

Abstract
Vascular endothelial growth factor (VEGF) is a potent inducer of inflammation, and we have shown that this latter effect is mediated through endothelial cell (EC) PAF synthesis. Since the phospholipid remodelling pathway enzymes (CoA-independent transacylase, CoA-IT; phospholipase A2, PLA2; and lyso-PAF acetyltransferase, lyso-PAF-AT) may participate in PAF synthesis, we assessed their contribution to VEGF-induced PAF synthesis in bovine aortic EC (BAEC) and human umbilical vein EC (HUVEC).
VEGF enhanced BAEC and HUVEC PAF synthesis by up to 28 and 4 fold above basal levels respectively.
A pretreatment with a CoA-IT and lyso-PAF-AT inhibitor (Sanguinarin; 500 nM) blocked VEGF-induced PAF synthesis by 95%, a specific CoA-IT inhibitor (SKF45905; 10 – 50 μM) was without effect, confirming the crucial role of the PLA2 and lyso-PAF-AT.
Treatment with secreted PLA2 (sPLA2) inhibitors which have been shown to inhibit both groups IIA and V sPLA2 (SB203347; 10 μM and LY311727; 100 μM) blocked EC PAF synthesis by up to 90%, whereas selective inhibition of group IIA sPLA2 (LY311727; 1 μM) had no significant effect.
RT – PCR and Western blot analyses demonstrated the presence of group V sPLA2 whereas group IIA sPLA2 was undetected in EC.
Treatment with cytosolic and calcium-independent PLA2 inhibitors (Arachidonyl trifluoromethyl ketone, Bromoenol lactone, Methyl arachydonyl fluorophosphate, up to 50 μM) did not prevent but rather potentiated the VEGF effect on EC PAF synthesis.
These results provide evidence that with VEGF activation of EC cells, the group V sPLA2 provides substrate for EC PAF formation.
Keywords: VEGF, PAF, PLA2, inflammation, angiogenesis
Introduction
Over the past 20 years, strong evidence has confirmed that endothelial cells (EC) represent a metabolically active tissue rather than a simple barrier between blood and interstitial fluid. The pluripotent ability of these cells allows them to respond to a wide range of stimuli known to play a critical role not only in the balance of vascular tone and permeability, but also in the pathogenesis of certain diseases such as tumor growth, diabetic retinopathy as well as psoriasis (Folkman, 1991; Folkman & Klagsbrun, 1997). Aberrant angiogenesis, characterized by uncontrolled growth of new capillaries from pre-existing blood vessels, has been found to be a crucial component of these pathologies (Folkman & Klagsbrun, 1997).
Several reports suggest that inflammation regularly and perhaps invariably precedes and/or accompanies angiogenesis (Jackson et al., 1997; 1998). Blood vessels in and around tumours display increased vascular permeability (Dvorak et al., 1988) and inflammatory monocytes/macrophages can be found at sites where angiogenesis is occurring in an abnormal environment (Sunderkotter et al., 1994). One likely candidate for the regulation of pathological angiogenesis is the vascular endothelial growth factor/vascular permeability factor (VEGF/VPF) (Ferrara, 1995). This is suggested in part by observations that high levels of VEGF are produced by various types of tumours (Kondo et al., 1994), and that tumour growth is attenuated in vivo by anti-VEGF antibodies (Kim et al., 1993). Although many growth factors such as VEGF can induce endothelial cell (EC) migration and proliferation in culture, VEGF is the only one capable of enhancing vascular permeability (Connolly et al., 1989; Folkman & Klagsbrun, 1997), and it is likely that its angiogenic properties are mediated in part by its ability to modulate fluid and protein extravasation. We have recently reported that VEGF-induced protein extravasation in vivo was abolished by a pretreatment with a selective platelet-activating factor (PAF) receptor antagonist, suggesting that VEGF effect on vascular permeability was mediated through PAF synthesis (Sirois & Edelman, 1997). This hypothesis was supported by the rapid induction of PAF synthesis in cultured bovine aortic endothelial cells (BAEC) treated with VEGF. In addition, we showed that VEGF effect on BAEC migration, proliferation and PAF synthesis was dependent on the activation of the receptor encoded by the Flk-1/KDR gene (Bernatchez et al., 1999). However, the intracellular events related to VEGF-induced PAF production remain unknown.
Two synthetic pathways for PAF synthesis have been described. One route, de novo synthesis, is thought to produce constitutively a small amount of PAF in tissues, which allows efficient homeostasis between the bloodstream and interstitial fluid (Venable et al., 1993). The dominant inflammatory mechanism of PAF biosynthesis in EC is thought to occur through a two step remodelling pathway where the acyl moiety of alkylacylglycerophosphorylcholine, a membrane-associated phospholipid, is initially removed by either the direct action of a phospholipase A2 (PLA2) or by a CoA-independent transacylase (CoA-IT) to form lyso-PAF. The final step, the acetylation of the lyso-PAF, is catalyzed by the acetylCoA:lyso-PAF acetyltransferase (lyso-PAF-AT) leading to PAF synthesis (Bussolino & Camussi, 1995; Snyder et al., 1996).
Several inflammatory mediators were shown to directly induce the activation of the remodelling route, leading to very-early, early and delayed PAF synthesis (Bussolino & Camussi, 1995). However, there are no data regarding the capacity of growth factors to activate this pathway. Consequently, we sought to determine whether the elevation of PAF synthesis elicited by VEGF might be related to an increased activity of the remodelling pathway.
Methods
Drugs
VEGF (human recombinant vascular endothelial growth factor, 165 amino acid peptide) was purchased from PeproTech (Rocky Hill, NJ, U.S.A.). Arachidonoyl trifluoromethyl ketone (AACOCF3) and Scalaradial were purchased from Calbiochem (La Jolla, CA, U.S.A.), Sanguinarin was purchased from Sigma (St-Louis, MO, U.S.A.), and Methyl arachydonyl fluorophosphate (MAFP) and Bromoenol lactone (BEL) were purchased from Cayman Chemicals (Ann Arbor, MI, U.S.A.). SB203347 and SKF45905 were donated by Dr James D. Winkler (SmithKline Beecham Pharmaceuticals; King of Prussia, PA, U.S.A.). LY311727 was kindly provided by Dr Jerome Fleisch (Lilly Research Laboratories, Indianapolis, IN, U.S.A.).
Cell culture
BAEC and human umbilical vein endothelial cells (HUVEC) were isolated from freshly harvested aorta or umbilical cords respectively, and cultured and characterized as described previously (Sirois & Edelman, 1997; Bernatchez et al., 1999). BAEC and HUVEC were not passaged for more than 10 and five passages respectively.
Measurement of PAF synthesis
PAF production by BAEC and HUVEC was measured by incorporation of 3H-acetate into lyso-PAF (Sirois & Edelman, 1997; Bernatchez et al., 1999). Confluent BAEC or HUVEC (6-well tissue culture plate) were rinsed with HBSS (Hank's balanced salt solution)/HEPES (10 mM; pH 7.4). Cells were then stimulated for 15 min in 1 ml of HBSS-HEPES (10 mM, pH 7.4)+CaCl2 (0 – 10 mM)+3H-acetate (25 μCi) plus the appropriate concentration of VEGF. Inhibitors were added 5 to 30 min prior to the addition of VEGF (1 nM). The reaction was stopped by addition of acidified methanol (50 mM acetic acid), the wells were scraped and added to chloroform (2.5 ml) and 0.1 M sodium acetate (1 ml) mixture. Culture plates were washed twice with 1 ml of methanol, added to the chloroform mixture, shaken vigorously and centrifuged for 2 min at 1700 r.p.m. The upper phase was discarded and the chloroform phase was washed twice with 2 ml of the organic phase of a HBSS-HEPES (10 mM)-methanol-chloroform-sodium acetate (0.1 M) solution (1 : 2.5 : 3.75 : 1). Isolated lipids were evaporated under a stream of N2 gas, resuspended in 175 μl of mobile phase solvent (water-chloroform-methanol 5 : 40 : 55) and purified by a silica-based normal-phase HPLC column (4.5×250 mm, 5 μm silica particle size; Varian, Harbour City, CA, U.S.A.) and eluted with the mobile phase solvent at a 0.5 ml min−1 flow rate. Fractions corresponding to 3H-alkyl-PAF were quantified by counting radioactivity with a β-counter. The authenticity of synthesized 3H-alkyl-PAF was confirmed by the similar HPLC elution pattern as standard 3H-alkyl-PAF (New England Nuclear, MA, U.S.A.), and by its ability to induce platelet aggregation as standard alkyl-PAF (Avanti Polar Lipids, Alabaster, AL, U.S.A.) (Sirois & Edelman, 1997).
Western blot analysis of secreted phospholipases A2 expression
Confluent BAEC and HUVEC (100 mm tissue culture plate) were rinsed with HBSS-HEPES (10 mM; pH 7.4). Cells were then stimulated for 15 min in 3 ml of HBSS-HEPES (10 mM, pH 7.4)+CaCl2 (10 mM)+acetate (6 nM) with or without VEGF (1 nM). The supernatants and the cells were collected. Total proteins were isolated by the addition of 500 μl of lysis buffer with PMSF 1 mM (Sigma), leupeptin 10 μg ml−1 (Sigma), aprotinin 30 μg ml−1 (Sigma) and NaVO3 1 mM (Sigma). Plates were scraped using a plastic policeman and the protein concentration was determined with a Bio-Rad protein assay kit (Bio-Rad, Hercules, CA, U.S.A.). Total protein (40 μg) was separated by a 10% tricine SDS – PAGE gel (Novex) and transblotted onto a Immobilon-P PVDF membrane (Millipore, Bedford, MA, U.S.A.). Membranes were blocked in immunoblot buffer (mM:Tris-HCl 10 (pH 8.0), EDTA 1, NaCl 150, 0.1% Triton X-100) with 5% bovine serum albumin and 5% nonfat dry milk for 1 h at room temperature with gentle agitation. Membranes were then incubated for 1 h in Immunoblot buffer containing 1% Bovine Serum Albumin and 1% nonfat dry milk with sPLA2 (human synovial) polyclonal antiserum (dilution 1 : 4000, Cayman Chemical) which recognizes both group IIA and group V PLA2. Membranes were washed three times with Immunoblot buffer and incubated with biotinylated goat anti-rabbit IgG antibodies (dilution 1 : 10,000, Vector Laboratories, Burlingame, CA, U.S.A.) for 30 min. Membranes were washed three times with Immunoblot buffer and incubated with horseradish peroxidase streptavidin (dilution 1 : 10,000, Vector Laboratories) for 30 min. Membranes were washed with immunoblot buffer and horseradish peroxidase was revealed by chemiluminesence (ECL kit, Amersham). Human synovial fluid group IIA sPLA2 (Cayman Chemicals) and P388D1 macrophage cell lysate were used as positive controls. Kaleidoscope molecular weight marker proteins were used as standards for SDS – PAGE.
Reverse transcriptase-polymerase chain reaction analysis of group IIA and V sPLA2 gene expression
Total RNA from BAEC and HUVEC was prepared using the Trizol reagent system (Life Technologies, Grand Island, NY, U.S.A.). Total RNA from human brain, heart and intestine was from Clonetech (Palo Alto, CA, U.S.A.). Single strand cDNA was synthesized in a reaction that contained 2 μg of total RNA with 0.5 μg random hexamers (Amersham Pharmacia, Uppsala, Sweden) and 200 units M-MLV reverse transcriptase (Promega, Madison, WI, U.S.A.) at 37°C for 1 h. 0.3 μg of cDNA was then utilized for PCR using Taq DNA polymerase (Promega). Reaction conditions were as follows: denature at 94°C for 40 s, anneal at 50°C for 1 min, extend at 72°C for 1 min for 30 cycles. The primers used to amplify group IIA sPLA2 were designed using the human sequence: 5′-CTT ACC ATG AAG ACC CTC CTA CTG TTG GCA-3′ and 5′-GAG GGG ACT CAG CAA CGA GGG GTG CT-3′. Primers for group V sPLA2 were from highly conserved regions of the human and mouse sequences: 5′-GGC TTC TAC GGC TGT TAC TG-3′ and 5′-GTA GAC GAG CTT CCG GTC AC-3′.
Statistical analysis
Data are mean±s.e.mean. Statistical comparisons were made by analysis of variance followed by an unpaired Student's t-test. Data were considered significantly different if values of P<0.05 were observed.
Results
Induction of endothelial cell PAF synthesis by VEGF is [Ca2+]-dependent
We recently reported that VEGF dose-dependently induced the synthesis of PAF in BAEC (Sirois & Edelman, 1997; Bernatchez et al., 1999). However, these experiments were performed exclusively in the presence of high extracellular Ca2+ concentration (10 mM). In the present study we assessed the contribution of extracellular Ca2+ for the induction of EC PAF production mediated by VEGF on BAEC. In the presence of 10 mM CaCl2, VEGF (1 nM) induced an increase in PAF synthesis which was considered maximal (100%) as compared to control buffer (0%) (Figure 1). However, when BAEC were incubated in the absence of extracellular CaCl2 (0 mM) or CaCl2 (0 mM)+EDTA (1 mM), VEGF failed to significantly increase the basal production of PAF by EC. We then sought to determine the Ca2+ concentration required by the BAEC to synthesize PAF in response to VEGF. Although an increase of CaCl2 to 1 and 10 μM concentrations did not modulate the basal PAF production, the application of CaCl2 to 100 μM and 1 mM induced a significant increase of PAF synthesis, which represented 1.2 and 67% of the maximal amount of PAF synthesized by BAEC when stimulated with VEGF (1 nM) in the presence of 10 mM CaCl2. As a result, the subsequent experiments were performed in the presence of 10 mM CaCl2.
Figure 1.
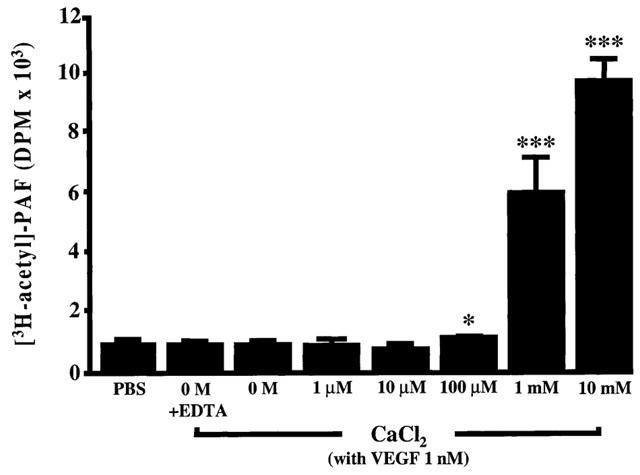
PAF biosynthesis induced by VEGF is Ca2+-dependent. Confluent BAEC (6-well tissue culture plate) were incubated with 3H-acetate and stimulated with VEGF (1 nM) for 15 min with various concentrations of CaCl2 (0 – 10 mM). The radioactive polar lipids samples were extracted by the Bligh and Dyer procedure. The samples were injected into a 4.6×250 mm Varian Si-5 column and eluted with a mobile phase (H2O:CHCl3:MeOH; 5 : 40 : 55; 0.5 ml min−1). Fractions were collected every min after injection and radioactivity was determined with a β-counter. The values are means of at least six experiments. *P<0.05 and ***P<0.001 as compared to control buffer (PBS) as determined by analysis of variance followed by an unpaired Student's t-test.
The effect of VEGF on EC PAF synthesis is attenuated by remodelling pathway and group V sPLA2 inhibitors
Since growing evidence indicates that the remodelling route is activated during inflammation and other hypersensitivity responses, we used a range of specific inhibitors of the aforementioned pathway to determine its possible involvement in VEGF-induced PAF production in EC from two different sources, namely BAEC and HUVEC (Figure 2). First, VEGF (1 nM) increased by 28 and 4 fold the basal production of PAF by BAEC and HUVEC as compared to PBS control buffer (P<0.001), respectively (Figure 3A,B). The addition of a potent inhibitor of both the lyso-PAF-AT and the CoA-IT activities (Sanguinarin; 500 nM) (Snyder et al., 1996) blocked the amount of PAF synthesized by BAEC and HUVEC by 95 and 97%, respectively, in response to VEGF treatment (1 nM) (Figure 3A,B). In contrast, a pretreatment with a selective CoA-IT inhibitor (SKF45905; 10 – 50 μM; IC50=6 μM) (Winkler et al., 1996a) did not attenuate PAF synthesis (Figure 3A,B), suggesting that CoA-IT is not involved in EC PAF synthesis.
Figure 2.
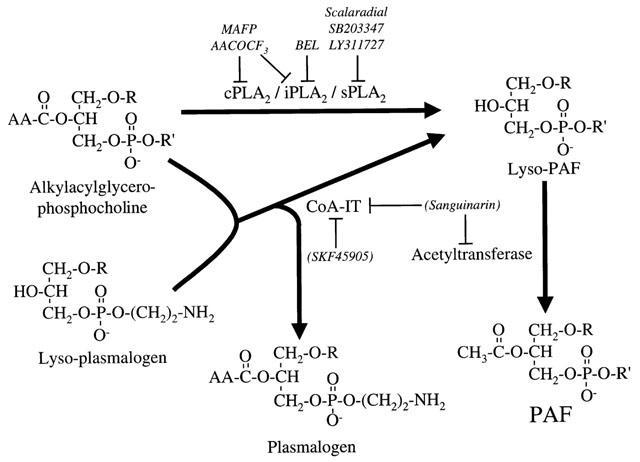
Biosynthesis of lyso-PAF and PAF via the remodelling pathway. The initial hydrolysis of the acyl moiety of alkylacylglycerophosphocholine to form lyso-PAF and arachidonic acid (AA) can be catalyzed by the action of a direct phospholipase A2 (PLA2) or a CoA-independent transacylase (CoA-IT). Lyso-plasmalogen and other lyso-glycerophospholipids can act as the acyl acceptor in the CoA-independent transacylase type of reaction. The lyso-PAF is then converted to PAF by the acetyl-CoA:lyso-PAF acetyltransferase. Above, are the names of selective inhibitors used to identify the enzymes involved in VEGF-mediated PAF synthesis upon remodelling pathway activation. R=(CH2)n-CH3 where n=15 to 17 and R′=(CH2)2N+(CH3)3.
Figure 3.
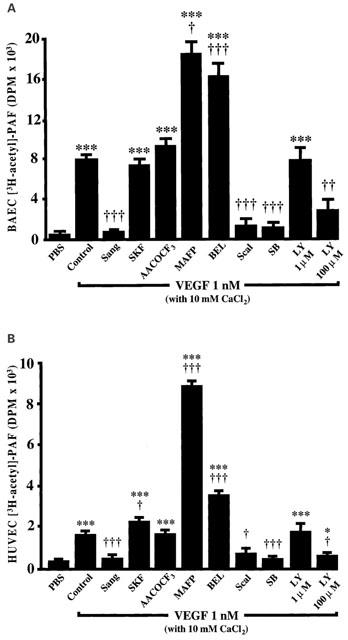
Effect of the remodelling pathway inhibitors on VEGF-induced PAF synthesis. (A) Confluent BAEC (6-well tissue culture plate) were pretreated 5 or 30 min with the remodelling pathway inhibitors Sanguinarine (Sang; 500 μM), SKF45905 (SKF; up to 50 μM), AACOCF3 (AACOCF3; 50 μM), MAFP (MAFP; 10 μM), BEL (BEL; 10 μM), Scalaradial (Scal; 10 μM), SB203347 (SB; 10 μM) and LY311727 (LY; up to 100 μM) stimulated with VEGF (1 nM)+3H-acetate+10 mM CaCl2, and the lipids were extracted and purified as described in Figure 1. The values are means of at least four experiments. (B) Confluent HUVEC were treated as described for BAEC. *P<0.05 and ***P<0.001 as compared to control buffer (PBS), and †P<0.05, ††P<0.01, †††P<0.001 as compared to VEGF (1 nM) as determined by analysis of variance followed by an unpaired Student's t-test.
Considering that PLA2 were also shown to be capable of mediating the production of lyso-PAF, we used a variety of PLA2 inhibitors. First, a pretreatment with a competitive inhibitor of the group IV 85 kDa cytosolic PLA2 (cPLA2) and group VI Ca2+-independent PLA2 (iPLA2) (AACOCF3; 10 – 50 μM; IC50=100 nM for human recombinant cPLA2 and 15 μM for semi-purified iPLA2) (Ackermann et al., 1995) did not reduce VEGF-induced EC PAF synthesis. In contrast, a potent irreversible inhibitor of both the cPLA2 and iPLA2 (MAFP; 10 μM; IC50=600 nM for recombinant cPLA2 and 500 nM for iPLA2) (Leslie, 1997; Balsinde et al., 1997; Fujishima et al., 1999) increased VEGF effect on BAEC and HUVEC PAF synthesis by 126 and 531%, respectively (Figure 3A,B). Interestingly, a specific inhibitor of the group VI iPLA2 (BEL; 10 μM; IC50=60 nM) (Ackermann et al., 1995) also potentiated VEGF effect, elevating PAF biosynthesis by 113 and 129% in BAEC and HUVEC respectively (Figure 3A,B). These results clearly show that both cPLA2 and iPLA2 activity are not directly involved in EC PAF synthesis upon VEGF stimulation as their inhibition did not prevent PAF synthesis. In contrast, a broad-range 14 kDa sPLA2 inhibitor (Scalaradial; 10 μM) (de carvalho & Jacobs, 1991; Marshall et al., 1994; 1995) which has been shown to block several sPLA2s attenuated by 87 and 73% the synthesis of PAF mediated by VEGF in BAEC and HUVEC respectively, thereby suggesting that this biological response is initiated by a sPLA2. Furthermore, the use of a specific group IIA and V sPLA2 inhibitor (SB203347; 10 μM; IC50=500 nM) (Marshall et al., 1995) blocked by 90% VEGF effect on both EC types (Figure 3A,B). Interestingly, the use of a structurally designed sPLA2 inhibitor (LY311727) at a concentration known to block specifically group IIA sPLA2 (1 μM; IC50=<1 μM for group IIA sPLA2) (Murakami et al., 1998) did not have any effect as well on PAF synthesis induced by VEGF, whereas it attenuated by 62 and 81% the synthesis of PAF in BAEC and HUVEC, respectively, when used at a concentration reported to block both groups IIA and V sPLA2 (100 μM; IC50=>50 μM for group V sPLA2) (Figure 3A,B) (Murakami et al., 1998).
BAEC and HUVEC express group V but not group IIA sPLA2
Considering that the addition of SB203347 (10 μM) and LY311727 (100 μM) attenuated PAF biosynthesis, we attempted to determine if BAEC and HUVEC express either group IIA and/or group V sPLA2. Confluent cells were stimulated with VEGF (1 nM) for 15 min, the supernatants and the cells were collected, the proteins were separated by SDS – PAGE and a Western blot analysis was performed with the use of an anti-human sPLA2 antibody which recognizes both group IIA and V sPLA2. Human synovial sPLA2 (group IIA sPLA2 positive control) and P388D1 (group V sPLA2 positive control) (Balboa et al., 1996) presented a different migration pattern from each other (Figure 4). Interestingly, the application of 40 μg of BAEC and HUVEC protein extracts that were stimulated with VEGF revealed that both EC types express group V sPLA2, whereas group IIA sPLA2 expression was not detected (Figure 4). In another experiment, we have observed a constitutive endogenous expression of group V sPLA2 in non-treated BAEC and HUVEC (data not shown). This enzyme remained cell-associated as we did not detect the presence of group V or group IIA sPLA2 in the supernatant of BAEC and HUVEC treated or not with VEGF (1 nM) for 15 min (data not shown).
Figure 4.
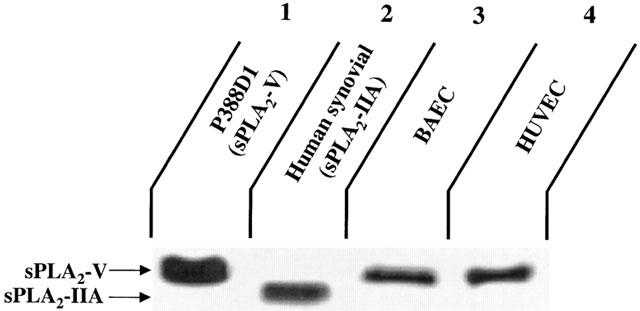
Expression of group IIA and V sPLA2 by BAEC and HUVEC. Confluent BAEC and HUVEC (100 mm tissue culture plate) were rinsed and stimulated with VEGF (1 nM) for 15 min. Cells were scraped and total proteins were isolated. Forty μg of crude proteins were separated by a 10% SDS – PAGE and transblotted onto a PVDF membrane. Proteins were detected by a sPLA2 (human synovial) polyclonal antiserum which recognizes both group IIA and group V sPLA2. Lane 1: P388D1 macrophage cell lysate (group V sPLA2 positive control). Lane 2: Human synovial fluid sPLA2 (group IIA sPLA2 positive control). Lane 3: BAEC lysate. Lane 4: HUVEC lysate.
HUVEC express group V but not group IIA sPLA2 mRNA
Since the data presented in Figure 4 demonstrate the presence of group V sPLA2 and the absence of group IIA sPLA2 in both BAEC and HUVEC, we then sought to confirm by RT – PCR if these two EC types synthesize constitutively group IIA and V sPLA2 mRNA. This approach allows a far more specific and sensitive analysis of the group IIA and V sPLA2 gene expression. The present RT – PCR experiments confirmed the presence of group IIA sPLA2 mRNA in human intestine RNA extracts (positive control), its absence in human brain RNA extracts (negative control) (450 base-pairs; Figure 5A) and the presence of group V sPLA2 in human brain and heart RNA extracts (positive controls) (225 base-pairs; Figure 5B). Interestingly, group IIA sPLA2 was not detected in both stimulated and non-stimulated BAEC and HUVEC (Figure 5A), whereas group V sPLA2 was detected in stimulated and non-stimulated HUVEC but not in BAEC (Figure 5B). As the bovine group V sPLA2 cDNA sequence is still unknown, we may hypothesize that this enzyme has not been detected in BAEC by RT – PCR because of possible mismatches between the human and mouse oligonucleotides used in these experiments, and the bovine group V sPLA2 cDNA sequences.
Figure 5.
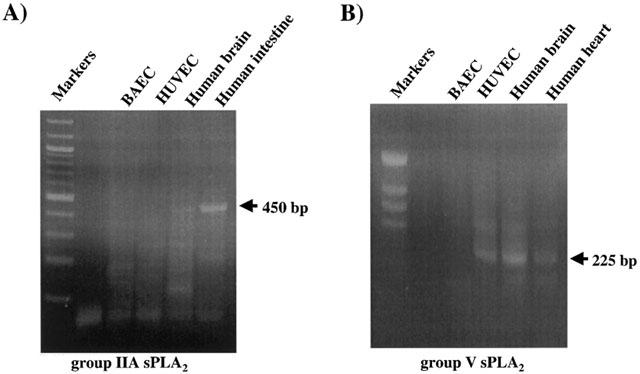
Expression of group IIA and V sPLA2 mRNA by BAEC and HUVEC. Confluent BAEC and HUVEC were lysed, total RNA was extracted and used for RT – PCR. The sizes of the expected amplified fragments are 450 base-pairs for group IIA sPLA2 (A) and 225 base-pairs for group V sPLA2 (B). Human brain and intestine mRNA were used as negative and positive controls respectively, for group IIA sPLA2 RT – PCR (A). Human brain and human heart mRNA were used as positive controls for group V sPLA2 RT – PCR (B).
Discussion
Vascular endothelial growth factor, unlike any other growth factors studied to date, is capable of inducing protein extravasation and it is likely that its angiogenic properties are mediated in large part through the induction of plasma protein leakage (Dvorak et al., 1988; Connolly et al., 1989). In the present study, we report that VEGF induces PAF synthesis by EC through the action of sPLA2, most likely group V sPLA2. These findings not only shed light on the intracellular events initiated by VEGF on the vascular endothelium, but also may enable rational interventions to regulate vascular diseases such as inflammation and possibly uncontrolled angiogenesis attributable to VEGF overexpression.
VEGF increased EC PAF synthesis in vitro
In a previous report we showed for the first time the effect of VEGF on mediating PAF synthesis in BAEC (Sirois & Edelman, 1997). More recently, others have confirmed the capacity of VEGF to elicit PAF synthesis in HUVEC (Montrucchio et al., 2000). It is of interest to note that BAEC are far more potent than HUVEC to synthesize PAF in response to VEGF. This observation is supported by previous studies that have shown that venous endothelial cells produce far less PAF as compared to arterial cells (Whatley et al., 1988). Thus, this difference of response to VEGF is most likely not attributable to differences in VEGF activity on BAEC and HUVEC, but rather to differences between the capacity of the enzymes to synthesize PAF in these two EC types.
VEGF stimulated the remodelling pathway through the action of group V sPLA2
Micromolar (μM) calcium levels are known to be a crucial cofactor for a wide range of intracellular events (Chakravarthy et al., 1999). However, our results showed that an extracellular millimolar (mM) Ca2+ concentration is required to fully elicit PAF synthesis. These results are in accordance with previous studies that showed that maximal BAEC PAF synthesis is directly dependent on millimolar extracellular Ca2+ concentration (Whatley et al., 1989). These authors furthermore demonstrated that this important Ca2+ requirement is attributable to the PLA2-mediated conversion of membrane phospholipids into lyso-PAF induced by various pro-inflammatory stimuli. Interestingly, the Ca2+-concentration dependence of enzymatic activity is one of the major defining characteristics for 14 kDa sPLA2s, since they were reported to bind millimolar Ca2+ within a highly conserved calcium binding loop in order to stabilize the transition state of the phospholipid substrate (Dennis, 1994; 1997; Tishfield, 1997). Hence, our results demonstrate that biosynthesis of PAF in BAEC through Flk-1/KDR tyrosine kinase receptor activation by VEGF is at least in part similar to the one observed following activation of G protein-coupled receptors and raise the possibility that sPLA2 may participate in mediating VEGF-induced EC PAF synthesis.
Sanguinarin has previously been shown to have a high degree of selectivity towards the inhibition of both the lyso-PAF-AT and CoA-IT activities (Snyder et al., 1996). Therefore, this inhibitor should be a valuable tool to assess the possible involvement of the remodelling pathway in numerous cellular events. Pretreatment of BAEC and HUVEC with Sanguinarin almost completely abolished the synthesis of PAF induced by VEGF, suggesting that VEGF is capable of inducing the activation of the remodelling pathway. To the best of our knowledge, these data are the first to show that a growth factor can mediate such activity, and also confirm that the remodelling route is influenced by proinflammatory stimuli.
A possible key player in the synthesis of PAF through the remodelling pathway is the CoA-IT. This enzyme transfers an sn-2 acyl group from a diacylglycerophosphatide to a lyso-phospholipid in the presence or absence of Coenzyme A (Snyder et al., 1992; Winkler et al., 1996a). However, a pretreatment with a specific CoA-IT inhibitor (SKF45905) (Winkler et al., 1996a) did not attenuate PAF synthesis in both EC types. This observation is in accordance with previous reports indicating that EC contain far less CoA-IT activity than inflammatory cells such as neutrophils and monocytes (Winkler & Chilton, 1995; Winkler et al., 1996b). Taken together, these observations demonstrate that the CoA-IT is unlikely to take part in EC lyso-PAF synthesis mediated by VEGF and suggest that lyso-PAF synthesis is initiated through the activation of a sPLA2. This hypothesis is relevant since others have shown that VEGF could promote the synthesis of prostaglandins in EC, which is recognized as a marker of PLA2 activity (Murohara et al., 1998).
Phospholipases A2 consist of a growing superfamily of enzymes, also capable of hydrolyzing membrane phospholipids with the concomitant production of lyso-phospholipids (Dennis, 1994; 1997; Tishfield, 1997). Though some of these enzymes are very unlikely to play a role in VEGF-induced PAF synthesis by BAEC, others may be directly involved in EC PAF synthesis. Since our results support that Ca2+ is a crucial cofactor for VEGF-induced PAF production by EC, likely candidates for mediating such activity are the groups IIA and V sPLA2. Nonetheless, the possible effects of other recently described sPLA2 (groups IID, IIE, IIF and X) cannot be ruled out (Six & Dennis, 2000), despite the fact that their expression have not been reported in EC yet.
Hence, we used different inhibitors of the groups IIA and V sPLA2. A pretreatment with a non-specific 14 kDa sPLA2 inhibitor (Scalaradial) previously reported to block several sPLA2s including group IIA and most probably group V sPLA2 (de carvalho & Jacobs, 1991; Marshall et al., 1994; 1995) blocked almost completely EC PAF synthesis. Moreover, an inhibitor of both groups IIA and V sPLA2 (SB203347) (Marshall et al., 1995) which possess a 40 fold specificity for groups IIA and V sPLA2 over group IV 85 kDa cPLA2 abrogated almost completely VEGF-induced EC PAF synthesis. In contrast, treatment with LY311727 (1 μM), another well-described sPLA2 inhibitor which at such concentration blocks group IIA sPLA2 activity, did not significantly affect VEGF induction of PAF synthesis in both BAEC in HUVEC. However, when used at a concentration reported to block both groups IIA and V sPLA2 (100 μM) (Murakami et al., 1998), LY311727 inhibited significantly VEGF-induced PAF synthesis. Taken together, these results demonstrate that group V sPLA2 is responsible for the synthesis of lyso-PAF induced by VEGF. Interestingly, several pieces of evidence support such hypothesis. Others have reported that group V sPLA2 can catalyze more efficiently the hydrolysis of phospholipid bilayers than group IIA sPLA2, liberating arachidonic acid and lyso-phospholipids, while group IIA sPLA2 appears to have a relatively low affinity for membrane phospholipids (Han et al., 1998).
Group IV cPLA2 and group VI iPLA2 are two cell-associated phospholipases both capable of influencing cellular fatty acid metabolism by increasing free arachidonic acid levels (Balsinde & Dennis, 1996; Leslie, 1997). Thus, these two enzymes could possibly be linked to the synthesis of lyso-PAF from membrane phospholipids initiated by VEGF. A specific inhibitor of both the group IV cPLA2 and VI iPLA2 (AACOCF3) (Ackermann et al., 1995, McNicol & Nickolaychuk, 1995; Leslie, 1997; Fujishima et al., 1999) did not significantly modulate PAF synthesis induced by VEGF. In contrast, an irreversible inhibitor of both the cPLA2 and iPLA2 with a greater effect on iPLA2 than cPLA2 (MAFP) (Leslie, 1997; Balsinde & Dennis, 1997; Fujishima et al., 1999) greatly potentiated VEGF effect. Moreover, a specific iPLA2 inhibitor (BEL) also increased the synthesis of PAF induced by VEGF in BAEC and HUVEC. Thus, this clearly demonstrates that cPLA2 and iPLA2 are not directly involved in EC lyso-PAF biosynthesis induced by VEGF, and that iPLA2 inhibition by either BEL or MAFP potentiates VEGF effect on EC PAF synthesis. This furthermore suggests that group VI iPLA2 may hydrolyze similar membrane phospholipids to the ones required to mediate EC PAF synthesis, and as a result may act in a competition fashion with the enzyme involved in PAF synthesis. Hence, by blocking the iPLA2, a pool of membrane phospholipids may be exclusively hydrolyzed by the group V sPLA2 leading to increased amount of lyso-PAF synthesized. This hypothesis is furthermore supported by the fact that these two enzymes do not demonstrate substrate selectivity, unlike cPLA2 which prefers arachidonic acid-containing phospholipids. Finally, these data indicate as well that cPLA2 is unlikely to take part in EC PAF production and evidence for this is starting to emerge. Indeed, several reports indicate that cPLA2 is not involved in monocyte or neutrophil PAF biosynthesis but rather in prostanoid production (Leslie, 1997; Marshall et al., 1997; Winkler et al., 1997). As a result, this enzyme is most probably not implicated in EC PAF synthesis. In addition, this enzyme is active at low micromolar Ca2+, and we showed that at such concentrations VEGF did not induce the synthesis of PAF.
It is interesting to note that previous studies have shown that cPLA2 activity is essential for the pro-inflammatory activity of group V sPLA2 (Balsinde et al., 1998; Cho, 2000). However, these studies investigate the implication of these two enzymes to prostaglandin synthesis. In contrast, we show that PAF synthesis mediated by group V sPLA2 is independent of cPLA2 activity. This is in agreement with a previous report that has also shown that PAF formation is dependent of sPLA2 and independent of cPLA2 (Marshall et al., 1999), one might hypothesize that cPLA2 activity is required for prostanoid synthesis, whereas cPLA2 appears not to be implicated in PAF synthesis. This hypothesis is furthermore supported by a recent report in which it has been shown that group V sPLA2 alone could mediate lyso-PAF synthesis (Murakami et al., 2001).
In order to support the hypothesis that group V sPLA2 mediates BAEC PAF synthesis, we then sought to determine if BAEC and HUVEC express a significant amount of group IIA and/or V sPLA2 enzymes. Western blot analysis indicated that both BAEC and HUVEC either stimulated or not with VEGF indeed express group V sPLA2 and not group IIA sPLA2, thereby confirming the possible involvement of group V sPLA2 in PAF synthesis. Finally, group V sPLA2 remained cell-associated and was not detected in the supernatant of BAEC and HUVEC challenged or not with VEGF. We confirmed also by RT – PCR the presence of group V sPLA2 and the absence of group IIA sPLA2 at least in HUVEC. Since our data support a direct role for group V sPLA2 in VEGF-induced PAF synthesis in BAEC and that Western blot analysis clearly shows that BAEC express group V sPLA2, one might hypothesize that the discrepancy observed between Western blot and RT – PCR analyses for BAEC expression of group V sPLA2 may be rationalized by the fact that the oligonucleotides used for RT – PCR were synthesized according to the human group V sPLA2 cDNA sequences, since the bovine gene has not yet been sequenced. As a result, mismatches between bovine group V sPLA2 mRNA and the RT – PCR oligomers used might have been present. Nonetheless, our RT – PCR experiments support the crucial role of group V sPLA2 in HUVEC, and completely dissipates any possible ambiguity regarding a role of group IIA sPLA2 in HUVEC PAF synthesis since this enzyme was not detected in HUVEC by Northern and Western blot analyses. Interestingly, others have shown that group IIA sPLA2 expression is inducible by pro-inflammatory agents (Murakami et al., 1997). Moreover, the fact that HUVEC do not express group IIA sPLA2 and that the blockade of this enzyme by LY311727 (1 μM) did not reduce PAF synthesis confirms that the inhibitory effect of the two specific inhibitors of groups IIA and V sPLA2, namely SB203347 and LY311727 (100 μM), on PAF synthesis induced by VEGF is not through the inhibition of group IIA sPLA2 but rather through the inhibition of group V sPLA2.
VEGF and group V sPLA2
Though group V sPLA2 has been cloned only recently (Chen and Dennis, 1998), a growing body of evidence has confirmed that this novel enzyme is constitutively expressed mainly in the cardiovascular system (Chen et al., 1994) and is indeed an active effector in fatty acid metabolism. It was shown to take part in arachidonic acid-mediated signal transduction in macrophage-like cell line P388D1 (Balboa et al., 1996), as well as in eicosanoid formation in mast cells (Reddy et al., 1997). Moreover, group V rather that group IIA as previously believed, is the primary 14 kDa PLA2 synthesized by P388D1 cells (Balboa et al., 1996). In the present study, we demonstrate that this newly characterized enzyme is likely responsible for VEGF-induced PAF synthesis, that it is expressed in EC from both bovine aortas and human umbilical veins, and that it remains associated with the cells that were challenged with VEGF. This is in agreement with a recent report that has shown that group V sPLA2 was cell associated and identified intracellularly (Bingham et al., 1999).
Previous investigations have proposed that an increase in microvascular permeability is a crucial step in angiogenesis associated with tumours and wounds (Connolly et al., 1989; Folkman & Klagsbrun, 1997). According to this hypothesis, a major function of VEGF is probably the induction of plasma protein leakage. We have shown earlier that VEGF-induced protein extravasation was abolished by a PAF receptor antagonist in vivo (Sirois & Edelman, 1997). Since this study supports the idea that group V sPLA2 is responsible for the PAF synthesis mediated by VEGF and that diverse studies suggest that PAF acts as a pro-angiogenic factor (Camussi et al., 1995; Bussolino & Camussi, 1995; Montrucchio et al., 2000), it would be of interest to investigate if the inhibition of this enzyme may interfere with VEGF inflammatory effect in vivo, as well as with its angiogenic potential.
In conclusion, our data showed that the production of PAF elicited by VEGF on BAEC and HUVEC is mediated by the activation of the remodelling pathway, and more specifically through the action of the group V sPLA2 and lyso-PAF-AT. As the inflammatory reaction mediated by VEGF might be an essential key factor of angiogenesis, the inhibition of PAF synthesis mediated by VEGF might prevent the deleterious inflammation and angiogenesis imputable to VEGF overexpression in vivo.
Acknowledgments
We wish to thank Maria Kotsiopriftis and Nadheige Lochard for their technical assistance. We would like to thank also Dr James D. Winkler for his scientific comments. This study was supported by grants from the Medical Research Council of Canada (MRCC) (MT-14378), Canadian Institutes of Health Research (CIHR) (MOP-43919) and the Heart and Stroke Foundation of Québec to Dr Sirois and by NIH grant HD26171-10 to Dr Dennis. Mr Bernatchez is a recipient of a studentship from the CIHR, Dr Winstead is supported by the National Institute of Health training grant (DK070202) and Dr Sirois is a recipient of a scholarship from the Heart and Stroke Foundation of Canada.
Abbreviations
- AACOCF3
arachidonyl trifluoromethyl ketone
- BAEC
bovine aortic endothelial cells
- BEL
bromoenol lactone
- CoA-IT
coenzyme A-independent transacylase
- DMEM
Dulbecco's modified Eagle's medium
- EC
endothelial cells
- HBSS
Hank's balanced salt solution
- HPLC
high performance liquid chromatography
- HUVEC
human umbilical vein endothelial cells
- iPLA2
calcium-independent phospholipase A2
- lyso-PAF-AT
lyso-PAF acetyltransferase
- MAFP
methyl arachidonyl fluorophosphate
- PAF
platelet-activating factor
- PGI2
prostacyclin
- RT – PCR
reverse transcriptase-polymerase chain reaction
- sPLA2
secreted phospholipase A2
- VEGF
vascular endothelial growth factor
References
- ACKERMANN E.J., CONDE-FRIEBOES K., DENNIS E.A. Inhibition of macrophage Ca(2+)-independent phospholipase A2 by bromoenol lactone and trifluoromethyl ketones. J. Biol. Chem. 1995;270:445–450. doi: 10.1074/jbc.270.1.445. [DOI] [PubMed] [Google Scholar]
- BALBOA M.A., BALSINDE J., WINSTEAD M.V., TISHFIELD J.A., DENNIS E.A. Novel group V phospholipase A2 involved in arachidonic acid mobilization in murine P388D1 macrophages. J. Biol. Chem. 1996;271:32381–32384. doi: 10.1074/jbc.271.50.32381. [DOI] [PubMed] [Google Scholar]
- BALSINDE J.J, , BALBOA M.A., DENNIS E.A. Antisense inhibition of Group VI Ca2+-independent phospholipase A2 blocks phospholipid fatty acid remodeling in murine P388D1 macrophages. J. Biol. Chem. 1997;272:29317–29321. doi: 10.1074/jbc.272.46.29317. [DOI] [PubMed] [Google Scholar]
- BALSINDE J.J, , BALBOA M.A., DENNIS E.A. Funtionnal coupling between secretory phospholipase A2 and cyclooxygenase-2 and its regulation by cytosolic group IV phospholipase A2. Proc. Natl. Acad. Sci. U.S.A. 1998;95:7951–7956. doi: 10.1073/pnas.95.14.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BALSINDE J., DENNIS E.A. Bromoenol lactone inhibits magnesium-dependent phosphatidate phosphohydrolase and blocks triacylglycerol biosynthesis in mouse P388D1 macrophages. J. Biol. Chem. 1996;271:31937–31941. doi: 10.1074/jbc.271.50.31937. [DOI] [PubMed] [Google Scholar]
- BALSINDE J., DENNIS E.A. Function and inhibition of intracellular calcium-independent phospholipase A2. J. Biol. Chem. 1997;272:16069–16072. doi: 10.1074/jbc.272.26.16069. [DOI] [PubMed] [Google Scholar]
- BERNATCHEZ P., SOKER S., SIROIS M.G. VEGF effect on endothelial cell proliferation migration and PAF synthesis is mediated through the activation of Flk-1 receptor. J. Biol. Chem. 1999;274:31047–31054. doi: 10.1074/jbc.274.43.31047. [DOI] [PubMed] [Google Scholar]
- BINGHAM C.O., FIJNEMAN R.J., FRIEND D.S., GODDEAU R.P., AUSTEN K.F., ARM J.P. Low molecular weight group IIA and group V phospholipase A2 enzymes have different intracellular locations in mouse bone marrow-derived mast cells. J. Biol. Chem. 1999;274:31476–31484. doi: 10.1074/jbc.274.44.31476. [DOI] [PubMed] [Google Scholar]
- BUSSOLINO F., CAMUSSI G. Platelet-activating factor produced by endothelial cells: a molecule with autocrine and paracrine properties. Eur. J. Biochem. 1995;229:327–337. [PubMed] [Google Scholar]
- CAMUSSI G., MONTRUCCHIO G., LUPIA E., DE MARTINO A., PERONA L., ARESE M., VERCELLONE A., TONIOLO A., BUSSOLINO F. Platelet-activating factor directly stimulates in vitro migration of endothelial cells and promotes in vivo angiogenesis. J. Immunol. 1995;154:6492–6501. [PubMed] [Google Scholar]
- CHAKRAVARTHY B., MORLEY P., WHITFIELD J. Ca2+-calmodulin and protein kinase C: a hypothetical synthesis of their conflicting convergences on shared substrate domains. Trends Neurol. Sci. 1999;22:12–16. doi: 10.1016/s0166-2236(98)01288-0. [DOI] [PubMed] [Google Scholar]
- CHEN Y., DENNIS E.A. Expression and characterization of human group V phospholipase A2. Biochim. Biophys. Acta. 1998;1394:57–64. doi: 10.1016/s0005-2760(98)00098-8. [DOI] [PubMed] [Google Scholar]
- CHEN J., ENGLE S.J., SEILHAMER J.J., TISHFIELD J.A. Cloning and recombinant expression of a nove human low molecular weight Ca2+-dependent phospholipase A2. J. Biol. Chem. 1994;269:2365–2368. [PubMed] [Google Scholar]
- CHO W. Structure, function and regulation of Group V phospholipase A2. Biochim. Biophys. Acta. 2000;1488:48–58. doi: 10.1016/s1388-1981(00)00109-8. [DOI] [PubMed] [Google Scholar]
- CONNOLLY D.T., HEUVELMAN D.M., NELSON R., OLANDER J.V., EPPLEY B.L., DELFINO J.J., SIEGEL N.R., LIMGRUBER R.M., FEDER J. Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J. Clin. Invest. 1989;84:1470–1478. doi: 10.1172/JCI114322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE CARVALHO M.S., JACOBS R.S. Two-step inactivation of bee venom phospholipase A2 by scalaradial. Biochem. Pharmacol. 1991;42:1621–1626. doi: 10.1016/0006-2952(91)90432-5. [DOI] [PubMed] [Google Scholar]
- DENNIS E.A. Diversity of group types, regulation, and function of phospholipase A2. J. Biol. Chem. 1994;269:13057–13060. [PubMed] [Google Scholar]
- DENNIS E.A. The growing phospholipase A2 superfamily of signal transduction enzymes. Trends Biochem. Sci. 1997;22:1–2. doi: 10.1016/s0968-0004(96)20031-3. [DOI] [PubMed] [Google Scholar]
- DVORAK H.F., NAGY J.A., DVORAK J.T., DVORAK A.M. Identification and characterization of the blood vesseles of solid tumors that are leaky to circulating macromolecules. Am. J. Pathol. 1988;133:95–109. [PMC free article] [PubMed] [Google Scholar]
- FERRARA N. Vascular endothelial growth factor. The trigger for neovascularization in the eye. Lab. Invest. 1995;72:615–618. [PubMed] [Google Scholar]
- FOLKMAN J. What is the evidence that tumors are angiogenesis-dependent. J. Natl. Cancer Inst. 1991;82:4–6. doi: 10.1093/jnci/82.1.4. [DOI] [PubMed] [Google Scholar]
- FOLKMAN J., KLAGSBRUN M. Angiogenic factors. Science. 1997;235:442–447. doi: 10.1126/science.2432664. [DOI] [PubMed] [Google Scholar]
- FUJISHIMA H., SANCHEZ MEJIA R.O., BINGHAM C.O., LAM B.K., SAPIRSTEIN A., BONVENTRE J.V., AUSTEN K.F., ARM J.P. Cytosolic phospholipase A2 is essential for both the immediate and delayed phases of eicosanoid generation in mouse bone marrow-derived mast cells. Proc. Natl. Acad. Sci. U.S.A. 1999;96:4803–4807. doi: 10.1073/pnas.96.9.4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAN S.-K., YOON E.T., WONHWA C. Bacterial expression and characterization of human secretory class V phospholipase A2. Biochem. J. 1998;331:353–357. doi: 10.1042/bj3310353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JACKSON J.R., BOLOGNESE B., MANGAR C.A., HUBBARD W.C., MARSHALL L.A., WINKLER J.D. The role of platelet activating factor and other lipid mediators in inflammatory angiogenesis. Biochim. Biophys. Acta. 1998;1392:145–152. doi: 10.1016/s0005-2760(98)00012-5. [DOI] [PubMed] [Google Scholar]
- JACKSON J.R., SEED M.P., KIRCHER C.H., WILLOUGHBY D.A., WINKLER J.D. The codependence of angiogenesis and chronic inflammation. FASEB J. 1997;11:457–465. [PubMed] [Google Scholar]
- KIM K.J., LI B., WINER J., ARMANINI M., GILLET N., PHILIPS H.S., FERRARA N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumor growth in vivo. Nature. 1993;362:841–844. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- KONDO S., ASANO M., OHMORI I., SUZUKI H. Vascular endothelial growth factor/vascular permeability factor is detectable in the sera of tumor bearing mice and cancer patients. Biochim. Biophys. Acta. 1994;1221:211–214. doi: 10.1016/0167-4889(94)90016-7. [DOI] [PubMed] [Google Scholar]
- LESLIE C.C. Properties and regulation of cytosolic phospholipase A2. J. Biol. Chem. 1997;272:16709–16712. doi: 10.1074/jbc.272.27.16709. [DOI] [PubMed] [Google Scholar]
- MARSHALL L.A., BOLOGNESE B., ROSHAK A. Respective roles of the 14 kDa and 85 kDa phospholipase A2 enzymes in human monocyte eicosanoid formation. Adv. Exp. Med. Biol. 1999;469:215–219. doi: 10.1007/978-1-4615-4793-8_32. [DOI] [PubMed] [Google Scholar]
- MARSHALL L.A., BOLOGNESE B., WINKLER J.D., ROSHAK J. Depletion of human monocyte 85-kDa phospholipase A2 does not alter leukotriene formation. J. Biol. Chem. 1997;272:759–765. doi: 10.1074/jbc.272.2.759. [DOI] [PubMed] [Google Scholar]
- MARSHALL L.A., HALL R.H., WINKLER J.D., BADGER A., BOLOGNESE B., ROSHAK A., FLAMBERG P.L., SUNG C.M., CHABOT-FLETCHER M., ADAMS J.L., MAYER R.J. SB 203347, an inhibitor of 14 kDa phospholipase A2, alters human neutrophil arachidonic acid release and metabolism and prolongs survival in murine endotoxic choc. J. Pharmacol. Exp. Ther. 1995;274:1254–1262. [PubMed] [Google Scholar]
- MARSHALL L.A., WINKLER J.D., GRISWOLD D.E., BOLOGNESE B., ROSHAK A., SUNG C.-M., WEBB E.F., JACOBS R. Effects of Scalaradial, a type II phospholipase A2 inhibitor, on human neutrophil arachidonic acid mobilization and lipid mediator formation. J. Pharmacol. Exp. Ther. 1994;268:709–717. [PubMed] [Google Scholar]
- MCNICOL A., NICKOLAYCHUK B.R. Inhibition of collagen-induced platelet activation by arachidonyl trifluoromethyl ketone. Biochem. Pharmacol. 1995;50:1795–1801. doi: 10.1016/0006-2952(95)02048-9. [DOI] [PubMed] [Google Scholar]
- MONTRUCCHIO G., LUPIA E., BATTAGLIA E., DEL SORBO L., BOCCELLINO M., BIANCONE, EMANUELLI G., CAMUSSI G. Platelet-activating factor enhances vascular endothelial growth factor-induced endothelial cell motility and neoangiogenesis in a murine matrigel model. Arterioscler. Thromb. Vasc. Biol. 2000;20:80–88. doi: 10.1161/01.atv.20.1.80. [DOI] [PubMed] [Google Scholar]
- MURAKAMI M., KODURI R.S., ENOMOTO A., SHIMBARA S., SEKI M., YOSHIHARA K., SINGERS A., VALENTIN E., GHOMASHCHI F., LAMBEAU G., GELB M.H., KUDO I. Distinct arachidonate-releasing functions of mammalian secreted phospholipase A2s in human embryonic kidney 293 and rat mastocytoma RBL-2H3 cells through heparan sulfate shuttling and external plasma membrane mechanisms. J. Biol. Chem. 2001;276:10083–10096. doi: 10.1074/jbc.M007877200. [DOI] [PubMed] [Google Scholar]
- MURAKAMI M., SHIMBARA S., KEMBE T., KUWATA H., WINSTEAD M.V., TISHFIELD J.A., KUDO I. Platelet-activating factor enhances vascular endothelial growth factor-induced endothelial cell motility and neoangiogenesis in a murine matrigel model. J. Biol. Chem. 1998;273:14411–14423. [Google Scholar]
- MURAKAMI M., TADA K., NAKAJIMA K.-I., KUDO I. The functions of five distinct mammalian phospholipase A2 in regulating arachidonic acid release. Type IIA and type V secretory phospholipase A2 are functionally redundant and act in concert with cytosolic phospholipase A2. Adv. Exp. Med. Biol. 1997;400:37–42. doi: 10.1074/jbc.273.23.14411. [DOI] [PubMed] [Google Scholar]
- MUROHARA T., HOROWITZ J.R., SILVER M., TSURUMI Y., CHEN D., SULLIVAN A., ISNER J.M. Vascular endothelial growth factor/vascular permeability factor enhances vascular permeability via nitric oxide and prostacyclin. Circulation. 1998;97:99–107. doi: 10.1161/01.cir.97.1.99. [DOI] [PubMed] [Google Scholar]
- REDDY S.T., WINSTEAD M.V., TISHFIELD J.A., HERSHMAN H.R. Analysis of the secretory phospholipase A2 that mediates prostaglandine production in mast cells. J. Biol. Chem. 1997;272:13591–13596. doi: 10.1074/jbc.272.21.13591. [DOI] [PubMed] [Google Scholar]
- SIROIS M.G., EDELMAN E.R. VEGF effect on vascular permeability is mediated by synthesis of platelet-activating factor. Am. J. Physiol. 1997;272:H2746–H2756. doi: 10.1152/ajpheart.1997.272.6.H2746. [DOI] [PubMed] [Google Scholar]
- SIX D.A., DENNIS E.A. The expanding superfamily of phospholipase A2 enzymes: classification and characterization. Biochim. Biophys. Acta. 2000;1488:1–19. doi: 10.1016/s1388-1981(00)00105-0. [DOI] [PubMed] [Google Scholar]
- SNYDER F., FITZGERALD V., BLANK M.L. Biosynthesis of Platelet-activating factor and enzyme inhibitors. Adv. Exp. Med. Biol. 1996;416:5–10. doi: 10.1007/978-1-4899-0179-8_2. [DOI] [PubMed] [Google Scholar]
- SNYDER F., LEE T.-C., BLANK M. The role of transacylase in the metabolism of arachidonate and platelet activating factor. Prog. Lipid Res. 1992;31:65–86. doi: 10.1016/0163-7827(92)90016-c. [DOI] [PubMed] [Google Scholar]
- SUNDERKOTTER C., STEINBRINK K., GOEBELER M., BHARDWAJ R., SORG C. Macrophages and angiogenesis. J. Leuk. Biol. 1994;55:410–422. doi: 10.1002/jlb.55.3.410. [DOI] [PubMed] [Google Scholar]
- TISHFIELD J.A. A reassessment of the low molecular weight phospholipase A2 gene family in mammals. J. Biol. Chem. 1997;272:17247–17250. doi: 10.1074/jbc.272.28.17247. [DOI] [PubMed] [Google Scholar]
- VENABLE M.E., ZIMMERMAN G.A., MCINTYRE T.M., PRESCOTT S.M. Platelet-activating factor: a phospholipid autacoid with diverse actions. J. Lipid Res. 1993;34:691–702. [PubMed] [Google Scholar]
- WHATLEY R.E., NELSON P., ZIMMERMAN G.A., STEVENTS D.L., PARKER C.J., MCINTYRE T.M., PRESCOTT S.M. The regulation of Platelet-activating Factor production in endothelial cells. J. Biol. Chem. 1989;264:6325–6333. [PubMed] [Google Scholar]
- WHATLEY R.E., ZIMMERMAN G.A., MCINTYRE T.M., PRESCOTT S.M. Endothelium from diverse vascular sources synthesizes platelet-activating factor. Arteriosclerosis. 1988;8:321–331. doi: 10.1161/01.atv.8.3.321. [DOI] [PubMed] [Google Scholar]
- WINKLER J.D., BOLOGNESE B., ROSHAK A.K., SUNG C.-M., MARSHALL L.A. Evidence that the 85 kDa phospholipase A2 is not linked to CoA-independent transacylase-mediated production of platelet-activating factor in human monocytes. Biochim. Biophys. Acta. 1997;1346:173–184. doi: 10.1016/s0005-2760(97)00032-5. [DOI] [PubMed] [Google Scholar]
- WINKLER J.D., CHILTON F.H. Inflammation: Mediators and Pathways. CRC Press; 1995. Pharmacology of CoA-independent transacylase; pp. 147–171. [Google Scholar]
- WINKLER J.D., ERIS T., SUNG C.-M., CHABOT-FLETCHER M., MAYER R.J., SURETTE M.E., CHILTON F.H. Inhibitors of coenzyme A-independent transacylase induce apoptosis in human HL-60. J. Pharmacol. Exp. Ther. 1996a;279:956–966. [PubMed] [Google Scholar]
- WINKLER J.D., SUNG C.-M., MARSHALL L.A., CHILTON F.H. Inhibitors of arachidonate metabolism and effects on PAF production. Adv. Exp. Med. Biol. 1996b;416:11–15. doi: 10.1007/978-1-4899-0179-8_3. [DOI] [PubMed] [Google Scholar]