

Abstract
Human isolated subcutaneous arteries were mounted in a myograph and isometric tension measured. In some experiments, intracellular calcium [Ca2+]i was also measured using fura-2.
Angiotensin II (100 pM – 1 μM) increased [Ca2+]i and tone in a concentration-dependent manner. The effects of angiotensin II (100 nM) were inhibited by an AT1-receptor antagonist, candesartan (100 pM).
Ryanodine (10 μM), had no effect on angiotensin II-induced responses, but removal of extracellular Ca2+ abolished angiotensin II-induced rise in [Ca2+]i and tone. Inhibition of Ca2+ entry by Ni2+ (2 mM), also inhibited angiotensin II responses. The dihydropyridine, L-type calcium channel antagonist, amlodipine (10 μM), only partially attenuated angiotensin II responses.
Inhibition of protein kinase C (PKC) by chelerythrine (1 μM), or by overnight exposure to a phorbol ester (PDBu; 500 nM) had no effect on angiotensin II-induced contraction.
Genistein (10 μM), a tyrosine kinase inhibitor, inhibited angiotensin II-induced contraction, but did not inhibit the rise in [Ca2+]i, suggesting that at this concentration it affected the calcium sensitivity of the contractile apparatus. Genistein did not affect responses to norepinephrine (NE) or high potassium (KPSS).
A selective MEK inhibitor, PD98059 (30 μM), inhibited both the angiotensin II-induced contraction and rise in [Ca2+]i, but had no effect on responses to NE or KPSS.
AT1 activation causes Ca2+ influx via L-type calcium channels and a dihydropyridine-insensitive route, but does not release Ca2+ from intracellular sites. Activation of tyrosine kinase(s) and the ERK 1/2 pathway, but not classical or novel PKC, also play a role in angiotensin II-induced contraction in human subcutaneous resistance arteries.
Keywords: Smooth muscle, artery, angiotensin II, AT1 receptors, tyrosine kinase, ERK, PKC, intracellular Ca2+
Introduction
The octapeptide angiotensin II plays a major role in the regulation of blood pressure and also influences vascular growth and remodelling (Timmermans et al., 1993). In most blood vessels, the vascular effects of angiotensin II are predominantly mediated by the AT1-receptor (Timmermans et al., 1993). The AT1-receptor has been reported to couple to numerous signal transduction pathways in a variety of cell types, including intracellular Ca2+[Ca2+]i, phospholipase C, PKC, tyrosine kinases, MAPK and JAK/STAT (reviewed in Hughes, 2000). In vascular smooth muscle a number of these pathways (e.g. tyrosine kinases, MAPK and JAK/STAT) have been implicated in angiotensin II-induced growth (Berk & Corson, 1997), but their possible importance in angiotensin II-induced vasoconstriction is less well explored. Changes in [Ca2+]i play a major role in smooth muscle contraction, but tyrosine kinase and MAPK pathways have also been shown to modulate contraction in some smooth muscle through a variety of mechanisms (Hollenberg, 1994; Hughes & Wijetunge, 1998; Matrougui et al., 2000; Watts, 1996).
There is also evidence that PKC contributes to agonist-induced contraction in vascular smooth muscle (Morgan & Leinweber, 1998). Angiotensin II-induced contraction in rat aortic rings has been reported to be blocked by the putative PKC inhibitors, piperazine (H7), piperazine (CL), staurosporin, calphostin-C (Oriji & Keiser, 1997), and the rise in intracellular Ca2+ in response to angiotensin II has also been shown to be blunted following inhibition of PKC with chelerythrine in isolated intact rabbit afferent arterioles (Salomonsson et al., 1997).
However, the relative importance of various signalling pathways varies considerably between arteries of different calibre or from different sites. Consequently, the object of these studies was to examine the mechanisms involved in the contractile action of angiotensin II in human isolated subcutaneous arteries of a size sufficiently small to play a role in the regulation of peripheral vascular resistance in man (Mulvany & Aalkjaer, 1990).
Methods
Human subcutaneous resistance arteries (internal diameter ∼200 – 400 μm) were obtained from tissue resected at surgery and mounted as ring segments in an isometric myograph (Mulvany & Halpern, 1977). Use of this tissue conformed to local Ethical Committee Guidelines. The myograph contained 10 ml of physiological saline solution (PSS) (mM: NaCl, 118; KCl, 4.7; CaCl2.6H2O, 2.5; MgSO4.7H2O, 1.17; NaHCO3, 25.0; NaH2PO4.2H2O, 1.0; Na2EDTA, 0.03; and glucose, 5.5) maintained at 37°C and aerated with 95% oxygen/5% carbon dioxide.
The vessels were allowed to equilibrate for 1 h and then set at a ‘normalized' internal circumference 0.9 L100 estimated to be 0.9 times the circumference they would maintain if relaxed and exposed to 100 mmHg transmural pressure. This was calculated for each individual vessel on the basis of the passive length-tension characteristics of the artery and the Laplace relationship (Mulvany & Halpern, 1977). This procedure optimized active force generation by these vessels. All experiments were started by repetitively stimulating vessels for ∼2 min with a high potassium solution (KPSS), comprising PSS with equimolar substution of NaCl by KCl, until reproducible contractions were elicited. Although tachyphylaxis to angiotensin II is not a major problem in these vessels (Garcha et al., 1999), the possibility of tachyphylaxis was minimized by allowing at least a 2 h interval between exposures to angiotensin II.
In some studies [Ca2+]i and force were measured simultaneously using the fluorescent calcium indicator fura-2, essentially as previously described (Jensen et al., 1992). Arteries were set up in a single-channel myograph dedicated to fluorescence measurements and incubated with 6 μM fura-2/AM for 2 h in PSS at 37°C. After loading, vessels were thoroughly washed to remove free fura-2AM and allowed to equilibrate in PSS for 30 min before fluorescence measurements were made. Fluorescence was measured using a Deltascan spectrofluorimeter (Photon Technology International Inc., South Brunswick, NJ, U.S.A.) connected to an Axiovert 35 fluorescence microscope (Carl Zeiss Oberkochen, Germany) using only quartz objectives (Ultrafluor×10). [Ca2+]i was assessed on the basis of the ratio of fluorescence emission measured at 510±5 nm which was evoked by excitation at 340 and 380 nm. Emission signals and force were measured simultaneously at 4 Hz and acquired on-line using an A/D interface (Photon Technology International Inc., South Brunswick, NJ, U.S.A.) connected to an IBM PC. Data were stored on an optical disc and later analysed off-line using commercially available software (Photon Technology International Inc., South Brunswick, NJ, U.S.A.). In view of difficulties in calibrating fluorescence intensity to absolute [Ca2+]i in the cytoplasm (Parkinson & Hughes, 1995), changes in [Ca2+]i in response to a stimulant were normalized by expressing them as percentage change in peak ratio of 340/380 nm signal induced by depolarization with KPSS as previously described (Garcha & Hughes, 1994; 1995).
PKC downregulation
PKC activity was downregulated by incubating 2 mm vessel segments with 500 nM phorbol 12, 13-dibutyrate (PdBu) in PSS for 20 h at room temperature using a modification of the technique described by Ohanian et al. (1996). Vessels were incubated in parallel with either PdBu or 500 nM 4α-phorbol 12, 13-didecanoate (4αPDD), an inactive phorbol ester. At the end of the incubation period vessels were mounted on wires in the myograph in PSS at 37°C as described previously. The vessels treated with PdBu and 4αPDD were compared in parallel in the same dual-channel myograph. The vessel treated with PdBu overnight was initially exposed to 2 μM PdBu to ensure there was no contraction and that PKC had been downregulated. Following washout and recovery (2 h), cumulative concentration-response curves to angiotensin II (100 pM – 100 nM) and NE (1 nM – 10 μM) and responses to KPSS were generated in both vessels.
Drugs
Candesartan was a gift from Takeda Chemical Industries, Osaka, Japan. Amlodipine was a gift from Pfizer (Sandwich, U.K.). Noradrenaline, BAPTA, Nickel chloride, PDBu and 4αPDD were purchased from Sigma (Poole, Dorset, U.K.). Human angiotensin II, genistein, daidzein, PD98059, chelerythrine and ryanodine were purchased from Calbiochem-Novabiochem (U.K.). Fura-2AM was purchased from Molecular Probes (Oregon, U.S.A.). KPSS was prepared by equimolar substitution of KCl for NaCl in PSS, giving 118 mM KCl.
All drugs were made up on the day of use. Fura-2AM was made up in 25 μl of DMSO/Cremophor-EL/Pluronic F-127. The final concentration of DMSO (0.5%), Cremophor-EL (0.1%) and Pluronic F-127 (0.02%) in PSS did not affect basal tone or the contractile response of vessels.
Statistics
Concentration-response data derived from each individual tissue were fitted separately to a logistic function by non-linear regression and EC50, the concentration of drug producing 50% of the maximal response to the same agent calculated. Non-linear regression was carried out on a PC using commercially available software (Inplot 4.0 and Prism 2.01, GraphPAD Software, CA, U.S.A.). Data are presented as means±s.e.mean calculated from n experiments in separate tissues. Concentration-response data were compared statistically in terms of −log(EC50) and maximum response using a Wilcoxon Paired rank test, a Mann-Whitney U-test or a Kruskal Wallis non-parametric analysis of variance (ANOVA) as appropriate. If ANOVA showed that a significant difference existed between groups it was followed by multiple comparisons of ranks to determine P for individual comparisons (Conover, 1980). A value of P<0.05 was considered significant.
Results
Responses to angiotensin II
Angiotensin II increased [Ca2+]i and induced contraction (Figure 1A). Both the rise in [Ca2+]i and force in response to angiotensin II were concentration-dependent (Figure 1B,C). Contraction to angiotensin II tended to be somewhat more variable than responses to NE or KPSS. Three out of a total of 32 arteries failed to respond to angiotensin II. These vessels were discarded. In all responsive vessels (n=29), maximal angiotensin II-induced contraction (5.8±0.6 N m−1) was similar to that induced by NE (5.8±0.6 N m−1) and slightly smaller than that induced by KPSS (6.3±0.6 N m−1). Pre-incubation with the selective AT1 receptor antagonist, candesartan (10 pM), for 30 min inhibited both the rise in [Ca2+]i and force in response to angiotensin II without affecting responses to NE or KPSS (n=4; P<0.05) (Figure 2). Although contraction in response to angiotensin II in these particular studies was smaller than in the majority of studies, this observation is consistent with a previous study showing insurmountable antagonism of contractile responses to angiotensin II by candesartan and the absence of endothelial or AT2 mediated responses to angiotensin II in the same preparation (Garcha et al., 1999).
Figure 1.
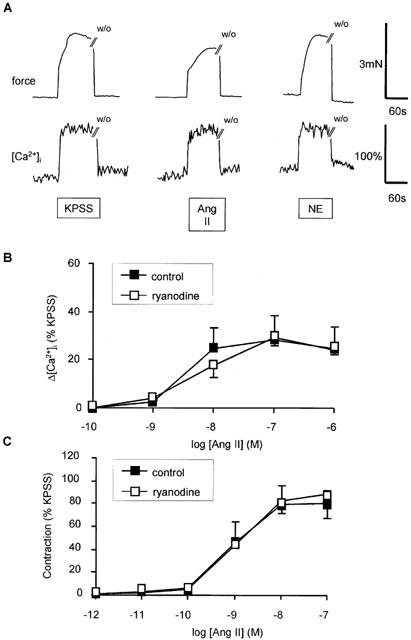
Effect of angiotensin II (AII) on intracellular Ca2+ and force production by human subcutaneous resistance arteries. (A) Traces showing effect of by KPSS, 100 nM angiotensin II, 10 μM NE on force (upper trace) and [Ca2+]i. Exposure to drugs is indicated by the bar and time of washout (w/o) is shown. (B) Concentration response relationship for peak increase in [Ca2+]i (Δ[Ca2+]i) in response to angiotensin II in the presence or absence of ryanodine (10 μM). Data represent mean±s.e.mean of per cent response to KPSS (n=4). (C) Concentration response relationship for contraction in response to angiotensin II in the presence or absence of ryanodine (10 μM). Data represent mean±s.e.mean of per cent response to KPSS (n=4).
Figure 2.
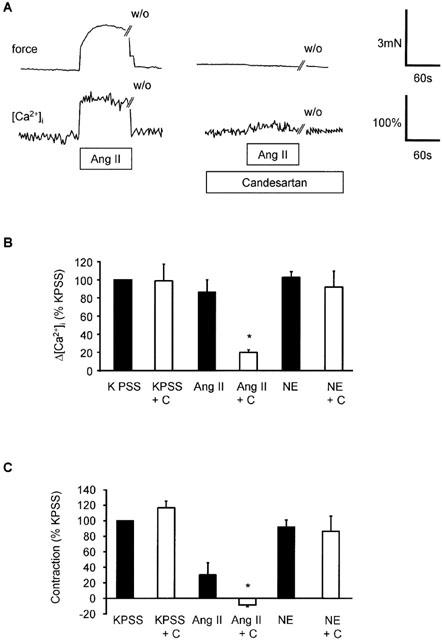
Effect of AT1 receptor antagonist, candesartan on [Ca2+]i and contraction induced by angiotensin II in human subcutaneous resistance arteries. (A) Traces showing the effect of candesartan (100 pM) on force (upper trace) and [Ca2+]i in the same vessel in response to angiotensin II (100 nM). Exposure to drugs is indicated by the bar and time of washout (w/o) is shown. (B) Comparison of peak increase in [Ca2+]i (Δ[Ca2+]i) in response to KPSS, angiotensin II (100 nM) and NE (10 μM) before and after candesartan (C; 100 pM). Bars represent mean±s.e.mean of per cent peak response to KPSS (n=4). *Indicates P<0.05. (C) Comparison of maximum increase in force in response to KPSS, angiotensin II (100 nM) and NE (10 μM) before and after candesartan (C; 100 pM). Bars represent mean±s.e.mean of per cent response to KPSS (n=4). *Indicates P<0.05.
Sources of Ca2+ mobilized by angiotensin II
In several blood vessels angiotensin II has been reported to increase [Ca2+]i by mobilizing Ca2+ from intracellular stores (Deth & Van Breemen, 1974; Morel et al., 1996; Satoh et al., 1987; Smith et al., 1984; Ushio-Fukai et al., 2000), so this possibility was investigated in human resistance arteries. We have previously shown that pre-incubation with ryanodine inhibits intracellular Ca2+ store release induced by NE and caffeine in these blood vessels (Parkinson & Hughes, 1995), therefore the effect of this agent on angiotensin II-induced responses was examined. Ryanodine (10 μM for 1 h) had no effect on either the rise in [Ca2+]i or force in response to increasing concentrations of angiotensin II when extracellular Ca2+ was present (Figure 1B,C). Responses to increasing concentrations of NE and KPSS responses were also not significantly affected by ryanodine (Figure 3) and the effect of ryanodine on NE was only evident when extracellular Ca2+ was removed. In subsequent studies, it was not possible to elicit responses to angiotensin II in the absence of extracellular Ca2+, so no further studies were performed with ryanodine.
Figure 3.
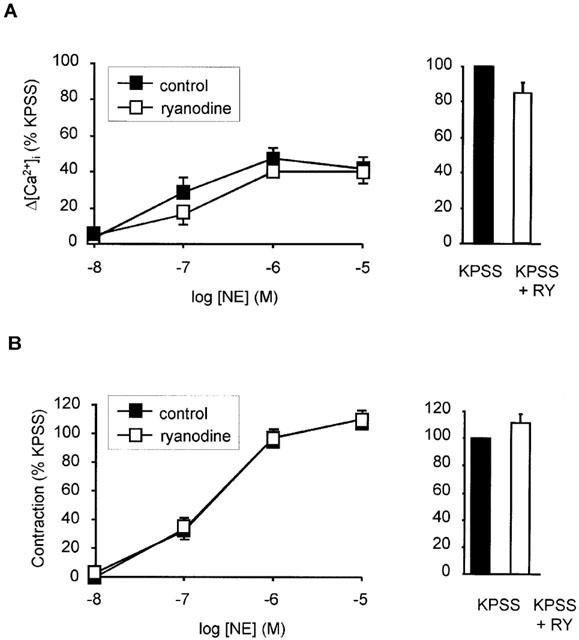
Effect of ryanodine on responses to noradrenaline (NE) and high potassium (KPSS). (A) Concentration response relationship for peak increase in [Ca2+]i (Δ[Ca2+]i) in response to NE in the presence and absence of ryanodine (10 μM). Effect of ryanodine on response to KPSS shown as inset. Data represent mean±s.e.mean of per cent response to KPSS (n=3). (B) Concentration response relationship for contraction in response to NE in the presence and absence of ryanodine (10 μM). Effect of ryanodine on response to KPSS shown as inset. Data represent mean±s.e.mean of per cent response to KPSS (n=3).
When Ca2+ was removed and 1 mM BAPTA added to the extracellular solution for 2 min prior to addition of agonist, angiotensin II (100 nM) failed to induce contraction or increase [Ca2+]i (Figure 4). In contrast, NE (10 μM) induced a transient contraction and rise in [Ca2+]i (Figure 4A), presumably as a result of mobilizing intracellularly stored Ca2+.
Figure 4.
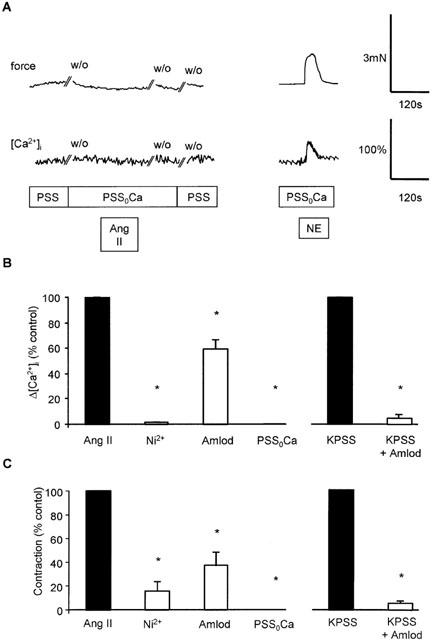
Effect of Ca2+-removal, Ni2+ and amlodipine, on responses to angiotensin II, NE and KPSS. (A) Traces showing the effect of a Ca2+ free solution (PSS0Ca) on contraction (upper trace) and [Ca2+]i in the same vessel in response to angiotensin II (100 nM). Exposure to drugs is indicated by the bar, and time of washout (w/o) is also shown. (B) Comparison of peak increase in [Ca2+]i (Δ[Ca2+]i) in response to angiotensin II following no treatment, Ni2+ (2 mM; n=4), amlodipine (10 μM; n=3) or Ca2+-free PSS (PSS0Ca; n=4). Right panel shows effect of amlodipine (10 μM; n=3) on peak increase in [Ca2+]i induced by KPSS. Bars represent mean±s.e.mean of per cent peak control response. *Indicates P<0.05. (C) Comparison of peak increase in force in response to angiotensin II following no treatment, Ni2+ (2 mM; n=4), amlodipine (10 μM; n=3) or Ca2+-free PSS (PSS0Ca; n=4). Right panel shows effect of amlodipine (10 μM; n=3) on peak increase in force induced by KPSS. Bars represent mean±s.e.mean of per cent peak control response. *Indicates P<0.05.
Pre-incubation with the inorganic Ca2+ entry blocker, Ni2+ (2 mM), also almost completely abolished the rise in [Ca2+]i and force in response to 100 nM angiotensin II in PSS (Figure 4B) confirming the absence of detectable release of Ca2+ from intracellular stores by angiotensin II. The Ca2+ influx pathway involved in response to angiotensin II was further characterized by pre-incubating vessels for 1 h with a dihydropyridine L-type Ca2+ channel antagonist, amlodipine (10 μM), prior to addition of angiotensin II. In contrast to the effect of Ni2+, this only partially inhibited the rise in [Ca2+]i and force induced by angiotensin II (Figure 4B). In contrast, responses to depolarization by KPSS were almost completely inhibited by this concentration of amlodipine.
Signalling pathways involved in angiotensin II-induced responses
AT1 receptor activation has been proposed to cause stimulation of PLC and consequent activation of PKC, therefore the role of classical PKC isoforms in the response to angiotensin II in human subcutaneous resistance arteries was examined. Pre-incubation of arteries for 10 min with the selective inhibitor, chelerythrine (1 μM), or downregulation of PKC by prolonged exposure to PDBu (500 nM) had no effect on angiotensin II-induced contraction (1 pM – 1 μM); compared to control and 4αPDD (500 nM) pre-treated (inactive analogue) respectively (n=5; NS) (Figure 5).
Figure 5.
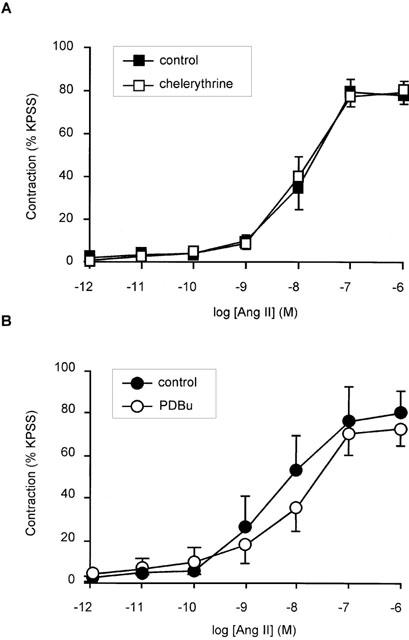
Effect of PKC inhibition with chelerythrine and PKC down-regulation with PDBu on angiotensin II-induced contraction in human subcutaneous resistance arteries. (A) Concentration response relationship for contraction in response to angiotensin II in the presence and absence of 1 μM chelerythrine). Points represent mean±s.e.mean of per cent response to KPSS (n=5). (B) Concentration response relationship between angiotensin II and contraction (expressed as per cent response to KPSS) in vessels treated overnight with 500 nM PDBu or with 500 nM 4αPDD (inactive control). Points represent mean±s.e.mean of per cent response to KPSS (n=5).
Activation of tyrosine kinases and MAPK has also been implicated in the growth promoting action of angiotensin II in cultured vascular smooth muscle cells, so the possible role of these pathways in contraction was also examined. Preincubation with genistein (10 μM) a selective tyrosine kinase inhibitor for 10 min, attenuated force responses to angiotensin II with the maximum being reduced from 75±13 to 18±6% (n=5; P<0.05) (Figure 6A). In contrast, the rise in [Ca2+]i to 100 nM angiotensin II was unaffected by genistein pre-treatment as was the rise in [Ca2+]i to 10 μM NE and KPSS (n=5) (Figure 6C). Genistein did not affect contractile responses to NE or KPSS (Figure 6B) and daidzein (10 μM), an analogue of genistein, which does not inhibit tyrosine kinases, had no effect on angiotensin II-induced contraction (1 pM – 1 μM) (Figure 7). Higher concentrations of genistein (30 – 100 μM) were found to further inhibit angiotensin II tone, but since responses to NE and KPSS were also reduced at these concentrations this action was considered to be non-selective and was not examined further (data not shown).
Figure 6.
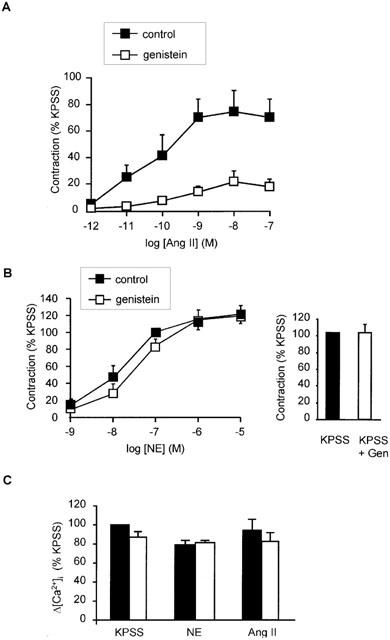
Effect of the tyrosine kinase inhibitor, genistein, on responses to angiotensin II, NE and KPSS. (A) Concentration-response relationship for contraction in response to angiotensin II in the presence or absence of genistein (10 μM). Points represent mean±s.e.mean of per cent response to KPSS (n=5). (B) Concentration-response relationship for contraction in response to NE in the presence or absence of genistein (10 μM). Points represent mean±s.e.mean of per cent response to KPSS (n=5). (C) Comparison of peak increase in [Ca2+]i (Δ[Ca2+]i) in response to angiotensin II (100 nM), NE (10 μM) and KPSS in the presence and absence of 10 μM genistein. Bars represent mean±s.e.mean of per cent peak response to KPSS (n=4 for all). *Indicates P<0.05.
Figure 7.
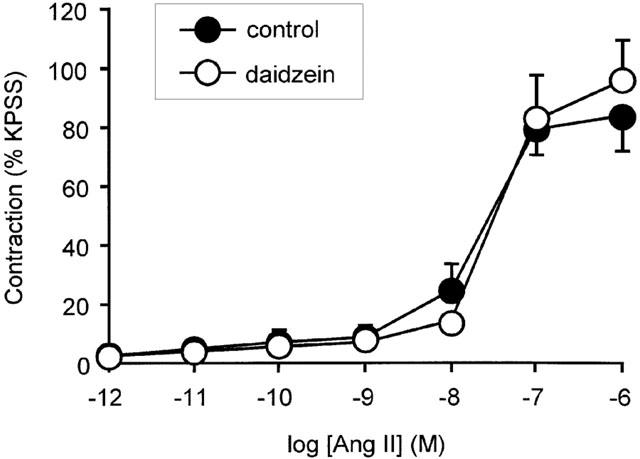
Effect of daidzein on contractile responses to angiotensin II. Concentration response relationship for contraction in response to angiotensin II in the presence or absence of daidzein (10 μM). Points represent mean±s.e.mean of per cent response to KPSS (n=4).
Pre-incubation with PD98059 (30 μM) for 10 min, a selective MEK inhibitor, also attenuated contraction in response to increasing concentrations of angiotensin II with the maximum response being reduced from 70±7 to 35±8% (n=5; P<0.05) (Figure 8A). In contrast to genistein pre-treatment, the rise in [Ca2+]i to 100 nM angiotensin II was also significantly inhibited by PD98059 pre-treatment (n=3; P<0.05) (Figure 8C). Contraction in response to NE and KPSS was unaffected by PD98059 pre-treatment (Figure 8B).
Figure 8.
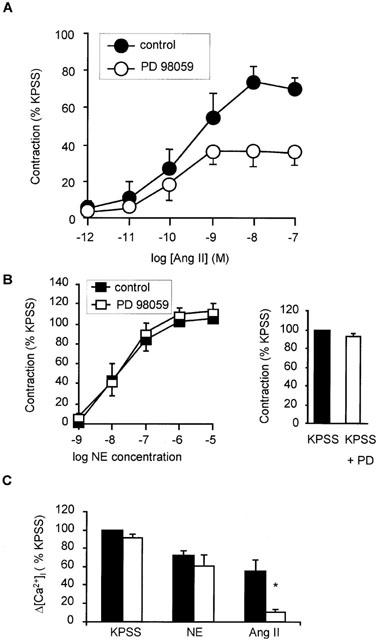
Effect of MEK inhibitor PD98059 on angiotensin II-induced contraction and peak increase in [Ca2+]i in response to angiotensin II, NE and KPSS. (A) Effect of PD98059 (30 μM; 10 min) on concentration response relationship for contraction in response to angiotensin II (n=5). (B) Effect of PD98059 (30 μM; 10 min) on concentration-response relationship for contraction in response to NE (n=5). Inset is bar graph showing effect of PD98059 (30 μM; 10 min) on contraction in response to KPSS. Bars represent mean±s.e.mean of five observations. (C) Comparison of peak increase in [Ca2+]i (Δ[Ca2+]i) in response to 100 nM angiotensin II, 10 μM NE and KPSS in the absence and presence of 30 μM PD98059 (n=3 for all). Bars represent mean±s.e.mean of per cent peak response to 118 mM K. *Indicates P<0.05.
Discussion
These data indicate that angiotensin II acts on AT1 receptors in human subcutaneous resistance arteries to increase [Ca2+]i and induce contraction. This finding is consistent with previous studies showing that the vasoconstrictor actions of angiotensin II are mediated by AT1 receptors in most blood vessels (Timmermans et al., 1993).
In the presence of extracellular Ca2+ responses to angiotensin II were unaffected by ryanodine, an agent which depletes Ca2+ from the sarcoplasmic reticulum stores (Kanmura et al., 1988). This observation contrasts with a previous study in rat portal vein myocytes, where ryanodine caused a 60% reduction in angiotensin II-evoked increases in [Ca2+]i (Morel et al., 1996). In these studies, although ryanodine was able to completely inhibit NE-induced release of intracellular Ca2+ (Parkinson & Hughes, 1995), ryanodine had no discernable effect on responses to NE in the presence of extracellular Ca2+. This contrasts with previous observations in rabbit ear artery (Kanmura et al., 1988), where ryanodine has been shown to inhibit responses to submaximal concentrations of NE, but is similar to previous studies using ryanodine in rat resistance arteries (Garcha & Hughes, 1995; Julou-Schaeffer & Freslon, 1988). These findings have been interpreted as indicating a more dominant role of Ca2+ influx in mediating responses in resistance arteries compared to larger arteries (Ashida et al., 1988; Garcha & Hughes, 1995). Further evidence regarding the contribution of intracellular Ca2+ stores to angiotensin II-induced responses came from studies where extracellular Ca2+ was removed. This completely abolished the rise in [Ca2+]i and contraction in response to angiotensin II, whereas transient responses to NE were demonstrable under these conditions confirming the existence of an intracellular Ca2+ store mobilized by NE in these arteries. Ni, an inorganic Ca2+ entry blocker also abolished the rise in [Ca2+]i and markedly inhibited contraction in response to angiotensin II. Taken together these observations suggest that release of intracellular Ca2+ does not play a discernable role in [Ca2+]i responses to angiotensin II in these blood vessels.
Unlike removal of extracellular Ca2+ or preincubation with Ni2+, inhibition of L-type calcium channels with amlodipine only partially inhibited angiotensin II-induced rises in [Ca2+]i and tension. This suggests that only part of the angiotensin II-induced Ca2+ influx occurs through L-type (voltage-operated) calcium channels. The identity of the dihydropyridine-insensitive route of Ca2+ entry is uncertain, but in rabbit ear artery, angiotensin II has been reported to activate channels permeable to cations, including Ca2+ (Hughes & Bolton, 1995). It is possible that a similar channel also accounts for the amlodipine-insensitive Ca2+ entry in human subcutaneous arteries. Our findings contrast with those of another study where the L-type Ca2+ channel blocker, nicardipine, almost completely inhibited angiotensin II-induced contraction in small arteries derived from human mammary tissue (Baan et al., 1999). Whether this difference reflects differences between tissues or non-specific effects of nicardipine (Cousins et al., 1995) is unknown.
Our observations provide no evidence that activation of phorbol ester-sensitive PKC contribute to angiotensin II-induced contraction. Studies in some other arteries (Oriji & Keiser, 1997; Salomonsson et al., 1997) have concluded that PKC is important in angiotensin II-induced contraction. Interestingly, Ohanian et al. (1996) also failed to inhibit contraction in response to NE, vasopressin or high potassium, following PKC downregulation with phorbol esters in small arteries derived from rat mesentery. This suggests possible differences in the importance of PKC in responses of various arteries, possibly depending on arterial calibre. Another possibility concerns the potential role of atypical PKC(s) in responses to angiotensin II. The PKC family has been divided into three subgroups: classical PKC (α, βI, βII and γ) that require Ca2+ and diacylglycerol (DAG) to be activated; novel PKC (δ, ε, ζ and θ) that require only DAG to be activated; and atypical PKC (ζ and λ/ι) that require neither Ca2+ nor DAG to be activated. Because atypical PKC lack a diglyceride-binding domain, phorbol esters do not downregulate this class. Consequently the studies with PDBu do not exclude a role of atypical PKC in responses to angiotensin II and it is noteworthy that an atypical PKC, PKC-zeta, has been reported to be responsible for ERK activation in cultured rat aortic smooth muscle cells (Liao et al., 1997). However, chelerythrine is an isoform non-selective PKC inhibitor that competitively interferes with the phosphate acceptor site and non-competitively inhibits the ATP binding site of PKC (Herbert et al., 1990) and would therefore be anticipated to inhibit all PKC isoforms, though possibly with different potency. Experimental data is inconclusive on this point as there are conflicting reports regarding the sensitivity of PKC-zeta to chelerythrine (Clement-Chomienne & Walsh, 1996; Thompson & Fields, 1996). Nevertheless, these observations with two different inhibitors of PKC would certainly seem to exclude involvement of classical or novel PKC in the responses to angiotensin II in these human resistance arteries.
In contrast, tyrosine kinases appear to play a major role in angiotensin II-induced contraction. Relatively low concentrations of the tyrosine kinase inhibitor genistein (Akiyama et al., 1987) selectively inhibited contractile responses to angiotensin II without affecting the angiotensin II-induced rise in [Ca2+]i. Other studies have also found that angiotensin II-induced contraction is more sensitive to selected tyrosine kinase inhibitors than α1-adrenoceptor-induced tone (Laniyonu et al., 1994; Malloy & Sauro, 1996; Saifeddine et al., 1992), but [Ca2+]i was not measured in any of these studies. However Toma et al. (1995) reported that genistein could affect contraction in response to calcium in α-toxin skinned rat mesenteric arteries, and Touyz & Schiffrin (1996) reported that genistein (1 μM) did not affect the peak rise in [Ca2+]i following angiotensin II stimulation of cultured vascular smooth muscle cells.
At higher concentrations genistein inhibited contraction to NE and KPSS. This was not investigated further, but is likely to be due to inhibition of L-type calcium channels as previously reported by Wijetunge et al. (1992).
Our data suggests that activation of a tyrosine kinase sensitive to low concentrations of genistein is involved in a process by which angiotensin II sensitizes the contractile machinery to a rise in [Ca2+]i. Calcium sensitization is now a well recognized mechanism contributing to receptor-induced contraction (Kitazawa et al., 1991) accounting for the increased force induced by an agonist for a given level of [Ca2+]i in comparison with a non-receptor stimulant such as potassium-depolarization, but the precise intracellular mechanisms involved in this process remain to be fully elucidated. It is particularly interesting that although most agonists, including NE, induce calcium sensitization the effect of genistein in this study was selective for angiotensin II. This could suggest a distinct tyrosine kinase-mediated pathway linked to angiotensin II-induced sensitization in human resistance arteries.
Inhibition of MEK with PD98059 selectively inhibited responses to angiotensin II. Unlike genistein, PD98059 attenuated both changes in [Ca2+]i and force. It is intriguing that PD98059 had no effect on NE or KPSS-induced tone. In human resistance arteries, angiotensin II acts exclusively via the AT1 receptor (Garcha et al., 1999), while NE acts largely via α1-adrenoceptors and α2-adrenoceptors to a lesser extent (Nielsen et al., 1990). All these receptor types couple to heterotrimeric G proteins and it is not clear why there should appear to be such a marked difference in post receptor signalling. It is possible that the difference relates to the subtype of G protein activated by the particular receptor in this tissue. In many tissues α1-adrenoceptors and AT1 receptors couple to Gq, which characteristically is linked to production of IP3 and release of intracellular Ca2+. NE does cause release of intracellular Ca2+ in human resistance arteries, but angiotensin II does not; this may indicate that the AT1 receptor does not act predominantly via Gq in this artery. In rat portal vein myocytes the AT1 receptor couples via G13 (Macrez-Lepretre et al., 1997a), while the α1-adrenoceptor is linked to Gq and G11 (Macrez-Lepretre et al., 1997b). Whether a similar difference in G protein coupling occurs in human resistance arteries and whether this could account for these observations remains to be established.
The difference between the effects of genistein and PD98059 implies that at the concentrations used in this study, these agents act on disparate elements of the angiotensin II-signalling pathway. The ERK1/2 pathway is involved in influencing Ca2+ influx, while the genistein-sensitive component contributes to Ca2+ sensitization. Previous studies in arterial smooth muscle have yielded conflicting information regarding the importance of the ERK1/2 pathway to contraction. A study in rat aorta found that angiotensin II-induced phosphorylation of ERK1/2, but that contraction was unaffected by PD98059 (Watts et al., 1998). In contrast, in pressurized rat mesenteric resistance arteries angiotensin II also induced activation of ERK1/2, but in this case contraction was partially inhibited by PD98059 (Matrougui et al., 2000). Interestingly, in this case activation of ERK was inhibited by the PKC inhibitors Ro-31-8220 and Go-6967 implying involvement of PKC in this effect. It is possible that the difference between these observations and ours reflects differences in methodology (e.g. pressurization) or species/tissue.
In smooth muscle cells cultured from human subcutaneous arteries, Touyz et al. (1999) reported that PD98059 inhibited the angiotensin II-induced rise in [Ca2+]i. Since PD98059 also inhibited angiotensin II induced contraction in isolated arteries, these authors suggested that MEK inhibition of angiotensin II-induced Ca2+ influx might account for its inhibitory effects on contraction. Our findings confirm this suggestion using simultaneous measurement of [Ca2+]i and force in isolated arteries.
Little is known about the processes by which ERK activation may regulate contraction. ERK has been reported to phosphorylate thin filament proteins, such as caldesmon (Adam et al., 1995) and calponin (Menice et al., 1997), but, as far as we are aware, no mechanism linking ERK activation to Ca2+ influx has been described. Further studies investigating the effect of ERK on Ca2+ influx pathways and calcium channels in particular would be of considerable interest.
In summary, we have shown that angiotensin II-induced rise in [Ca2+]i and consequent contraction in human subcutaneous resistance arteries is mediated through activation of an AT1 receptor and is dependent on Ca2+ influx. AT1-induced Ca2+ influx occurs through L-type calcium channels in addition to a dihydropyridine-insensitive route. Responses to angiotensin II are unaffected by inhibition of classical or novel PKC, but inhibition of MEK reduces both the rise in [Ca2+]i and force. In addition, angiotensin II appears to activate a tyrosine kinase sensitive to low concentrations of genistein that may contribute to sensitization of the contractile machinery to [Ca2+]i by angiotensin II.
Acknowledgments
This work was supported by an educational grant from Takeda (GmbH). We are grateful to the cardiac surgeons and theatre staff at St. Mary's Hospital, London for their assistance with this study.
Abbreviations
- 4αPDD
4α-phorbol 12,13-didecanoate
- ANOVA
analysis of variance
- [Ca2+]i
intracellular Ca2+
- EC50
concentration of drug producing 50% maximum response
- ERK
extracellular signal-regulated kinase
- GPCR
G-protein-coupled receptor
- JAK
Janus family kinase
- KPSS
modified physiological saline containing 118 mM KCl
- L100
circumference at 100 mmHg transmural pressure
- MAPK
mitogen-activated protein kinase
- MEK
MAPK kinase
- NE
noradrenaline
- PDBu
phorbol-12,13-dibutyrate
- PKC
protein kinase C
- PSS
physiological saline solution
- STAT
signal transducers and activators of transcription
References
- ADAM L.P., FRANKLIN M.T., RAFF G.J., HATHAWAY D.R. Activation of mitogen-activated protein kinase in porcine carotid arteries. Circ. Res. 1995;76:183–190. doi: 10.1161/01.res.76.2.183. [DOI] [PubMed] [Google Scholar]
- AKIYAMA T., ISHIDA J., NAKAGAWA S., OGAWARA H., WATANABE S., ITOH N., SHIBUYA M., FUKAMI Y. Genistein, a specific inhibitor of tyrosine-specific protein kinases. J. Biol. Chem. 1987;262:5592–5595. [PubMed] [Google Scholar]
- ASHIDA T., SCHAEFFER J., GOLDMAN W.F., WADE J.B., BLAUSTEIN M.P. Role of sarcoplasmic reticulum in arterial contraction: comparison of ryanodine's effect in a conduit and a muscular artery. Circ. Res. 1988;62:854–863. doi: 10.1161/01.res.62.4.854. [DOI] [PubMed] [Google Scholar]
- BAAN J., JR, PFAFFENDORF M., VAN DER WAL A.C., CHANG P.C., VAN ZWIETEN P.A. Influence of losartan and nicardipine on the contractile responses of human subcutaneous arteries and veins to angiotensin II. Fundam. Clin. Pharmacol. 1999;13:43–49. doi: 10.1111/j.1472-8206.1999.tb00319.x. [DOI] [PubMed] [Google Scholar]
- BERK B.C., CORSON M.A. Angiotensin II signal transduction in vascular smooth muscle: role of tyrosine kinases. Circ. Res. 1997;80:607–616. doi: 10.1161/01.res.80.5.607. [DOI] [PubMed] [Google Scholar]
- CLEMENT-CHOMIENNE O., WALSH M.P. Identification of protein kinase C isoenzymes in smooth muscle: partial purification and characterization of chicken gizzard PKC zeta. Biochem. Cell. Biol. 1996;74:51–65. doi: 10.1139/o96-006. [DOI] [PubMed] [Google Scholar]
- CONOVER W.J. Practical Nonparametric Statistics 1980New York: John Wiley and Sons; 2nd edition [Google Scholar]
- COUSINS H.M., EDWARDS F.R., HIRST G.D.S. Neuronally released and applied acetylcholine on the longitudinal muscle of the guinea-pig ileum. Neuroscience. 1995;65:193–207. doi: 10.1016/0306-4522(94)00466-i. [DOI] [PubMed] [Google Scholar]
- DETH R., VAN BREEMEN C. Relative contributions of Ca2+ influx and cellular Ca2+ release during drug induced activation of the rabbit aorta. Pflugers Arch. 1974;348:13–22. doi: 10.1007/BF00587735. [DOI] [PubMed] [Google Scholar]
- GARCHA R.S., HUGHES A.D. Action of heparin and ruthenium red on responses of reversibly- permeabilised rat mesenteric arteries. Eur. J. Pharmacol. 1994;268:319–325. doi: 10.1016/0922-4106(94)90056-6. [DOI] [PubMed] [Google Scholar]
- GARCHA R.S., HUGHES A.D. Inhibition of norepinephrine and caffeine-induced activation by ryanodine and thapsigargin in rat mesenteric arteries. J. Cardiovasc. Pharmacol. 1995;25:840–846. doi: 10.1097/00005344-199505000-00022. [DOI] [PubMed] [Google Scholar]
- GARCHA R.S., SEVER P.S., HUGHES A.D. Action of AT1 receptor antagonists on angiotensin II-induced tone in human isolated subcutaneous resistance arteries. Br. J. Pharmacol. 1999;127:1876–1882. doi: 10.1038/sj.bjp.0702722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HERBERT J.M., AUGEREAU J.M., GLEYE J., MAFFRAND J.P. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem. Biophys. Res. Commun. 1990;172:993–999. doi: 10.1016/0006-291x(90)91544-3. [DOI] [PubMed] [Google Scholar]
- HOLLENBERG M.D. Tyrosine kinase pathways and the regulation of smooth muscle contractility. T.I.P.S. 1994;15:108–114. doi: 10.1016/0165-6147(94)90046-9. [DOI] [PubMed] [Google Scholar]
- HUGHES A.D. AT1-signalling in vascular smooth muscle. J.R.A.A.S. 2000;1:125–130. doi: 10.3317/jraas.2000.014. [DOI] [PubMed] [Google Scholar]
- HUGHES A.D., BOLTON T.B. Action of angiotensin II, 5-hydroxytryptamine and adenosine triphosphate on ionic currents in single ear artery cells of the rabbit. Br. J. Pharmacol. 1995;116:2148–2154. doi: 10.1111/j.1476-5381.1995.tb16424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUGHES A.D., WIJETUNGE S. Role of tyrosine phosphorylation in excitation-contraction coupling in vascular smooth muscle. Acta Physiol. Scand. 1998;164:457–469. doi: 10.1046/j.1365-201X.1998.00446.x. [DOI] [PubMed] [Google Scholar]
- JENSEN P.E., MULVANY M.J., AALKJAER C. Endogenous and exogenous agonist-induced changes in the coupling between [Ca2+]i and force in rat resistance arteries. Pflugers Arch. 1992;420:536–543. doi: 10.1007/BF00374630. [DOI] [PubMed] [Google Scholar]
- JULOU-SCHAEFFER G., FRESLON J.L. Effects of ryanodine on tension development in rat aorta and mesenteric resistance vessels. Br. J. Pharmacol. 1988;95:605–613. doi: 10.1111/j.1476-5381.1988.tb11682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KANMURA Y., MISSIAEN L., RAEYMAEKERS L., CASTEELS R. Ryanodine reduces the amount of calcium in intracellular stores of smooth-muscle cells of the rabbit ear artery. Pflugers Arch. 1988;413:153–159. doi: 10.1007/BF00582525. [DOI] [PubMed] [Google Scholar]
- KITAZAWA T., GAYLINN B.D., DENNEY G.H., SOMLYO A.P. G-protein- mediated Ca2+ sensitization of smooth muscle contraction through myosin light chain phosphorylation. J. Biol. Chem. 1991;266:1708–1715. [PubMed] [Google Scholar]
- LANIYONU A.A., SAIFEDDINE M., YANG S.G., HOLLENBERG M.D. Tyrosine kinase inhibitors and the contractile action of G-protein- linked vascular agonists. Can. J. Physiol. Pharmacol. 1994;72:1075–1085. doi: 10.1139/y94-150. [DOI] [PubMed] [Google Scholar]
- LIAO D.F., MONIA B., DEAN N., BERK B.C. Protein kinase C-zeta mediates angiotensin II activation of ERK1/2 in vascular smooth muscle cells. J. Biol. Chem. 1997;272:6146–6150. doi: 10.1074/jbc.272.10.6146. [DOI] [PubMed] [Google Scholar]
- MACREZ-LEPRETRE N., KALKBRENNER F., MOREL J.L., SCHULTZ G., MIRONNEAU J. G protein heterotrimer Galpha13beta1gamma3 couples the angiotensin AT1A receptor to increases in cytoplasmic Ca2+ in rat portal vein myocytes. J. Biol. Chem. 1997;272:10095–10102. doi: 10.1074/jbc.272.15.10095. [DOI] [PubMed] [Google Scholar]
- MACREZ-LEPRETRE N., KALKBRENNER F., SCHULTZ G., MIRONNEAU J. Distinct functions of Gq and G11 proteins in coupling alpha1-adrenoreceptors to Ca2+ release and Ca2+ entry in rat portal vein myocytes. J. Biol. Chem. 1997;272:5261–5268. doi: 10.1074/jbc.272.8.5261. [DOI] [PubMed] [Google Scholar]
- MALLOY L.G., SAURO M.D. Tyrosine kinase inhibition suppresses angiotensin contraction in hypertensive and normotensive small resistance arteries. Life Sci. 1996;58:L317–L324. doi: 10.1016/0024-3205(96)00147-6. [DOI] [PubMed] [Google Scholar]
- MATROUGUI K., ESKILDSEN-HELMOND Y.E., FIEBELER A., HENRION D., LEVY B.I., TEDGUI A., MULVANY M.J. Angiotensin II stimulates extracellular signal-regulated kinase activity in intact pressurized rat mesenteric resistance arteries. Hypertension. 2000;36:617–621. doi: 10.1161/01.hyp.36.4.617. [DOI] [PubMed] [Google Scholar]
- MENICE C.B., HULVERSHORN J., ADAM L.P., WANG C.A., MORGAN K.G. Calponin and mitogen-activated protein kinase signaling in differentiated vascular smooth muscle. J. Biol. Chem. 1997;272:25157–25161. doi: 10.1074/jbc.272.40.25157. [DOI] [PubMed] [Google Scholar]
- MOREL J.L., MACREZ-LEPRETRE N., MIRONNEAU J. Angiotensin II-activated Ca2+ entry-induced release of Ca2+ from intracellular stores in rat portal vein myocytes. Br. J. Pharmacol. 1996;118:73–78. doi: 10.1111/j.1476-5381.1996.tb15368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORGAN K.G., LEINWEBER B.D. PKC-dependent signalling mechanisms in differentiated smooth muscle. Acta Physiol. Scand. 1998;164:495–505. doi: 10.1046/j.1365-201X.1998.00445.x. [DOI] [PubMed] [Google Scholar]
- MULVANY M.J., AALKJAER C. Structure and function of small arteries. Physiol. Rev. 1990;70:921–961. doi: 10.1152/physrev.1990.70.4.921. [DOI] [PubMed] [Google Scholar]
- MULVANY M.J., HALPERN W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ. Res. 1977;41:19–26. doi: 10.1161/01.res.41.1.19. [DOI] [PubMed] [Google Scholar]
- OHANIAN V., OHANIAN J., SHAW L., SCARTH S., PARKER P.J., HEAGERTY A.M. Identification of protein kinase C isoforms in rat mesenteric small arteries and their possible role in agonist-induced contraction. Circ. Res. 1996;78:806–812. doi: 10.1161/01.res.78.5.806. [DOI] [PubMed] [Google Scholar]
- ORIJI G.K., KEISER H.R. Protein kinase C mediates angiotensin II-induced contractions and the release of endothelin and prostacyclin in rat aortic rings. Prostaglandins Leukot. Essent. Fatty Acids. 1997;57:135–141. doi: 10.1016/s0952-3278(97)90003-x. [DOI] [PubMed] [Google Scholar]
- NIELSEN H., MORTENSEN F.V., MULVANY M.J. Responses to noradrenaline in human subcutaneous resistance arteries are mediated by both alpha 1- and alpha 2-adrenoceptors. Br. J. Pharmacol. 1990;99:31–34. doi: 10.1111/j.1476-5381.1990.tb14649.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARKINSON N.A., HUGHES A.D. The mechanism of action of alpha 2-adrenoceptors in human isolated subcutaneous resistance arteries. Br. J. Pharmacol. 1995;115:1463–1468. doi: 10.1111/j.1476-5381.1995.tb16638.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAIFEDDINE M., LANIYONU A.A., YANG S.G., HOLLENBERG M.D. Tyrosine kinase inhibitors and the contractile action of angiotensin II in vascular tissue. Pharmacol. Commun. 1992;1:177–184. [Google Scholar]
- SALOMONSSON M., KORNFELD M., GUTIERREZ A.M., MAGNUSSON M., PERSSON A.E. Effects of stimulation and inhibition of protein kinase C on the cytosolic calcium concentration in rabbit afferent arterioles. Acta Physiol. Scand. 1997;161:271–279. doi: 10.1046/j.1365-201X.1997.d01-1962.x. [DOI] [PubMed] [Google Scholar]
- SATOH S., ITOH T., KURIYAMA H. Actions of angiotensin II and noradrenaline on smooth muscle cells of the canine mesenteric artery. Pflugers Arch. 1987;410:132–138. doi: 10.1007/BF00581905. [DOI] [PubMed] [Google Scholar]
- SMITH J.B., SMITH L., BROWN E.R., BARNES D., SABIR M.A., DAVIS J.S., FARESE R.V. Angiotensin II rapidly increases phosphatidate-phosphoinositide synthesis and phosphoinositide hydrolysis and mobilizes intracellular calcium in cultured arterial muscle cells. Proc. Natl. Acad. Sci. U. S. A. 1984;81:7812–7816. doi: 10.1073/pnas.81.24.7812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOMPSON L.J., FIELDS A.P. betaII protein kinase C is required for the G2/M phase transition of cell cycle. J. Biol. Chem. 1996;271:15045–15053. doi: 10.1074/jbc.271.25.15045. [DOI] [PubMed] [Google Scholar]
- TIMMERMANS P.B., WONG P.C., CHIU A.T., HERBLIN W.F., BENFIELD P., CARINI D.J., LEE R.J., WEXLER R.R., SAYE J.A., SMITH R.D. Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacol. Rev. 1993;45:205–251. [PubMed] [Google Scholar]
- TOMA C., JENSEN P.E., PRIETO D., HUGHES A., MULVANY M.J., AALKJAER C. Effects of tyrosine kinase inhibitors on the contractility of rat mesenteric resistance arteries. Br. J. Pharmacol. 1995;114:1266–1272. doi: 10.1111/j.1476-5381.1995.tb13342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOUYZ R.M., HE G., DENG L.Y., SCHIFFRIN E.L. Role of extracellular signal-regulated kinases in angiotensin II- stimulated contraction of smooth muscle cells from human resistance arteries. Circulation. 1999;99:392–399. doi: 10.1161/01.cir.99.3.392. [DOI] [PubMed] [Google Scholar]
- TOUYZ R.M., SCHIFFRIN E.L. Tyrosine kinase signaling pathways modulate angiotensin II-induced calcium ([Ca2+]i) transients in vascular smooth muscle cells. Hypertension. 1996;27:1097–1103. doi: 10.1161/01.hyp.27.5.1097. [DOI] [PubMed] [Google Scholar]
- USHIO-FUKAI M., YAMAMOTO H., TOYOFUKU K., NISHIMURA J., HIRANO K., KANAIDE H. Changes in the cytosolic Ca2+ concentration and Ca2+-sensitivity of the contractile apparatus during angiotensin II-induced desensitization in the rabbit femoral artery. Br. J. Pharmacol. 2000;129:425–436. doi: 10.1038/sj.bjp.0703092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WATTS S.W. Serotonin activates the mitogen-activated protein kinase pathway in vascular smooth muscle: use of the mitogen-activated protein kinase kinase inhibitor PD098059. J. Pharmacol. Exp. Ther. 1996;279:1541–1550. [PubMed] [Google Scholar]
- WATTS S.W., FLORIAN J.A., MONROE K.M. Dissociation of angiotensin II-stimulated activation of mitogen- activated protein kinase kinase from vascular contraction. J. Pharmacol. Exp. Ther. 1998;286:1431–1438. [PubMed] [Google Scholar]
- WIJETUNGE S., AALKJAER C., SCHACHTER M., HUGHES A.D. Tyrosine kinase inhibitors block calcium channel currents in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 1992;189:1620–1623. doi: 10.1016/0006-291x(92)90262-j. [DOI] [PubMed] [Google Scholar]