

Abstract
Scald injury in Sv129+C57BL/6 mice induced a temperature and time dependent oedema formation as calculated by the extravascular accumulation of [125I]-albumin. Oedema formation was suppressed in NK1 knockout mice compared to wildtypes at 10 (P<0.01) and 30 min (P<0.001). However, at 60 min a similar degree of extravasation was observed in the two groups.
Kinin B1 (des-Arg10 Hoe 140; 1 μmol kg−1) and B2 (Hoe 140; 100 nmol kg−1) antagonists caused an inhibition of oedema in wildtype mice at 10 and 30 min (P<0.001), but not at 60 min or at 30 min in NK1 receptor knockout mice.
The inhibition of thermic oedema by des-Arg10 Hoe 140 was reversed by des-Arg9 bradykinin (0.1 μmol kg−1; P<0.01) and also observed with a second B1 receptor antagonist (des-Arg9 Leu8 bradykinin; 3 μmol kg−1; P<0.01). Furthermore des-Arg10 Hoe 140 had no effect on capsaicin (200 μg ear−1) ear oedema, but this was significantly reduced with Hoe 140 (P<0.05).
Scalding induced a large neutrophil accumulation at 4 h, as assessed by myeloperoxidase assay (P<0.001). This was not suppressed by NK1 receptor deletion or kinin antagonists.
These results confirm an essential role for the NK1 receptor in mediating the early, but not the delayed phase of oedema formation or neutrophil accumulation in response to scalding. The results also demonstrate a pivotal link between the kinins and sensory nerves in the microvascular response to burn injury, and for the first time show a rapid involvement of the B1 receptor in murine skin.
Keywords: Thermal injury, NK1 receptor, kinins, oedema, neutrophils
Introduction
Swelling, due to the formation of tissue oedema, is observed soon after noxious heat stimulation of the skin, for example after accidental scalding or burn injury. Several lines of evidence support the suggestion that tachykinin NK1 receptors play a major role in the early acute oedema formation. NK1 receptor antagonists and capsaicin depletion of sensory nerves substantially inhibit the oedema formation in a variety of noxious heat models in the rat (Saria, 1984; Jonsson et al., 1986; Lofgren et al., 1999). However, our recent studies suggest that NK1 receptor antagonists attenuate the early, but not the later stages of the ongoing inflammatory swelling (Siney & Brain, 1996) and have little effect on the subsequent neutrophil accumulation (Pinter et al., 1999).
The mouse is now routinely used in animal models of inflammation because of the comparative ease of genetic manipulation in this species. Tachykinin NK1 receptor knockout mice have been developed, and study of these animals has confirmed the importance of NK1 receptors in inflammation (Bozic et al., 1996; Ahluwalia et al., 1998). In our laboratory, using wildtype and NK1 receptor knockout mice, we have confirmed that substance P increases microvascular permeability via the NK1 receptor (Cao et al., 1999). However, interestingly neither substance P nor selective NK1 agonists were able to induce neutrophil accumulation in the naïve untreated normal skin of the wildtype mouse (Cao et al., 2000). By comparison, we found that if microvascular inflammation was induced in skin, then neutrophil accumulation was NK1 receptor dependent, as observed in experiments using NK1 antagonists or NK1 knockout mice (Cao et al., 2000), indicating that the NK1 receptor contributes to neutrophil accumulation, in addition to oedema formation in the inflamed microvasculature.
Bradykinin has been suggested to be involved in the inflammatory oedema observed in response to noxious heat stimulation in the rat as determined by the study of normal and kininogen-deficient rats (Yonehara et al., 1995). Furthermore in this study, it was suggested that kinin generation was linked to the release of substance P from sensory nerves. We have also observed a link between the kinin and tachykinin system in our NK1 knockout mice, in that both B1 and B2 receptor antagonists reduced neutrophil accumulation in response to carrageenan in wildtype mice. However in NK1 receptor knockout mice, the kinin antagonists were without effect (Cao et al., 2000). Indeed, evidence for a link between the tachykinin and kinin receptors has been produced in several models of inflammation (Ricciardolo et al., 1994; Lindstrom & Andersson, 1997; Schuligoi et al., 1998; Ferreira et al., 2000). In addition, studies in the microvasculature of neutral endopeptidase knockout mice have revealed that spontaneous plasma leakage is inhibited by treatment with NK1 and kinin B2 antagonists (Lu et al., 1997).
Thus, NK1 receptors contribute to the vascular and cellular phases of inflammation and have differential effects in inflamed skin and naïve skin. In addition, a link between the inflammatory activities of the tachykinin NK1 and kinin, B1 and B2 receptors is suggested from a range of studies. We therefore decided to examine the contribution that these receptors play in mediating the vascular and cellular responses to noxious heat using NK1 receptor knockout mice and selective kinin antagonists. In addition, the potential for interaction between the tachykinin NK1 and kinin B1 receptors in thermal injury was investigated.
Methods
Induction of thermal injury
All experiments were performed in accordance with the Animal (Scientific Procedures) Act 1986. Male/female Sv129+C57BL/6 wildtype and NK1 knockout mice (20 – 30 g) were generated at the Perlmutter Laboratory, Children's Hospital, Boston, U.S.A. (Bozic et al., 1996) and bred at King's College London. The mice were housed in a light (07:00 – 19:00) and temperature controlled (18 – 22°C) environment, and food and water were provided for consumption ad libitum. The animals were anaesthetized with urethane (2.5 μg g−1; i.p.; Sigma, U.K.) and the depth of anaesthesia was assessed by the pedal reflex with maintenance doses administered as required. Initially, a local cutaneous thermal injury was induced by submersion of one ear in ultrapure water heated to 40 – 60°C for 10 s to obtain a temperature-response curve. For subsequent experiments, a temperature of 55°C was selected as this was observed to induce a highly significant, but non-maximal increase in plasma extravasation. The control, contralateral ear was submersed in ultrapure water at room temperature for the same period. Ears selected for thermal injury or collection of control tissue were alternated to remove any bias relating to skin heterogeneity (e.g. thickness, vascular density). At 10 min – 4 h post-burn the animals were killed by cervical dislocation and the ears removed for analysis and weighing (wet weight).
Capsaicin-induced ear oedema
Animals were prepared as described above. Capsaicin (Sigma, U.K.) was dissolved in ethanol to give a stock solution of 10 μg μl−1. Ten μl of stock solution was topically applied to each side of the ear selected for capsaicin treatment to give a total dose of 200 μg ear−1. The control, contralateral ear was treated with ethanol in the same manner. Ears selected for capsaicin or vehicle treatment were alternated from animal to animal. At 30 min after application of capsaicin or vehicle, the mice were killed by cervical dislocation and the ears removed for analysis.
Measurement of plasma extravasation
Plasma extravasation was measured as previously described (Cao et al., 1999). Briefly, the mice received [125I]-albumin (45 kBq; ICN Biochemicals, U.S.A.) i.v. at least 5 min prior to the induction of thermal injury or capsaicin treatment. Microvascular extravasation in the ear following thermal injury or capsaicin treatment was calculated by comparison to a known volume of blood plasma which was collected via cardiac puncture immediately prior to cervical dislocation.
Measurement of neutrophil accumulation
The ears were snap frozen and stored at −70°C prior to homogenization and subsequent analysis. The tissues were homogenized in ice cold phosphate buffer (pH 6.0) containing 0.5% hexadecyltrimethylammonium bromide (HTAB). The resulting homogenate was centrifuged at 25,000×g for 25 min at 4°C. Neutrophil accumulation in tissues was determined via myeloperoxidase (MPO) activity (Schierwagen et al., 1990; Cao et al., 2000) which was calculated via the hydrogen peroxide (H2O2) dependent oxidation of 3,3′,5,5′-tetramethylbenzidine (TMB). The assay was performed in a 96 well microtitre plate. Tissue homogenates were diluted 1 : 2 in phosphate buffer (pH 6.0) to give a total volume of 50 μl. The resultant mixture was incubated with 100 μl of ‘K-Blue' (a commercial preparation of H2O2 and TMB; Bionostics, U.K.) for 60 min at room temperature. Following incubation, the absorbance of the plate was recorded at 620 nm. The neutrophil content in the homogenates was calculated by comparing their absorbance with that of a standardized preparation of mouse neutrophils. All samples were assayed in duplicate and the data expressed as neutrophils g tissue−1 (106).
Preparation of mouse neutrophil standards
Mixed leukocytes were collected from the peritoneal cavity of Sv129+C57BL/6 mice by inducing a sub-acute peritonitis (Moroney et al., 1988; Cao et al., 2000). For this, 6% oyster glycogen (Sigma, U.K.) was dissolved in 1 ml isotonic saline and injected i.p. After 16 h, the mice were killed by cervical dislocation and 5 ml ice-cold modified HBSS (Hank's buffered salt solution) free of Ca2+ and Mg2+ (Sigma, U.K.) was injected into the peritoneal cavity. After 1 min of massage, the peritoneal fluid was aspirated and centrifuged at 4°C for 10 min at 400×g. The supernatant was removed and erythrocytes lysed by brief exposure to hypotonic saline (0.2% sodium chloride). After tonicity was restored, the preparation was centrifuged in a similar manner and the supernatant discarded. The pellet was dissolved in 2 ml HBSS containing 1.26 mM Ca2+ and 0.9 mM Mg2+. A sample of the resultant mixture was diluted 1 : 10 with complete HBSS and exposed to 0.4% Trypan blue prior to haemocytometry to determine the total leukocyte count in the preparation. For determination of the number of neutrophils present, cell smears were prepared by cytospin (Shandon Scientific, U.K.) and slides were fixed in acetone, air dried and exposed to Mayer's haematoxylin (BDH, U.K.) for 10 min. The slides were washed with tap water and placed in 1% chromotrope 2R, 1% phenol. Standard mouse neutrophil preparations (0.078 – 2.5×106 cells ml−1) were aliquoted and stored at −70°C prior to use.
Drug treatments
The tachykinin NK1 antagonist, SR140333 ((S)1-(2-[3-(3,4-dichlorophenyl)-1-(3-isopropoxyphenylacetyl) piperidin-3-yl]ethyl)-4-phenyl-1-azoniabicyclo[2.2.2]octane chloride) was a gift from Dr X. Emonds-Alt (Sanofi Recherche, France). The drug was dissolved in a minimal volume of ethanol and diluted to a final volume with physiological saline. SR140333 was administered at a dose of 480 nmol kg−1. The kinin B1 receptor antagonists des-Arg10 Hoe 140 and des-Arg9 Leu8 bradykinin were purchased from Sigma, U.K. and Bachem, U.K. respectively. Both drugs were dissolved in physiological saline. des-Arg10 Hoe 140 was administered at a dose of 1 μmol kg−1 and des-Arg9 Leu8 bradykinin at 3 μmol kg−1. The B2 receptor antagonist, Hoe 140 was obtained from Sigma, U.K., dissolved in physiological saline and administered at a dose of 100 nmol kg−1. The B1 receptor agonist, des-Arg9 bradykinin was purchased from Bachem, U.K., dissolved in physiological saline and administered at a dose of 0.1 μmol kg−1. All drugs were administered to mice as i.v. pretreatments, 5 min prior to the induction of thermal injury or the topical application of capsaicin.
Statistical analysis
All data are presented as mean±s.e.mean and was analysed by ANOVA and the Bonferroni's test of multiple comparisons where appropriate. Plasma extravasation is expressed as ll g tissue-1 and neutrophil accumulation as cells g tissue−1. P<0.05 was taken as being significant.
Results
Effect of scald injury on plasma extravasation: role of NK1 receptors
Scalding of the ears of wildtype mice induced plasma extravasation which was seen to be both dependent on the temperature of burn and time post-burn. Thermic oedema was raised compared to controls at 45°C and became significantly different at 55 (Figure 1; P<0.001) and 60°C (Figure 1; P<0.001). Plasma extravasation did not change in the contralateral, control ears at any of the temperatures (Figure 1) or timepoints investigated (Figure 2). Oedema formation was rapid in wildtype animals and was significantly greater than controls at 10 min post-burn (Figure 2; P<0.05). Oedema formation continued to increase at 30 (Figure 2; P<0.001) and 60 min (Figure 2; P<0.001) post-burn. In contrast, scald oedema formation was markedly suppressed in NK1 knockout mice compared to wildtype animals at 10 (Figure 2; P<0.05) and 30 min (Figure 2; P<0.01) post-burn. However, at 60 min the degree of plasma extravasation in knockout mice was similar to that seen in wildtype mice (Figure 2). To confirm a role for the NK1 receptor in mediating the early phase of vascular leakage described, wildtype mice were pretreated (−5 min) with the NK1 antagonist, SR140333 (480 nmol kg−1, i.v.;) and oedema assessed at 10 min post-burn. SR140333 pretreated mice exhibited a similar pattern of plasma extravasation to that seen in NK1 knockout animals and were significantly different from vehicle treated controls (Figure 3; P<0.05).
Figure 1.
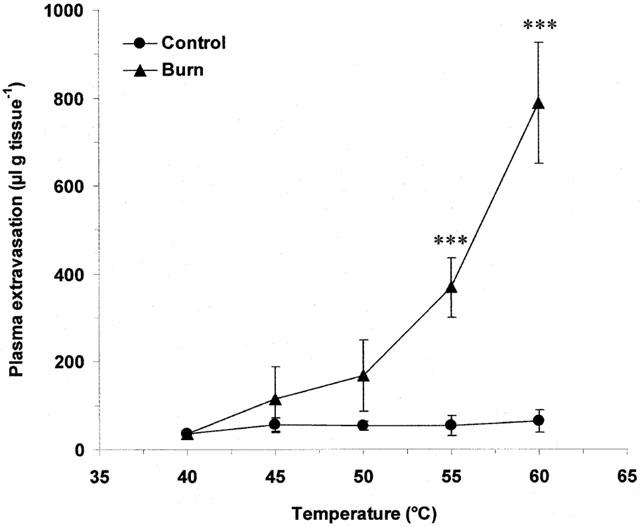
Temperature-dependency of thermic oedema. Plasma extravasation in the ears of wildtype mice at 60 min following thermal injury (40 – 60°C). All data are presented as mean±s.e.mean. ***P<0.001 vs control at the same temperature; n=4 – 5.
Figure 2.
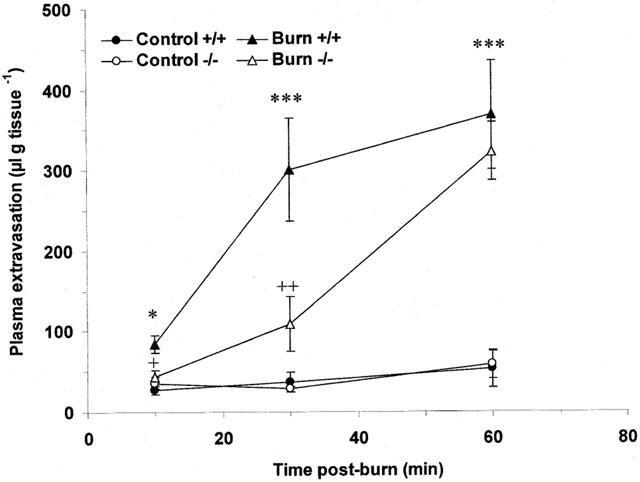
Comparison of thermic oedema in wildtype and NK1 knockout mice. Plasma extravasation in the ears of wildtype and knockout mice over time (10 – 60 min) following thermal injury (55°C). All data are presented as mean±s.e.mean. *P<0.05 vs control in the same animal at the same time point, ***P<0.001 vs control in the same animal at the same time point, +P<0.05 vs wildtype burn at the same time point, ++P<0.01 vs wildtype burn at the same time point; n=6 – 8.
Figure 3.
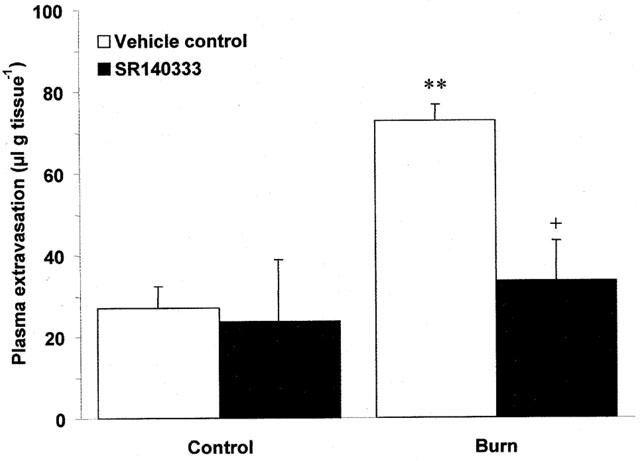
Effect of the NK1 antagonist, SR140333 on thermic oedema. Plasma extravasation in the ears of wildtype mice pretreated (−5 min; i.v.) with SR140333 (480 nmol kg−1) or vehicle control at 10 min following thermal injury (55°C). All data are presented as mean±s.e.mean. **P<0.01 vs control in the same animal, +P<0.05 vs burn in animals treated with vehicle; n=6.
Effect of kinin antagonists on scald and capsaicin induced ear oedema
Scald induced plasma extravasation in wildtype mice pretreated with the B1 (des-Arg10 Hoe 140; 1 μmol kg−1, i.v.) and B2 (Hoe 140; 100 nmol kg−1, i.v.) receptor antagonists progressed in a manner akin to that observed in NK1 knockout animals. Both antagonists significantly reduced vascular leakage at 10 (Figure 4a; P<0.001) and 30 min (Figure 4b; P<0.001) post-burn, but were ineffective at 60 min post-burn (Figure 4c). In addition, the inhibition of thermic oedema by des-Arg10 Hoe 140 was reversed by des-Arg9 bradykinin (0.1 μmol kg−1) when compared to animals pretreated with des-Arg10 Hoe 140 alone at 30 min post-burn (266.15±30.18 vs 149.74±24.22, n=4; P<0.01). A second B1 receptor antagonist, des-Arg9 Leu8 bradykinin (3 μmol kg−1) was applied to the model and was also seen to significantly attenuate post-burn oedema formation at 30 min when compared to vehicle treated controls (152.06±20.49 vs 333.61±61.10, n=6; P<0.01). In an effort to learn more about the role that kinin receptors play in mediating plasma extravasation in the mouse ear, the effect of des-Arg10 Hoe 140 (1 μmol kg−1) and Hoe 140 (100 nmol kg−1) on capsaicin ear oedema was examined at 30 min (Figure 5). Topical application of capsaicin was observed to cause a significant rise in plasma extravasation when compared to vehicle treated control ears in the same animal (P<0.001). This was significantly reduced by pretreatment with Hoe 140 (P<0.001), but not des-Arg10 Hoe 140 when compared to vehicle treated controls. In contrast to the profound effect of the kinin antagonists on thermic oedema in wildtype mice at 30 min, neither des-Arg10 Hoe 140 (1 μmol kg−1) or Hoe 140 (100 nmol kg−1) had any effect on modulating plasma extravasation caused by scald injury in NK1 knockout mice (Figure 6).
Figure 4.
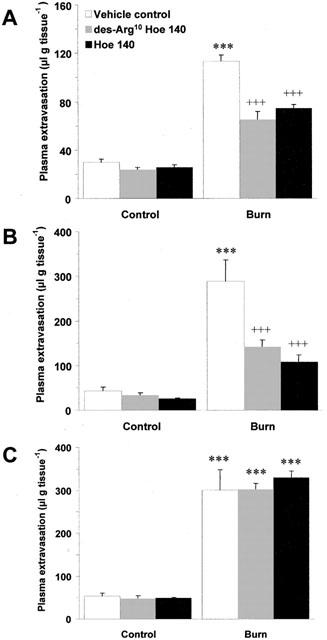
Effect of kinin antagonists on thermic oedema. Plasma extravasation in the ears of wildtype mice pretreated (−5 min; i.v) with vehicle control, des-Arg10 Hoe 140 (1 μmol kg−1) or Hoe 140 (100 nmol kg−1) at (A) 10 min, (B) 30 min and (C) 60 min post-burn (55°C). All data are presented as mean±s.e.mean. ***P<0.001 vs control in the same animal at the same time point, +++P<0.001 vs burn in animals treated with vehicle; n=5.
Figure 5.
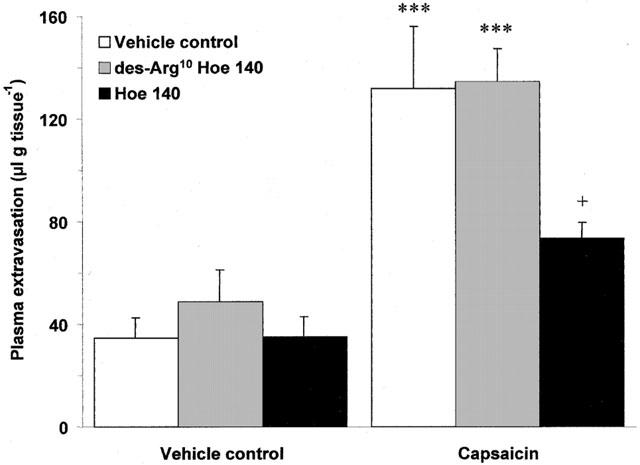
Effect of kinin antagonists on capsaicin ear oedema. Plasma extravasation in the ears of wildtype mice pretreated (−5 min; i.v) with vehicle control, des-Arg10 Hoe 140 (1 μmol kg−1) or Hoe 140 (100 nmol kg−1) at 30 min following topical application of capsaicin (200 μg ear−1). All data are presented as mean±s.e.mean. ***P<0.001 vs topical vehicle control in the same animal, +P<0.05 vs animals treated with topical capsaicin and i.v. vehicle control; n=5.
Figure 6.
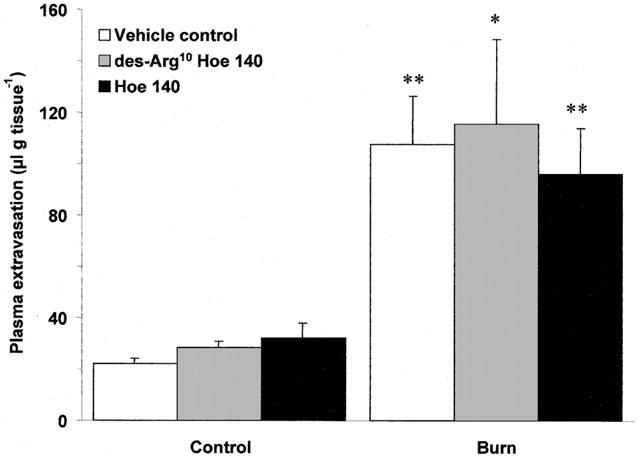
Effect of kinin antagonists on thermic oedema in NK1 knockout mice. Plasma extravasation in the ears of NK1 knockout mice pretreated (−5 min; i.v) with vehicle control, des-Arg10 Hoe 140 (1 μmol kg−1) or Hoe 140 (100 nmol kg−1) at 30 min post-burn (55°C). All data are presented as mean±s.e.mean. *P<0.05 vs control in the same animal, **P<0.01 vs control in the same animal; n=5.
Role of NK1 and kinin receptors in mediating scald induced neutrophil accumulation
Induction of thermal injury caused a significant increase in neutrophils accumulation in wildtype mice when assessed at 4 h post-burn (Figure 7a; P<0.001). However, no significant difference was observed between wildtype and NK1 knockout mice (Figure 7a). Pretreatment with des-Arg10 Hoe 140 (1 μmol kg−1) and Hoe 140 (100 nmol kg−1) also failed to prevent neutrophil accumulation in wildtype animals when compared to vehicle treated controls (Figure 7b). Similarly, neither NK1 receptor deletion (26.46±3.56 vs 28.69±3.16, n=5), treatment with des-Arg10 Hoe 140 (13.63±0.46 vs 13.94±1.17, n=5) or Hoe 140 (16.04±2.3 vs 13.94±1.17, n=5) had any effect on neutrophil depletion compared to wildtype mice and vehicle treated controls respectively at 30 min post-burn.
Figure 7.
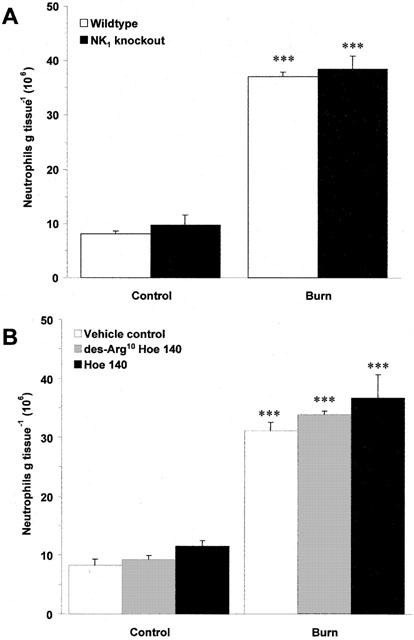
Effect of NK1 deletion and kinin antagonists on thermic neutrophil accumulation. (A) Neutrophil accumulation, as assessed by MPO assay in the ears of wildtype and NK1 knockout mice at 4 h post-burn (55°C). All data are presented as mean±s.e.mean. ***P<0.001 vs wildtype control; n=11. (B) Neutrophil accumulation in the ears of wildtype mice pretreated (−5 min; i.v) with vehicle control, des-Arg10 Hoe 140 (1 μmol kg−1) or Hoe 140 (100 nmol kg−1) at 4 h post-burn (55°C). ***P<0.001 vs control in the same animal; n=5.
Discussion
Previous studies have shown that local plasma extravasation following thermal injury is dependent on the temperature of burn and occurs in a manner progressive with time (Arturson & Jakobsson, 1985; Haegerstrand et al., 1987). In keeping with these studies, we have described a murine model of cutaneous thermal injury that has been applied to the study of tachykinins and kinins involved in mediating acute burn oedema. Initially, temperature-response curves were obtained. It was observed that plasma extravasation in the ears of mice was dependent on the temperature of exposure and that a threshold of 55°C caused a significant increase in plasma leakage compared to controls in this model. The degree of plasma extravasation following a 55°C burn was also seen to be dependent on time post-burn. It should be noted that plasma extravasation did not change in the contralateral, control ears at any of the time points or temperatures investigated, indicating that the injury induced was localized and did not lead to the release of circulating factors that may have affected the vascular permeability of sites remote to the burn.
Current evidence suggests that the neuropeptide, substance P is released from sensory neurones and acts on endothelial NK1 receptors to mediate plasma extravasation following thermal injury. Early studies showed that post-burn oedema formation was suppressed in rats pretreated with capsaicin to cause selective degeneration of sensory C-fibres (Saria, 1984; Yonehara et al., 1987) and a number of reports show that substance P release is increased in both experimental (Jonsson et al., 1986; Yonehara et al., 1987; Hu et al., 1996) and clinical thermal injury (Onuoha & Alpar, 2001). In addition, selective NK1 receptor antagonists have been shown to be of benefit in reducing post-burn oedema in the rat (Siney & Brain, 1996; Lofgren et al., 1999). The initial aim of this study was to confirm the importance of NK1 receptors in mediating thermic oedema via the use of NK1 receptor knockout mice. Our results show that in comparison to the very large and rapid plasma extravasation seen in wildtype mice following scald injury, there was a marked depression of this parameter in NK1 knockout mice at 10 and 30 min post-burn. However, at the later time point of 60 min, the degree of plasma extravasation in the two populations was comparable, confirming that the NK1 receptor plays an essential role in mediating the early, but not the late phase of plasma extravasation following thermal injury. To validate these findings, the selective NK1 receptor antagonist, SR140333 was administered to our model in a dosing regime identical to that described by Cao et al. (2000) where a similar reduction in the inflammatory response was seen in wildtype mice treated with SR140333 and NK1 receptor knockouts following an intradermal injection of carrageenan. In this study, SR140333 completely ablated thermic oedema in wildtype animals at 10 min post-burn. These results are directly comparable to those of Siney & Brain (1996) who reported that SR140333 blocked thermal skin oedema in the rat at 5 – 35 min, but not at 65 – 95 min post-burn.
The kinins are formed from kininogen precursors by a group of serine proteases collectively known as the kallikreins (Burch et al., 1990). The biologically active peptide bradykinin and its metabolite, des-Arg9 bradykinin are generated via activation of this system. In inflammation, bradykinin acts primarily through constitutive B2 receptors to cause vasodilation and increased vascular permeability, while des-Arg9 bradykinin is thought to have a greater selectivity for B1 receptors (Hall, 1992). The kinins have been implicated in mediating post-burn plasma extravasation. Although the role of the B2 receptor in mediating thermic oedema is well established (Wirth et al., 1992; Nwariaku et al., 1996), the relative contribution of the B1 receptor is not known. As such, the second aim of this study was to investigate the effect of B1 and B2 receptor antagonists in a murine model of thermal injury. The B1 antagonist, des-Arg10 Hoe 140 administered at a dose reported to selectively inhibit allergen-induced bronchial hyperresponsiveness in the rat (Huang et al., 1999) was seen to significantly reduce oedema formation in wildtype mice at 10 and 30 min post-burn, but not at 60 min. A second B1 antagonist, des-Arg9 Leu8 bradykinin, used at a dose previously shown to attenuate carrageenan induced neutrophil accumulation in Sv129+C57BL/6 mice (Cao et al., 2000), also inhibited plasma extravasation at 30 min post-burn indicating a role for B1 receptor. To confirm that the doses chosen to antagonize the B1 receptor were acting in a selective manner, des-Arg10 Hoe 140 was co-administered with a dose of the B1 agonist, des-Arg9 bradykinin previously reported to exacerbate adjuvant induced arthritis in the rat (Rupniak et al., 1997). des-Arg9 bradykinin prevented the inhibition of plasma extravasation seen with des-Arg10 Hoe 140 at 30 min post-burn. Taken together, the results suggest that kinin B1 receptors mediate at least the early phase of thermic oedema in Sv129+C57BL/6 mice. The effect of the B2 antagonist, Hoe 140 in this model was also examined. A dose of Hoe 140 previously reported to prevent carrageenan induced neutrophil accumulation in Sv129+C57BL/6 mice (Cao et al., 2000) suppressed the early phase of oedema formation, but not the later phase, indicating that as expected, the B2 receptor mediates post-burn plasma extravasation.
These findings were of interest for a number of reasons. Firstly, to our knowledge this is the first time that kinin B1 receptors have been demonstrated to have an active role in the burn wound. Secondly, the rapid activation of B1 receptors seen in this model (e.g. 10 min post-injury) would indicate that they are present in the murine microvasculature in a constitutive form. This is interesting because B1 receptors are generally not considered to be constitutively expressed in naïve tissues and are commonly reported to be upregulated in experimental animals following sub-acute exposure to proinflammatory cytokines (Campos et al., 1999) and bacterial endotoxins (Campos et al., 1996). However, there are a number of exceptions to this supposition. Functional studies indicate that constitutive B1 receptors are present in the rat (Boschcov et al., 1984; Calixto & Medeiros, 1992; Campos & Calixto, 1994; Wotherspoon & Winter, 2000) and more recently it has been suggested that they mediate experimental pleurisy (Vianna & Calixto, 1998) and neuronal activity (Maas et al., 1995; Pesquero et al., 2000) in the mouse. In keeping with these studies, we surmise that constitutive B1 receptors are activated by endogenous ligands to mediate the early stages of plasma extravasation following thermal injury. Nonetheless, it should be noted that the B1 receptor antagonists des-Arg10 Hoe 140 and des-Arg9 Leu8 bradykinin had no effect on plasma extravasation at 60 min post-burn in our model. This is perplexing, since even in models where constitutive B1 receptors are present, upregulation is still thought to occur following exposure to pro-inflammatory stimuli (Pesquero et al., 2000) and as such, it could be assumed that the B1 component of response would increase with time, especially as the B1 receptor is not prone to desensitization (Austin et al., 1997). However, it is well established that thermal injury results in the activation of many inflammatory pathways (Arturson, 1996) and at the later time point, it is conceivable that any B1 receptor mediated effects could be masked by the involvement of other mediators.
It was observed that the pattern and time course of plasma extravasation following thermal injury was very similar in wildtype mice treated with kinin antagonists and NK1 receptor knockout mice. To further investigate a link between these receptor systems, kinin receptor antagonists were administered to NK1 receptor knockout mice prior to the induction of thermal injury. Neither B1 or B2 receptor blockade had any additional inhibitory effect on oedema formation at 30 min post-burn suggesting that the kinins interact with NK1 related pathways in this model. In accordance with this concept, Yonehara et al. (1995) reported that plasma extravasation and substance P release was suppressed in burn wounds in kininogen-deficient rats. Furthermore, in other models of experimental inflammation, it has been reported that inducible B1 (Ferreira et al., 2000) and constitutive B2 receptors (Ricciardolo et al., 1994; Lindstrom & Andersson, 1997; Schuligoi et al., 1998) mediate tachykinin release from afferent sensory neurons. Taken together, we conclude that a similar interaction may occur in thermally injured tissues. In an effort to further understand this mechanism, the effect of the kinin antagonists on capsaicin induced ear oedema was examined in Sv129+C57BL/6 wildtype mice. It was observed that the B2 (Hoe 140), but not the B1 antagonist (des-Arg10 Hoe 140) attenuated plasma extravasation induced by capsaicin. A conceivable explanation for this is that the B1 and B2 receptors are found in different locations within the microvasculature. It is possible, that in murine skin, B1 receptors are located prejunctionally on the sensory neurones since their blockade does not affect oedema induced by capsaicin, but it does prevent oedema induced by endogenous kinins (e.g. generated following thermal injury), which we suggest interact with sensory neurones to cause substance P release. In contrast, it would appear that a population of B2 receptors are located postjunctionally on the post-capillary venule endothelial cells since their blockade can prevent sensory neuropeptide mediated plasma extravasation induced by capsaicin. Alternatively, the results may simply reflect the fact that the burn wound is an environment highly favourable to the production of the endogenous B1 receptor agonist, des-Arg9 bradykinin via the action of carboxypeptidase N on bradykinin, whereas other degradation pathways (e.g. plasma proteases) maybe of greater importance in removing bradykinin from dermal tissues acutely exposed to capsaicin. In addition, the differential effects of des-Arg10 Hoe 140 and Hoe 140 in the capsaicin ear oedema model would indicate that the doses chosen were selective to the kinin B1 and B2 receptors respectively.
In a separate series of experiments, the effects of NK1 receptor deletion and kinin antagonists on neutrophil accumulation following thermal injury were examined. The relative importance of the NK1 receptor in mediating neutrophil accumulation remains to be determined. Bozic et al. (1996) reported that neutrophil accumulation in the lung was suppressed in NK1 knockout mice following immune complex challenge and exogenous substance P has been shown to cause neutrophil accumulation in a variety of animal models (Perretti et al., 1993; Baluk et al., 1995; Walsh et al., 1995). Furthermore, in the murine air pouch model, neutrophil accumulation stimulated by IL-1β is attenuated by both NK1 antagonists (Ahluwalia et al., 1998). However, activation of NK1 receptors does not induce neutrophil accumulation in naïve rat (Pinter et al., 1999) or mouse (Cao et al., 2000) skin. In agreement with these findings, it was observed that post-burn neutrophil accumulation in NK1 receptor knockout mice was akin to that seen in wildtype mice at both 30 min and 4 h post-burn. As such, it would appear that the NK1 receptor does not play a critical role in mediating neutrophil accumulation following cutaneous thermal injury. Similarly, neither the B1 antagonist, des-Arg10 Hoe 140 or the B2 antagonist, Hoe 140 had any effect on neutrophil accumulation in this model indicating that like the NK1 receptor, the kinin receptors are not critical for neutrophil infiltration into the burn wound. This result was in direct contrast to the findings reported in other models of experimental inflammation where it has been shown that both the B1 (Bozic et al., 1996; Ahluwalia & Perretti, 1996; Cao et al., 2000; McLean et al., 2000) and the B2 receptor (Cao et al., 2000) can mediate leukocyte accumulation. However, as previously discussed, thermal injury initiates the activation of an array of inflammatory pathways and can lead to the production of cytokines (Ono et al., 1995), complement (Oldham et al., 1988) and leukotrienes (Dobke et al., 1987), all of which are potent neutrophil chemoattractants and/or activators. Thus, it is possible that kinin mediated neutrophil accumulation is masked by the involvement of other mediators in this model.
In conclusion, a murine model of thermal injury has been described and applied to the investigation of tachykinin and kinin involvement in the burn wound. We have demonstrated that tachykinin NK1 receptors and the kinin B1 and B2 receptors play an essential role in mediating the early, but not the delayed phase of plasma extravasation following thermal injury. This is the first time that functional B1 receptors have been shown to play a role in the burn wound and we propose that the kinin receptors are able to modulate the activity of tachykinins by stimulating their release from sensory neurones under these circumstances. Furthermore, we have shown that constitutive B1 receptors are present in the murine microvasculature and suggest that des-Arg9 bradykinin is generated in the burn wound. In addition, the possibility remains that the kinin B1 and B2 receptors are expressed in anatomically distinct regions of the murine microvasculature, the former being present on sensory neurones and the latter on the endothelium of post-capillary venules. If such mechanisms exist in man, it is possible that combinational therapy of tachykinin and kinin antagonists may be of therapeutic value in the treatment of accidental burns and other types of dermal trauma.
Acknowledgments
A. Rawlingson is in receipt of a Medical Research Council (U.K.) Ph.D. Studentship. The authors would like to thank Mr A.D. Grant for his assistance in maintaining the Sv129+C57BL/6 mouse colony.
Abbreviations
- HBSS
Hank's buffered salt solution
- H2O2
hydrogen peroxide
- HTAB
hexadecyltrimethylammonium
- MPO
myeloperoxidase
- NK1
neurokinin 1
- SR14033
((S)1-(2-[3-(3,4-dichlorophenyl)-1-(3-isopropoxyphenylacetyl) piperidin-3-yl]ethyl)-4-phenyl-1-azoniabicyclo[2.2.2]octane chloride)
- TMB
tetramethylbenzidine
References
- AHLUWALIA A., DE FELIPE C., O'BRIEN J., HUNT S.P., PERRETTI M. Impaired IL-1beta-induced neutrophil accumulation in tachykinin NK1 receptor knockout mice. Br. J. Pharmacol. 1998;124:1013–1015. doi: 10.1038/sj.bjp.0701978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AHLUWALIA A., PERRETTI M. Involvement of bradykinin B1 receptors in the polymorphonuclear leukocyte accumulation induced by IL-1 beta in vivo in the mouse. J. Immunol. 1996;156:269–274. [PubMed] [Google Scholar]
- ARTURSON G. Pathophysiology of the burn wound and pharmacological treatment. The Rudi Hermans Lecture, 1995. Burns. 1996;22:255–274. doi: 10.1016/0305-4179(95)00153-0. [DOI] [PubMed] [Google Scholar]
- ARTURSON G., JAKOBSSON O.P. Oedema measurements in a standard burn model. Burns Incl. Therm. Inj. 1985;12:1–7. doi: 10.1016/0305-4179(85)90176-7. [DOI] [PubMed] [Google Scholar]
- AUSTIN C.E., FAUSSNER A., ROBINSON H.E., CHAKRAVARTY S., KYLE D.J., BATHON J.M., PROUD D. Stable expression of the human kinin B1 receptor in Chinese hamster ovary cells. Characterization of ligand binding and effector pathways. J. Biol. Chem. 1997;272:11420–11425. doi: 10.1074/jbc.272.17.11420. [DOI] [PubMed] [Google Scholar]
- BALUK P., BERTRAND C., GEPPETTI P., MCDONALD D.M., NADEL J.A. NK1 receptors mediate leukocyte adhesion in neurogenic inflammation in the rat trachea. Am. J. Physiol. 1995;268:L263–L269. doi: 10.1152/ajplung.1995.268.2.L263. [DOI] [PubMed] [Google Scholar]
- BOSCHCOV P., PAIVA A.C., PAIVA T.B., SHIMUTA S.I. Further evidence for the existence of two receptor sites for bradykinin responsible for the diphasic effect in the rat isolated duodenum. Br. J. Pharmacol. 1984;83:591–600. doi: 10.1111/j.1476-5381.1984.tb16523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOZIC C.R., LU B., HOPKEN U.E., GERARD C., GERARD N.P. Neurogenic amplification of immune complex inflammation. Science. 1996;273:1722–1725. doi: 10.1126/science.273.5282.1722. [DOI] [PubMed] [Google Scholar]
- BURCH R.M., FARMER S.G., STERANKA L.R. Bradykinin receptor antagonists. Med. Res. Rev. 1990;10:237–269. doi: 10.1002/med.2610100204. [DOI] [PubMed] [Google Scholar]
- CALIXTO J.B., MEDEIROS Y.S. Bradykinin-induced biphasic response in the rat isolated stomach fundus: functional evidence for a novel bradykinin receptor. Life Sci. 1992;50:L47–L52. doi: 10.1016/0024-3205(92)90395-6. [DOI] [PubMed] [Google Scholar]
- CAMPOS A.H., CALIXTO J.B. Mechanisms involved in the contractile responses of kinins in rat portal vein rings: mediation by B1 and B2 receptors. J. Pharmacol. Exp. Ther. 1994;268:902–909. [PubMed] [Google Scholar]
- CAMPOS M.M., SOUZA G.E., CALIXTO J.B. Upregulation of B1 receptor mediating des-Arg9-BK-induced rat paw oedema by systemic treatment with bacterial endotoxin. Br. J. Pharmacol. 1996;117:793–798. doi: 10.1111/j.1476-5381.1996.tb15262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAMPOS M.M., SOUZA G.E., CALIXTO J.B. In vivo B1 kinin-receptor upregulation. Evidence for involvement of protein kinases and nuclear factor kappaB pathways. Br. J. Pharmacol. 1999;127:1851–1859. doi: 10.1038/sj.bjp.0702715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAO T., GERARD N.P., BRAIN S.D. Use of NK(1) knockout mice to analyze substance P-induced edema formation. Am. J. Physiol. 1999;277:R476–R481. doi: 10.1152/ajpregu.1999.277.2.R476. [DOI] [PubMed] [Google Scholar]
- CAO T., PINTER E., AL RASHED S., GERARD N., HOULT J.R., BRAIN S.D. Neurokinin-1 receptor agonists are involved in mediating neutrophil accumulation in the inflamed, but not normal, cutaneous microvasculature: an in vivo study using neurokinin-1 receptor knockout mice. J. Immunol. 2000;164:5424–5429. doi: 10.4049/jimmunol.164.10.5424. [DOI] [PubMed] [Google Scholar]
- DOBKE M.K., HAYES E.C., BAXTER C.R. Leukotrienes LTB4 and LTC4 in thermally injured patients' plasma and burn blister fluid. J. Burn Care Rehabil. 1987;8:189–191. doi: 10.1097/00004630-198705000-00002. [DOI] [PubMed] [Google Scholar]
- FERREIRA P.K., CAMPOS M.M., CALIXTO J.B. The role of sensorial neuropeptides in the edematogenic responses mediated by B(1) agonist des-Arg(9)-BK in rats pre-treated with LPS. Regul. Pept. 2000;89:29–35. doi: 10.1016/s0167-0115(00)00094-x. [DOI] [PubMed] [Google Scholar]
- HAEGERSTRAND A., DALSGAARD C.J., JONSSON C.E. Effects of capsaicin pretreatment on the inflammatory response to scalding injury in the rat. Acta Physiol Scand. 1987;130:345–348. doi: 10.1111/j.1748-1716.1987.tb08146.x. [DOI] [PubMed] [Google Scholar]
- HALL J.M. Bradykinin receptors: pharmacological properties and biological roles. Pharmacol. Ther. 1992;56:131–190. doi: 10.1016/0163-7258(92)90016-s. [DOI] [PubMed] [Google Scholar]
- HU D., CHEN B., LIN S., TANG C. Changes in substance P in the jejuna of rats after burns. Burns. 1996;22:463–467. doi: 10.1016/0305-4179(95)00179-4. [DOI] [PubMed] [Google Scholar]
- HUANG T.J., HADDAD E.B., FOX A.J., SALMON M., JONES C., BURGESS G., CHUNG K.F. Contribution of bradykinin B(1) and B(2) receptors in allergen-induced bronchial hyperresponsiveness. Am. J. Respir. Crit Care Med. 1999;160:1717–1723. doi: 10.1164/ajrccm.160.5.9901029. [DOI] [PubMed] [Google Scholar]
- JONSSON C.E., BRODIN E., DALSGAARD C.J., HAEGERSTRAND A. Release of substance-P-like immunoreactivity in dog paw lymph after scalding injury. Acta Physiol Scand. 1986;126:21–24. doi: 10.1111/j.1748-1716.1986.tb07783.x. [DOI] [PubMed] [Google Scholar]
- LINDSTROM E.G., ANDERSSON R.G. Neurokinin A-LI release after antigen challenge in guinea-pig bronchial tubes: influence of histamine and bradykinin. Br. J. Pharmacol. 1997;122:417–422. doi: 10.1038/sj.bjp.0701382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOFGREN O., QI Y., LUNDEBERG T. Inhibitory effects of tachykinin receptor antagonists on thermally induced inflammatory reactions in a rat model. Burns. 1999;25:125–129. doi: 10.1016/s0305-4179(98)00125-9. [DOI] [PubMed] [Google Scholar]
- LU B., FIGINI M., EMANUELI C., GEPPETTI P., GRADY E.F., GERARD N.P., ANSELL J., PAYAN D.G., GERARD C., BUNNETT N. The control of microvascular permeability and blood pressure by neutral endopeptidase. Nat. Med. 1997;3:904–907. doi: 10.1038/nm0897-904. [DOI] [PubMed] [Google Scholar]
- MAAS J., RAE G.A., HUIDOBRO-TORO J.P., CALIXTO J.B. Characterization of kinin receptors modulating neurogenic contractions of the mouse isolated vas deferens. Br. J. Pharmacol. 1995;114:1471–1477. doi: 10.1111/j.1476-5381.1995.tb13372.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCLEAN P.G., AHLUWALIA A., PERRETTI M. Association between kinin B(1) receptor expression and leukocyte trafficking across mouse mesenteric postcapillary venules. J. Exp. Med. 2000;192:367–380. doi: 10.1084/jem.192.3.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORONEY M.A., ALCARAZ M.J., FORDER R.A., CAREY F., HOULT J.R. Selectivity of neutrophil 5-lipoxygenase and cyclo-oxygenase inhibition by an anti-inflammatory flavonoid glycoside and related aglycone flavonoids. J. Pharm. Pharmacol. 1988;40:787–792. doi: 10.1111/j.2042-7158.1988.tb05173.x. [DOI] [PubMed] [Google Scholar]
- NWARIAKU F.E., SIKES P.J., LIGHTFOOT E., MILESKI W.J., BAXTER C. Effect of a bradykinin antagonist on the local inflammatory response following thermal injury. Burns. 1996;22:324–327. doi: 10.1016/0305-4179(95)00130-1. [DOI] [PubMed] [Google Scholar]
- OLDHAM K.T., GUICE K.S., TILL G.O., WARD P.A. Activation of complement by hydroxyl radical in thermal injury. Surgery. 1988;104:272–279. [PubMed] [Google Scholar]
- ONO I., GUNJI H., ZHANG J.Z., MARUYAMA K., KANEKO F. A study of cytokines in burn blister fluid related to wound healing. Burns. 1995;21:352–355. doi: 10.1016/0305-4179(95)00005-4. [DOI] [PubMed] [Google Scholar]
- ONUOHA G.N., ALPAR E.K. Levels of vasodilators (SP, CGRP) and vasoconstrictor (NPY) peptides in early human burns. Eur. J. Clin. Invest. 2001;31:253–257. doi: 10.1046/j.1365-2362.2001.00787.x. [DOI] [PubMed] [Google Scholar]
- PERRETTI M., AHLUWALIA A., FLOWER R.J., MANZINI S. Endogenous tachykinins play a role in IL-1-induced neutrophil accumulation: involvement of NK-1 receptors. Immunology. 1993;80:73–77. [PMC free article] [PubMed] [Google Scholar]
- PESQUERO J.B., ARAUJO R.C., HEPPENSTALL P.A., STUCKY C.L., SILVA J.A., JR, WALTHER T., OLIVEIRA S.M., PESQUERO J.L., PAIVA A.C., CALIXTO J.B., LEWIN G.R., BADER M. Hypoalgesia and altered inflammatory responses in mice lacking kinin B1 receptors. Proc. Natl. Acad. Sci. U.S.A. 2000;97:8140–8145. doi: 10.1073/pnas.120035997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PINTER E., BROWN B., HOULT J.R., BRAIN S.D. Lack of evidence for tachykinin NK1 receptor-mediated neutrophil accumulation in the rat cutaneous microvasculature by thermal injury. Eur. J. Pharmacol. 1999;369:91–98. doi: 10.1016/s0014-2999(99)00054-0. [DOI] [PubMed] [Google Scholar]
- RICCIARDOLO F.L., NADEL J.A., GRAF P.D., BERTRAND C., YOSHIHARA S., GEPPETTI P. Role of kinins in anaphylactic-induced bronchoconstriction mediated by tachykinins in guinea-pigs. Br. J. Pharmacol. 1994;113:508–512. doi: 10.1111/j.1476-5381.1994.tb17018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUPNIAK N.M., BOYCE S., WEBB J.K., WILLIAMS A.R., CARLSON E.J., HILL R.G., BORKOWSKI J.A., HESS J.F. Effects of the bradykinin B1 receptor antagonist des- Arg9[Leu8]bradykinin and genetic disruption of the B2 receptor on nociception in rats and mice. Pain. 1997;71:89–97. doi: 10.1016/s0304-3959(97)03343-5. [DOI] [PubMed] [Google Scholar]
- SARIA A. Substance P in sensory nerve fibres contributes to the development of oedema in the rat hind paw after thermal injury. Br. J. Pharmacol. 1984;82:217–222. doi: 10.1111/j.1476-5381.1984.tb16461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHIERWAGEN C., BYLUND-FELLENIUS A.C., LUNDBERG C. Improved method for quantification of tissue PMN accumulation measured by myeloperoxidase activity. J. Pharmacol. Methods. 1990;23:179–186. doi: 10.1016/0160-5402(90)90061-o. [DOI] [PubMed] [Google Scholar]
- SCHULIGOI R., PESKAR B.A., DONNERER J., AMANN R. Bradykinin-evoked sensitization of neuropeptide release from afferent neurons in the guinea-pig lung. Br. J. Pharmacol. 1998;125:388–392. doi: 10.1038/sj.bjp.0702079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SINEY L., BRAIN S.D. Involvement of sensory neuropeptides in the development of plasma extravasation in rat dorsal skin following thermal injury. Br. J. Pharmacol. 1996;117:1065–1070. doi: 10.1111/j.1476-5381.1996.tb16698.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VIANNA R.M., CALIXTO J.B. Characterization of the receptor and the mechanisms underlying the inflammatory response induced by des-Arg9-BK in mouse pleurisy. Br. J. Pharmacol. 1998;123:281–291. doi: 10.1038/sj.bjp.0701590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALSH D.T., WEG V.B., WILLIAMS T.J., NOURSHARGH S. Substance P-induced inflammatory responses in guinea-pig skin: the effect of specific NK1 receptor antagonists and the role of endogenous mediators. Br. J. Pharmacol. 1995;114:1343–1350. doi: 10.1111/j.1476-5381.1995.tb13354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WIRTH K.J., ALPERMANN H.G., SATOH R., INAZU M. The bradykinin antagonist Hoe 140 inhibits carrageenan- and thermically induced paw oedema in rats. Agents Actions Suppl. 1992;38 (Pt 3):428–431. [PubMed] [Google Scholar]
- WOTHERSPOON G., WINTER J. Bradykinin B1 receptor is constitutively expressed in the rat sensory nervous system. Neurosci. Lett. 2000;294:175–178. doi: 10.1016/s0304-3940(00)01561-5. [DOI] [PubMed] [Google Scholar]
- YONEHARA N., SAITO K., OH-ISHI S., KATORI M., INOKI R. Contribution of bradykinin to heat-induced substance P release in the hind instep of rats. Life Sci. 1995;56:1679–1688. doi: 10.1016/0024-3205(95)98574-y. [DOI] [PubMed] [Google Scholar]
- YONEHARA N., SHIBUTANI T., INOKI R. Contribution of substance P to heat-induced edema in rat paw. J. Pharmacol. Exp. Ther. 1987;242:1071–1076. [PubMed] [Google Scholar]