-MARIANI M.M., RIBES G. Evidence for a direct stimulatory effect of cibenzoline on insulin secretion in rats. Eur. J. Pharmacol. 1992;214:159–163. doi: 10.1016/0014-2999(92)90113-i. [DOI] [PubMed] [Google Scholar]
- BRIVING C., ANDERSSON B.M., NORDBERG P., WALLMARK B. Inhibition of gastric H+/K+-ATPase by substituted imidazo[1,2-a]pyridines. Biochim. Biophys. Acta. 1988;946:185–192. doi: 10.1016/0005-2736(88)90391-4. [DOI] [PubMed] [Google Scholar]
- CHUTKOW W.A., SIMON M.C., LE BEAU M.M., BURANT C.F. Cloning, tissue expression, and chromosomal localization of SUR2, the putative drug-binding subunit of cardiac, skeletal muscle, and vascular KATP channels. Diabetes. 1996;45:1439–1445. doi: 10.2337/diab.45.10.1439. [DOI] [PubMed] [Google Scholar]
- DUNNE M.J. Block of ATP-regulated potassium channels by phentolamine and other alpha-adrenoceptor antagonists. Br. J. Pharmacol. 1991;103:1847–1850. doi: 10.1111/j.1476-5381.1991.tb12340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRYKLUND J., GEDDA K., WALLMARK B. Specific labeling of gastric H+,K+-ATPase by omeprazole. Biochem. Pharmacol. 1988;37:2543–2549. doi: 10.1016/0006-2952(88)90244-4. [DOI] [PubMed] [Google Scholar]
- FORTE J.G., MACHEN T.E., OBRINK K.J. Mechanisms of gastric H+ and Cl− transport. Ann. Rev. Physiol. 1980;42:111–126. doi: 10.1146/annurev.ph.42.030180.000551. [DOI] [PubMed] [Google Scholar]
- GACHOT B.A., BEZIER M., CHERRIER J.F., DAUBEZE J. Cibenzoline and hypoglycaemia. Lancet. 1988;2:280. doi: 10.1016/s0140-6736(88)92570-6. [DOI] [PubMed] [Google Scholar]
- HARRON D.W., BROGDEN R.N., FAULDS D., FITTON A. Cibenzoline. A review of its pharmacological properties and therapeutic potential in arrhythmias. Drugs. 1992;43:734–759. doi: 10.2165/00003495-199243050-00008. [DOI] [PubMed] [Google Scholar]
- HOLCK M., OSTERRIEDER W. Inhibition of the myocardial Ca2+ inward current by the class I antiarrhythmic agent, cibenzoline. Br. J. Pharmacol. 1986;87:705–711. doi: 10.1111/j.1476-5381.1986.tb14588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HORIE M., HAYASHI S., YUZUKI Y., SASAYAMA S. Comparative studies of ATP sensitive potassium channels in heart and pancreatic beta cells using Vaughan-Williams class Ia antiarrhythmics. Cardiobasc. Res. 1992;26:1087–1094. doi: 10.1093/cvr/26.11.1087. [DOI] [PubMed] [Google Scholar]
- HORIE M., WATANUKI M., TSUJI K., ISHIDA H., ISHIDA-TAKAHASHI A., YUZUKI Y., SEINO Y., SASAYAMA S. Blockade of cardiac ATP-sensitive K+ channel by cibenzoline targets its pore-forming subunit. J. Cardiovasc. Pharmacol. 2000;35:434–442. doi: 10.1097/00005344-200003000-00014. [DOI] [PubMed] [Google Scholar]
- INAGAKI N., GONOI T., CLEMENT J.P., 4TH, NAMBA N., INAZAWA J., GONZALEZ G., AGUILAR-BRYAN L., SEINO S., BRYAN J. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270:1166–1170. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- ISHIDA-TAKAHASHI A., HORIE M., TSUURA Y., ISHIDA H., AI T., SASAYAMA S. Block of pancreatic ATP-sensitive K+ channels and insulinotrophic action by the antiarrhythmic agent, cibenzoline. Br. J. Pharmacol. 1996;117:1749–1755. doi: 10.1111/j.1476-5381.1996.tb15349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JEANDEL C., PREISS M.A., PIERSON H., PENIN F., CUNY G., BANNWARTH B., NETTER P. Hypoglycaemia induced by cibenzoline. Lancet. 1988;1:1232–1233. doi: 10.1016/s0140-6736(88)92058-2. [DOI] [PubMed] [Google Scholar]
- JONAS J.C., PLANT T.D., HENQUIN J.C. Imidazoline antagonists of alpha 2-adrenoceptors increase insulin release in vitro by inhibiting ATP-sensitive K+ channels in pancreatic beta-cells. Br. J. Pharmacol. 1992;107:8–14. doi: 10.1111/j.1476-5381.1992.tb14456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAKEI M., NAKAZAKI M., KAMISAKI T., NAGAYAMA I., FUKAMACHI Y., TANAKA H. Inhibition of the ATP-sensitive potassium channel by class I antiarrhythmic agent, cibenzoline, in rat pancreatic beta-cells. Br. J. Pharmacol. 1993;109:1226–1231. doi: 10.1111/j.1476-5381.1993.tb13753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KEELING D.J., LAING S.M., SENN-BILFINGER J. SCH 28080 is a luminally acting, K+-site inhibitor of the gastric (H++K+)-ATPase. Biochem. Pharmacol. 1988;37:2231–2236. doi: 10.1016/0006-2952(88)90586-2. [DOI] [PubMed] [Google Scholar]
- KEELING D.J., MALCOLM R.C., KAING S.M., LEACH C.A., IFE R.J. SK&F96067 is a reversible luminally acting inhibitor of the gastric H+,K+-ATPase. Biochem. Pharmacol. 1991;42:123–130. doi: 10.1016/0006-2952(91)90690-7. [DOI] [PubMed] [Google Scholar]
- LE BRIGAND L., VIRSOLVY A., MANECHEZ D., GODFROID J.J., GUARDIOLA-LEMAITRE B., GRIBBLE F.M., ASHCROFT F.M., BATAILLE D. In vitro mechanism of action on insulin release of S-22068, a new putative antidiabetic compound. Br. J. Pharmacol. 1999;128:1021–1026. doi: 10.1038/sj.bjp.0702883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE J., SIMPSON G., SCHOLES P. An ATPase from dog gastric mucosa: changes of outer pH in suspensions of membrane vesicles accompanying ATP hydrolysis. Biochem. Biophys. Res. Commun. 1974;60:825–832. doi: 10.1016/0006-291x(74)90315-5. [DOI] [PubMed] [Google Scholar]
- MASSARELLA J.W., KHOO K.C., SZUNA A.J., SANDOR D.A., MORGANROTH J., AOGAICHI K. Pharmacokinetics of cibenzoline after single and repetitive dosing in healthy volunteers. J. Clin. Pharmacol. 1986;26:125–130. doi: 10.1002/j.1552-4604.1986.tb02920.x. [DOI] [PubMed] [Google Scholar]
- MILLAR J.S., WILLIAMS E.M.V. Effects on rabbit nodal, atrial ventricular and purkinje cell potentials of a new antiarrhythmic drug, cibenzoline, which protects against action potential shortening in hypoxia. Br. J. Pharmacol. 1982;75:469–478. doi: 10.1111/j.1476-5381.1982.tb09163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORII M., ISHIMURA N., TAKEGUCHI N. Quasi-elastic light-scattering studies of conformational states of the H+,K+-ATPase. Intervesicular aggregation of gastric vesicles by disulfide cross-linking. Biochemistry. 1984;23:6816–6821. doi: 10.1021/bi00321a083. [DOI] [PubMed] [Google Scholar]
- MORII M., TAKATA H., FUJISAKI H., TAKEGUCHI N. The potency of substituted benzimidazoles such as E3810, omeprazole, Ro18-5364 to inhibit gastric H+,K+-ATPase is correlated with the rate of acid-activation of the inhibitor. Biochem. Pharmacol. 1990;39:661–667. doi: 10.1016/0006-2952(90)90143-9. [DOI] [PubMed] [Google Scholar]
- MUKAI E., ISHIDA H., HORIE M., NOMA A., SEINO Y., TAKANO M. The antiarrhythmic agent cibenzoline inhibits KATP channels by binding to Kir6.2. Biochem. Biophys. Res. Commun. 1998a;251:477–481. doi: 10.1006/bbrc.1998.9492. [DOI] [PubMed] [Google Scholar]
- MUKAI E., ISHIDA H., KATO S., TSUURA Y., FUJIMOTO S., ISHIDA-TAKAHASHI A., HORIE M., TSUDA K., SEINO Y. Metabolic inhibition impairs ATP-sensitive K+channel block by sulfonylurea in pancreatic beta-cells. Am. J. Physiol. 1998b;274:E38–E44. doi: 10.1152/ajpendo.1998.274.1.E38. [DOI] [PubMed] [Google Scholar]
- OGATA T., YAMASAKI Y. Scanning EM of resting gastric parietal cells reveals a network of cytoplasmic tubules and cisternae connected to the intracellular canaliculus. Anat. Rec. 2000;258:15–24. doi: 10.1002/(SICI)1097-0185(20000101)258:1<15::AID-AR2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- PLANT T.D., HENQUIN J.C. Phentolamine and yohimbine inhibit ATP-sensitive K+ channels in mouse pancreatic beta-cells. Br. J. Pharmacol. 1990;101:115–120. doi: 10.1111/j.1476-5381.1990.tb12099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PROKS P., ASHCROFT F.M. Phentolamine block of KATP channels is mediated by Kir6.2. Proc. Natl. Acad. Sci. USA. 1997;94:11716–11720. doi: 10.1073/pnas.94.21.11716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RABON E., TAKEGUCHI N., SACHS G. Water and salt permeability of gastric vesicles. J. Membr. Biol. 1980;53:109–117. doi: 10.1007/BF01870579. [DOI] [PubMed] [Google Scholar]
- RICHARDSON P., HAWKEY C.J., STACK W.A. Proton pump inhibitors. Pharmacology and rationale for use in gastrointestinal disorders. Drugs. 1998;56:307–335. doi: 10.2165/00003495-199856030-00002. [DOI] [PubMed] [Google Scholar]
- SACHS G., CHANG H.J., RABON E., SCHACKMAN R., LEWIN M., SACCOMANI G. A nonelectrogenic H pump in plasma membranes of hog stomach. J. Biol. Chem. 1976;251:7690–7698. [PubMed] [Google Scholar]
- SACHS G., SHIN J.M., BRIVING C., WALLMARK B., HERSEY S. The pharmacology of the gastric acid pump: the H+,K+-ATPase. Annu. Rev. Pharmacol. Toxicol. 1995;35:277–305. doi: 10.1146/annurev.pa.35.040195.001425. [DOI] [PubMed] [Google Scholar]
- SACCOMANI G., STEWART H.B., SHAW D., LEWIN M., SACHS G. Characterization of gastric mucosal membranes. IV. Fractionation and purification of K+-ATPase-containing vesicles by zonal centrifugation and free-flow electrophoresis technique. Biochim. Biophys. Acta. 1977;465:311–330. doi: 10.1016/0005-2736(77)90081-5. [DOI] [PubMed] [Google Scholar]
- SCOTT C.K., SUNDELL E., CASTROVILLY L. Studies on the mechanism of action of the gastric microsomal (H++K+)-ATPase inhibitors SCH 32651 and SCH 28080. Biochem. Pharmacol. 1987;36:97–104. doi: 10.1016/0006-2952(87)90386-8. [DOI] [PubMed] [Google Scholar]
- SHULL G.E., LINGREL J.B. Molecular cloning of the rat stomach (H++K+)-ATPase. J. Biol. Chem. 1986;261:16788–16791. [PubMed] [Google Scholar]
- TAKEGUCHI N., JOSHIMA R., INOUE Y., KASHIWAGURA T., MORII M. Effects of Cu2+-o-phenanthroline on gastric (H++K+)-ATPase. Evidence for opening of a closed anion conductance by S-S cross-linking. J. Biol. Chem. 1983;258:3094–3098. [PubMed] [Google Scholar]
- TAKEGUCHI N., YAMAZAKI Y. Disulfide cross-linking ATPase opens Cl− conductance, triggering proton uptake in gastric vesicles. J. Biol. Chem. 1986;261:2560–2566. [PubMed] [Google Scholar]
- TOUBOUL P., ATALLAH G., KIRKORIAN G., DE ZULOAGA C., DUFOUR A., AYMARD M.F., LAVAUD P., MOLEUR P. Electro physiologic effects of cibenzoline in humans related to dose and plasma concentration. Am. Heart J. 1986;112:333–339. doi: 10.1016/0002-8703(86)90271-1. [DOI] [PubMed] [Google Scholar]
- WALLMARK B., LARSSON H., HUMBLE L. The relationship between gastric acid secretion and gastric H+,K+-ATPase activity. J. Biol. Chem. 1985;260:13681–13684. [PubMed] [Google Scholar]
- WALLMARK B., BRIVING C., FRYKLUND J., MUNSON K., JACKSON R., MEDLEIN J., RABON E., SACHS G. Inhibition of gastric H+,K+-ATPase and acid secretion by SCH 28080, a substituted pyridyl(1,2a)imidazole. J. Biol. Chem. 1987;262:2077–2084. [PubMed] [Google Scholar]
- WOLOSIN J.M., FORTE J.G. Changes in the membrane environment of the (K++H+)-ATPase following stimulation of the gastric oxyntic cell. J. Biol. Chem. 1981;256:3149–3152. [PubMed] [Google Scholar]
- YODA A., HOKIN L.E. On the reversibility of binding of cardiotonic steroids to a partially purified (Na+K)-activated adenosinetriphosphatase from beef brain. Biochem. Biophys. Res. Commun. 1970;40:880–886. doi: 10.1016/0006-291x(70)90985-x. [DOI] [PubMed] [Google Scholar]