

Abstract
It has been reported that co-administration of fluoxetine with 3,4-methylenedioxymethamphetamine (MDMA, ‘ecstasy') prevents MDMA-induced degeneration of 5-HT nerve endings in rat brain. The mechanisms involved have now been investigated.
MDMA (15 mg kg−1, i.p.) administration produced a neurotoxic loss of 5-HT and 5-hydroxyindoleacetic acid (5-HIAA) in cortex, hippocampus and striatum and a reduction in cortical [3H]-paroxetine binding 7 days later. Fluoxetine (10 mg kg−1, i.p., ×2, 60 min apart) administered concurrently with MDMA or given 2 and 4 days earlier provided complete protection, and significant protection when given 7 days earlier. Fluvoxamine (15 mg kg−1, i.p., ×2, 60 min apart) only produced neuroprotection when administered concurrently.
Fluoxetine (10 mg kg−1, ×2) markedly increased the KD and reduced the Bmax of cortical [3H]-paroxetine binding 2 and 4 days later. The Bmax was still decreased 7 days later, but the KD was unchanged. [3H]-Paroxetine binding characteristics were unchanged 24 h after fluvoxamine (15 mg kg−1, ×2).
A significant cerebral concentration of fluoxetine plus norfluoxetine was detected over the 7 days following fluoxetine administration. The fluvoxamine concentration had decreased markedly by 24 h.
Pretreatment with fluoxetine (10 mg kg−1, ×2) failed to alter cerebral MDMA accumulation compared to saline pretreated controls.
Neither fluoxetine or fluvoxamine altered MDMA-induced acute hyperthermia.
These data demonstrate that fluoxetine produces long-lasting protection against MDMA-induced neurodegeneration, an effect apparently related to the presence of the drug and its active metabolite inhibiting the 5-HT transporter. Fluoxetine does not alter the metabolism of MDMA or its rate of cerebral accumulation.
Keywords: 3,4-methylenedioxymethamphetamine; ecstasy; neuroprotection; 5-hydroxytryptamine; fluoxetine; norfluoxetine; fluvoxamine
Introduction
The recreationally used drug 3,4-methylenedioxymethamphetamine (MDMA or ‘ecstasy') has two distinct neurochemical effects on the concentration of 5-hydroxytryptamine (5-HT) in the brain of rats. The first effect is a rapid and dramatic increase in the extracellular concentration of 5-HT. This results from the ability of MDMA to stimulate 5-HT release (Gough et al., 1991; Brodkin et al., 1993), inhibit uptake (Steele et al., 1987; Johnson et al., 1991) and block metabolism of 5-HT by MAO-A (Leonardi & Azmitia, 1994). MDMA binds with high affinity to the 5-HT transporter protein (Poblete et al., 1989), reverses the direction of neurotransmitter flow (Rudnick & Wall, 1992) and exerts a pronounced stimulatory effect on 5-HT release in synaptosomes (Nichols et al., 1982; McKenna et al., 1991; Berger et al., 1992), brain slices (Johnson et al., 1986; Sprouse et al., 1989; Fitzgerald & Reid, 1993) and cultured neurones (Azmitia et al., 1990; Gu & Azmitia, 1993). 5-HT release has also been demonstrated in vivo by the use of intracerebral microdialysis (Schmidt et al., 1987; Gough et al., 1991; Gudelsky & Nash, 1996). The acute 5-HT releasing effect of MDMA involves both a calcium-dependent mechanism (Crespi et al., 1997) and a calcium independent process mediated by the transporter (Johnson et al., 1986). In fact, the release of 5-HT induced by MDMA in vivo is mostly attenuated by drugs that bind to the 5-HT transporter (fluoxetine, citalopram) thereby inhibiting the 5-HT uptake into presynaptic terminals (Schmidt, 1987; Schmidt et al., 1987; Azmitia et al., 1990; Hekmatpanah & Peroutka, 1990; Gudelsky & Nash, 1996; Mechan et al., 2000). The effect of MDMA on 5-HT release is reversible, the 5-HT extracellular concentration returning to control values 3 – 4 h after drug administration (Gudelsky & Nash, 1996; Mechan et al., 2000). Simultaneously with the 5-HT release, rats display both the 5-HT behavioural syndrome and hyperthermia (Slikker et al., 1989; Colado et al., 1993).
The second action of MDMA is that of producing, in several areas of the brain, a long-term loss of fine 5-HT axon terminals arising primarily from the dorsal raphe nucleus (O'hearn et al., 1988). The degeneration has been demonstrated both histologically (O'hearn et al., 1988; Molliver et al., 1990) and biochemically and is reflected in a substantial decrease in the concentration of 5-HT and its metabolite, 5-hydroxyindoleacetic acid (5-HIAA), and a reduction in the density of 5-HT uptake sites labelled with [3H]-paroxetine (Sharkey et al., 1991; Hewitt & Green, 1994; Colado et al., 1997b).
The mechanisms involved in producing this neurodegeneration are not fully understood at present, but recent data indicate that a major mechanism by which MDMA induces damage to 5-HT containing neurones in rat brain is by increasing the formation of hydroxyl free radicals (Colado & Green, 1995; Colado et al., 1997b; Yeh, 1999) which probably result from MDMA metabolism (Colado et al., 1995). The use of intracerebral microdialysis has demonstrated that systemic administration of MDMA increases the formation of 2,3-dihydroxybenzoic acid (2,3-DHBA) from salicylate in hippocampal and striatal dialysates (Colado et al., 1997b; Shankaran et al., 1999), a conversion that only occurs in the presence of a high concentration of free radicals. The fact that this effect was absent in rats with a lesion of the 5-HT nerve terminals indicates that these reactive species are probably formed in 5-HT nerve endings (Colado et al., 1997b). Additional evidence supporting the existence of an oxidative stress includes the report that the hydroxyl radical scavenger alpha-phenyl-N-tert-butyl nitrone (PBN) is neuroprotective (Colado & Green, 1995; Colado et al., 1997b; Yeh, 1999) and also abolishes the MDMA-induced rise in 2,3-DHBA (Colado et al., 1997b), the observation that MDMA increases lipid peroxidation in the brain (Sprague & Nichols, 1995; Colado et al., 1997a) and the fact that transgenic mice overexpressing CuZn superoxide dismutase are resistant to the neurotoxic actions of the drug (Cadet et al., 1995).
Initial studies based on both the pharmacology of MDMA and putative neuroprotective compounds, suggested a key role for dopamine in the expression of MDMA-induced neurotoxicity (Stone et al., 1988; Schmidt et al., 1990; Hewitt & Green, 1994). Most of these studies were performed in the absence of measures of body temperatures and consequently, a re-evaluation of the earlier reports was necessary. There is now evidence that α-methy-p-tyrosine and haloperidol only prevent MDMA-induced damage because they either induce hypothermia (Malberg et al., 1996) or prevent acute hyperthermia (Colado et al., 1999) and that the enhancing effect of L-DOPA on MDMA-induced neurodegeneration (Schmidt et al., 1991) might result from its ability to increase the MDMA-induced hyperthermia (Colado et al., 1999). In addition, the MAO-B inhibitor deprenyl has been shown to possess free radical scavenger activity (Thomas et al., 1997) which could explain its neuroprotective effect against MDMA toxicity (Sprague & Nichols, 1995).
There is a large amount of evidence indicating the involvement of the 5-HT transporter in both the acute and in the long-term effects induced by MDMA. Fluoxetine, a selective 5-HT uptake inhibitor (Wong et al., 1983), when given with MDMA, prevents both the acute increase in hydroxyl radical formation which follows administration of the amphetamine derivative (Shankaran et al., 1999) and the long-lasting neurotoxic effects (Schmidt, 1987; Malberg et al., 1996). The neuroprotective effect of fluoxetine does not depend on an effect on body temperature since animals treated with fluoxetine and MDMA displayed a hyperthermic response similar to that observed when MDMA is given alone (Malberg et al., 1996; Mechan et al., 2000). Since MDMA interacts with the 5-HT transporter protein (Poblete et al., 1989) it seems reasonable to propose that fluoxetine may be preventing the uptake of MDMA or most likely, of a neurotoxic metabolite of MDMA into the 5-HT nerve endings (Malberg et al., 1996; Esteban et al., 2001).
Fluoxetine is extensively biotransformed (N-demethylation) by the hepatic cytochrome P450 2D6 enzyme system to norfluoxetine (Bergstrom et al., 1992; Hamelin et al., 1996), an active metabolite which is eliminated much more slowly than fluoxetine by both rats and humans (Gardier et al., 1994; Hamelin et al., 1996; Holladay et al., 1998). The plasma elimination half-lives of fluoxetine and norfluoxetine are respectively in the range 1 – 3 days and 7 – 15 days (Lemberger et al., 1985; Benfield et al., 1986; Stanford, 1996; Preskorn, 1997; Sanchez & Hyttel, 1999). Both compounds are highly lipophilic and accumulate in the brain where their concentrations are higher than those found in plasma (Gardier et al., 1994; Lefebvre et al., 1999). Norfluoxetine is as potent as fluoxetine in inhibiting 5-HT uptake and it is also more selective (Bolden-Watson & Richelson, 1993; Sanchez & Hyttel, 1999). Its presence in vivo therefore probably contributes substantially to the pharmacological and therapeutic effects of the parent compound (Horng & Wong, 1976; Fuller et al., 1978; Schmidt, 1987).
Fluvoxamine is also a selective 5-HT uptake inhibitor but with a different pharmacokinetic profile. Fluvoxamine undergoes oxidative metabolism but, in contrast to fluoxetine, it generates several metabolites, none of which appear to be pharmacologically active. In addition, CYP2D6 isoenzymes do not play a significant role in its metabolism (van harten, 1995; Preskorn, 1997).
The aim of this current study was to examine the mechanisms involved in the neuroprotective effect of fluoxetine against MDMA-induced neurotoxic loss of 5-HT in rat brain. To do this, MDMA was either co-administered with fluoxetine or given several days (2, 4 and 7 days) later. In addition, the study also investigated whether the long-term neuroprotective effect of fluoxetine was due to changes in the 5-HT transporter produced by the presence of either fluoxetine or norfluoxetine or, alternatively, it resulted from a pharmacokinetic interaction between fluoxetine and MDMA since, in rats, MDMA is metabolized to 3,4-dihydroxymethamphetamine (DHMA) by CYP2D1 and fluoxetine and norfluoxetine are potent inhibitors of CYP2D1. This investigation has also examined whether the neuroprotection induced by fluoxetine can be extended to other 5-HT selective uptake inhibitors such as fluvoxamine.
Methods
Animals, drug administration and experimental protocol
All experimental procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication No. 85-23, revised 1985).
Male Dark Agouti rats (175 – 200 g, Harlan Iberica, Barcelona) were used. They were housed in groups of five, in conditions of constant temperature (21±2°C) and a 12 h light/dark cycle (lights on: 0700 h) and given free access to food and water. (±) MDMA (obtained from the Ministry of Health, Spain), fluoxetine (Lilly) and fluvoxamine (Tocris) were dissolved in saline (0.9% NaCl) and given i.p. in a volume of 1 ml kg−1 for MDMA and fluvoxamine and 2 ml kg−1 for fluoxetine. Doses are reported in terms of the base.
Fluoxetine (10 mg kg−1, i.p., twice, 60 min apart) was given 2, 4 and 7 days before MDMA (15 mg kg−1, i.p.). In order to compare the neuroprotective effect of fluoxetine after these pretreatment periods with that when the drug was co-administered with MDMA, a further group of animals received fluoxetine 5 min before and 55 min after MDMA.
Fluvoxamine (15 mg kg−1, i.p., twice, 60 min apart) was given either 24 h before MDMA (15 mg kg−1, i.p) or 5 min before and 55 min after MDMA. Animals were always sacrificed 7 days after MDMA administration.
Rats were given a single administration of MDMA at the dose of 15 mg kg−1 since previous studies had demonstrated that this regimen produced an approximately 50% loss of several 5-HT parameters (O'shea et al., 1998). Fluoxetine and fluvoxamine were administered at doses altering the extracellular concentration of 5-HT (Bosker et al., 1995; Mechan et al., 2000).
Measurement of rectal temperature
Temperature was measured during the 6 h following MDMA administration by means of a digital readout thermocouple (Type K thermometer, Portec, U.K.) with a resolution of±0.1°C and accuracy of±0.2°C attached to a CAC-005 Rodent Sensor which was inserted 2.5 cm into the rectum of the rat, the animal being lightly restrained by holding in the hand. A steady readout was obtained within 10 s of probe insertion.
Measurement of 5-HT and its metabolite, 5-HIAA, in cerebral tissue
Rats were killed by cervical dislocation and decapitation, the brains rapidly removed and cortex, hippocampus and striatum dissected out on ice. Tissue was homogenized and 5-HT and 5-HIAA measured by high performance liquid chromatography (h.p.l.c.). Briefly, the mobile phase consisted of KH2PO4 (0.05 M), octanesulphonic acid (0.16 mM), ethylenediaminetetraacetic acid (EDTA, 0.1 mM) and methanol (16%), and was adjusted to pH 3 with phosphoric acid, filtered and degassed. The flow rate was 1.3 ml min−1 and the working electrode potential was set at +0.8 V.
The h.p.l.c. system consisted of a pump (Waters 510) linked to an automatic sample injector (Loop 200 μl, Waters 712 WISP), a stainless steel reversed-phase column (Spherisorb ODS2, 5 μm, 150×4.6 mm) with a precolumn and an amperometric detector (Waters M460). The current produced was monitored by using an integrator (Waters M745).
Measurement of fluoxetine, norfluoxetine, fluvoxamine and MDMA in cerebral tissue
Cortical tissue was homogenized in ice-cold sodium carbonate-sodium bicarbonate buffer (pH: 11.5) using an ultrasonicator. The homogenate was centrifuged at 18,000 r.p.m. for 20 min at 4°C. The supernatant was applied to a 145 mg C8 endcapped SPE Light column (International Sorbent Technology, Waters). The column was washed with methanol (2 ml) followed by distilled H2O (2 ml) before sample (400 μl supernatant+250 μl distilled H2O+100 μl protriptiline (5 μg ml−1) or 100 μl fluoxetine (10 μg ml−1) or 100 μl 3,4-methylenedioxyethylamphetamine (10 μg ml−1) as internal standards for fluoxetine, fluvoxamine and MDMA, respectively) was applied. The column was washed with H2O (2 ml) before selective elution of fluoxetine, norfluoxetine, fluvoxamine or MDMA with methanol (1 ml).
An aliquot (20 μl) of the resulting eluate was injected into a Waters h.p.l.c. system which consisted of a pump (Waters 510) linked to a manual sample injector (Loop 20 μl, Rheodyne), a stainless steel column (RP 18, 5 μm, 150×4.6 mm, XTerra) with a precolumn (RP 18, 5 μm, 20×3.9 mm, XTerra) and an ultraviolet/visible detector (Waters 2487). The current produced was monitored by using an integrator (Waters M745). For fluoxetine and norfluoxetine analysis the mobile phase consisted of 20 mM potassium dihydrogen phosphate (65%) and acetonitrile (35%, pH 2.5); the flow rate was set to 1.5 ml min−1 and u.v. absorption was measured at 210 nm. The retention times were 6.1 min for protriptiline (internal standard), 8.4 min for norfluoxetine and 9.6 min for fluoxetine. For fluvoxamine analysis, the experimental conditions were the same as above and the flow rate was set to 1 ml min−1. The retention times were 10.6 min for fluvoxamine and 17.8 min for fluoxetine (internal standard). For MDMA analysis the mobile phase consisted of 20 mM potassium dihydrogen phosphate (75%) and acetonitrile (25%, pH 2.5); the flow rate was set to 0.8 ml min−1 and u.v. absorption measured at 235 nm. The retention times were 4.3 min for MDMA and 5.2 min for 3,4-methylenedioxyethylamphetamine (internal standard). In each run all the peaks were completely resolved without any interference from endogenous compounds. Recoveries were determined by comparing peak height ratios of the extracted standards with those obtained by direct injection of the same amount of compounds ranged from 5 – 40 μg ml−1 for fluoxetine and norfluoxetine, from 2.5 – 40 μg ml−1 for fluvoxamine and from 1.25 – 80 μg ml−1 for MDMA. The limit of detection under the described conditions was 10 pg μl−1 for all the compounds with a signal to noise ratio of 3. Intra-assay variation was less than 5% and recovery greater than 90% except for MDMA (75%).
[3H]-Paroxetine binding in tissue homogenates
[3H]-Paroxetine binding was measured by the method described in detail by Hewitt & Green (1994). The animals were killed, the brain rapidly removed and dissected on ice within 2 min. Cortex from individual animals was homogenized in ice-cold Tris-HCl (50 mM; pH 7.4) containing (in mM): NaCl 120 and KCl 5, using an Ultra-Turrax. The homogenate was centrifuged at 30,000×g for 10 min at 4°C. The supernatant was discarded and the wash procedure repeated twice more. The pellet was finally resuspended in the Tris buffer at a concentration of 5 mg tissue ml−1. Aliquots of this membrane suspension were incubated for 90 min at room temperature with [3H]-paroxetine (specific activity 19.7 Ci mmol−1; Du Pont) in the absence or presence of 5-HT for determination of non specific binding. The assay solution (1 ml) contained 100 μl of [3H]-paroxetine (0.02 – 4 nM) and 800 μl tissue preparation with the addition of either 100 μl 5-HT (100 μM) or 100 μl of buffer. After incubation, assay was terminated by rapid filtration through Whatman GF/B filters pretreated with 0.05% polyethylenimine using the Skatron Cell Harvester. The radioactivity was counted by scintillation spectrometry. Protein concentration was measured by the method of Lowry et al. (1951).
Statistics
Data from the monoamine and [3H]-paroxetine binding experiments were analysed using one-way analysis of variance (ANOVA) followed by the Newman-Keuls multiple comparison test when a significant F value was obtained (GraphPad Prism, San Diego, CA, U.S.A.).
The affinity constant (KD) and maximal number of binding sites (Bmax) for [3H]-paroxetine were calculated from saturation binding data with non-linear regression analysis fitted to a one-site binding model using the Prism software package. The analyses of KD and Bmax were performed using an unpaired t-test.
Statistical analysis of the temperature measurements were performed using the statistical computer package BMDP/386 Dynamic (BMDP Statistical Solutions, Cork, Eire). Data were analysed by means of two-way ANOVA with repeated measures (program 2 V) or, where missing values occurred, an unbalanced repeated measure model (program 5 V) was used. Both used treatment as the between subjects factor and time as the repeated measure. ANOVA was performed on both pre-treatment and post-treatment data.
Results
Effect of fluoxetine and fluvoxamine on the changes induced by MDMA in brain indole content and cortical [3H]-paroxetine binding
A single injection of MDMA (15 mg kg−1, i.p.) produced a marked decrease in the concentration of 5-HT and 5-HIAA in the cortex, hippocampus and striatum (Figure 1) and a similar reduction in the cortical [3H]-paroxetine binding 7 days later (Figure 1). Two injections of fluoxetine (10 mg kg−1, i.p., 5 min prior to and 55 min after the MDMA), prevented the loss of indole content and attenuated the decrease in 5-HT uptake sites (Figure 1). Complete neuroprotection was observed when fluoxetine (2 injections, 60 min apart) was administered 2 and 4 days before MDMA (Figure 1). When fluoxetine was given 7 days before MDMA it still afforded a modest but significant protection against the MDMA-induced damage to 5-HT nerve endings (Figure 1).
Figure 1.

Effect of fluoxetine on the 3,4-methylenedioxymethamphetamine (MDMA)-induced decrease in indole concentration in (a) cortex, (b) hippocampus, (c) striatum and (d) density of [3H]-paroxetine labelled 5-HT uptake sites in cortex. Two injections of fluoxetine (10 mg kg−1, i.p) with an interval of 60 min were given 2, 4 or 7 days before MDMA (15 mg kg−1, i.p.), rats being sacrificed 7 days later. A group of animals received fluoxetine 5 min before and 55 min after MDMA. Results shown as mean±s.e.mean (n=6 – 16). Different from saline: *P<0.001. Different from MDMA: ▿P<0.05, ▵P<0.001.
While fluvoxamine (15 mg kg−1, i.p., 5 min prior to and 55 min after the MDMA) also completely prevented the subsequent MDMA-induced reduction in brain indole content and [3H]-paroxetine binding density, it had no neuroprotective effect when given 24 h prior to the MDMA (Figure 2).
Figure 2.

Effect of fluvoxamine on the 3,4-methylenedioxymethamphetamine (MDMA)-induced decrease in indole concentration in (a) cortex, (b) hippocampus, (c) striatum and (d) density of [3H]-paroxetine labelled 5-HT uptake sites in cortex. Two injections of fluvoxamine (15 mg kg−1, i.p) with an interval of 60 min were given 24 h before MDMA (15 mg kg−1, i.p.), rats being sacrificed 7 days later. A group of animals received fluvoxamine 5 min before and 55 min after MDMA. Results shown as mean±s.e.mean (n=6 – 16). Different from saline: *P<0.05; **P<0.001. Different from MDMA: ▵P<0.01, ▵▵P<0.001.
Effect of fluoxetine and fluvoxamine on saturation binding isotherms of [3H]-paroxetine in the cortex
Figure 3 shows saturation binding isotherms of [3H]-paroxetine in the cortex of saline and fluoxetine treated rats. In the saline groups, a non linear regression analysis of specific binding revealed a single saturable site (r2 0.99) with a KD of 0.203 – 0.256 nM and a Bmax of 72 – 87 fmol mg−1 protein.
Figure 3.

Saturation-binding analysis of [3H]-paroxetine to the cortex of saline and fluoxetine injected rats 2 (a), 4 (b) and 7 (c) days after treatment. Rats received two doses of fluoxetine (10 mg kg−1, i.p.) with an interval of 60 min. A saturation isotherm obtained from 4 – 8 experiments per group in triplicate is shown. Bmax and KD of cortical [3H]-paroxetine binding in saline- and fluoxetine-treated rats is shown as mean±s.e.mean. The data were best fitted to a single site model (r2=0.99). Different from saline: *P<0.05, **P<0.01, ***P<0.001.
Administration of two doses of fluoxetine (10 mg kg−1, 60 min apart) produced a pronounced increase in the dissociation constant (KD) and a reduction in the maximal number of binding sites (Bmax) for [3H]-paroxetine in the cerebral cortex both 2 and 4 days later (Figure 3). The Bmax was still decreased 7 days later, although there was no change in the KD value (Figure 3).
Twenty-four hours after fluvoxamine (15 mg kg−1, i.p., two doses, 60 min apart) no significant change in the [3H]-paroxetine binding properties was observed in the drug-treated animals (KD: 0.215±0.05 nM, n=4; Bmax: 82±4.8 fmol mg−1 protein, n=4) compared with saline injected control rats (KD: 0.237±0.06 nM, n=4; Bmax: 87±5.4 fmol mg−1 protein, n=4).
Brain concentrations of fluoxetine, norfluoxetine and fluvoxamine
Following two doses of fluoxetine (10 mg kg−1, i.p., 60 min apart) the levels of both fluoxetine and its main metabolite, norfluoxetine, were determined in cortex over the next 7 days. At the intervals examined, the peak concentration of fluoxetine occurred within the first 30 min interval declining progressively thereafter, reaching a low and constant concentration between 4 – 7 days after dosing (Figure 4). Norfluoxetine concentrations rose over the first 24 – 48 h, the concentration then diminishing slowly over the next 5 days (Figure 4).
Figure 4.

Time course of fluoxetine and norfluoxetine concentrations in cortex after administration of two doses of fluoxetine (10 mg kg−1, i.p) with an interval of 60 min. Levels of the parent compound and its metabolite were measured at 0.5, 1, 4, 8, 24, 48, 96 and 168 h after last dose of fluoxetine. Results shown as mean±s.e.mean (n=5 at each time point).
The concentration of fluvoxamine 30 min after the second of two doses of the drug (15 mg kg−1, i.p. 60 min apart) was 35±6 nmol g−1 (n=4). The concentration then decreased rapidly, being 6±0.03 nmol g−1 (n=4) at 6 h and 3±0.2 nmol g−1 (n=4) at 24 h.
Effect of fluoxetine on indole concentration in the brain
Fluoxetine (10 mg kg−1, two injections, 60 min apart) had no effect on 5-HT concentration at any time examined (2, 4, 7 and 14 days) following administration (Figure 5). However, there was a significant decrease in 5-HIAA concentration in cortex, hippocampus and striatum at both 2 and 4 days. No change in the brain 5-HIAA concentration was observed after that time (Figure 5).
Figure 5.

Effect of fluoxetine on indole concentration in (a) cortex, (b) hippocampus and (c) striatum 2, 4, 7 and 14 days after administration. Rats received two injections of fluoxetine (10 mg kg−1, i.p) with an interval of 60 min. Results shown as mean±s.e.mean (n=6 – 12). Different from saline: *P<0.001.
Effect of fluoxetine on brain levels of MDMA
In order to investigate the possible effect of fluoxetine on the concentration of MDMA in the brain, rats were given fluoxetine (10 mg kg−1, i.p., two doses, 60 min apart). Two days later the animals received a single dose of MDMA (15 mg kg−1, i.p.) and the cortical concentration of MDMA was measured 60 min later. This time was chosen because the cerebral concentration of MDMA normally peaks 60 min after this dose of MDMA (Esteban et al., 2001). There was no difference between MDMA levels found in rats pretreated with fluoxetine (66±9 nmol g−1 tissue, n=4) and those injected with saline (59±4 nmol g−1 tissue, n=4).
Effect of fluoxetine and fluvoxamine on rectal temperature
Administration of MDMA (15 mg kg−1, i.p.) produced a clear and sustained hyperthermia (Figure 6a,b). Fluoxetine (10 mg kg−1, i.p.) either co-administered with MDMA (Figure 6a) or given 2, 4 (Figure 6a) or 7 days earlier failed to prevent the rise in temperature. Similarly, fluvoxamine (15 mg kg−1, i.p.) given 5 min before and 55 min after MDMA did not modify the hyperthermic response (Figure 6b). Administration of fluoxetine or fluvoxamine alone (two doses 60 min apart) had no effect on rectal temperature (data not shown).
Figure 6.
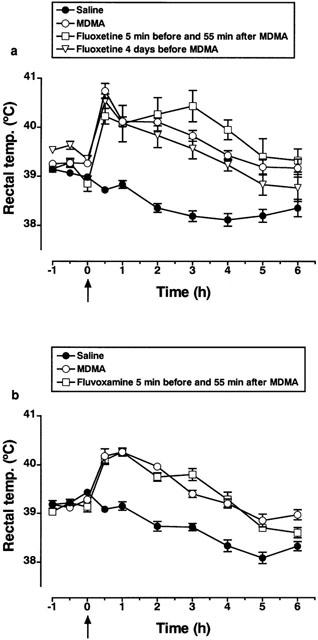
Effect of fluoxetine or fluvoxamine on the 3,4-methylenedioxymethamphetamine (MDMA)-induced hyperthermia. (a) Rectal temperature of rats injected with fluoxetine (10 mg kg−1, i.p., two doses, 60 min apart) or saline 4 days before MDMA (15 mg kg−1, i.p.) or 5 min before and 55 min after MDMA. Arrow indicates MDMA injection. MDMA significantly increased temperature compared to saline treatment (F(1,21)=116.4; P<0.001). Fluoxetine either co-administered with MDMA or injected 4 days before did not modify the hyperthermic response (F(1,13)=0.283 and F(1,15)=2.61, respectively). (b) Rectal temperature of rats injected with fluvoxamine (15 mg kg−1, i.p.) or saline 5 min before and 55 min after MDMA. MDMA significantly increased temperature compared to saline treatment (F(1,10)=133.1; P<0.001). Co-administration of fluvoxamine with MDMA did not modify the hyperthermic response (F(1,10)=0.409). Results shown as mean and vertical lines indicate s.e.mean (n=6 – 12).
Discussion
This study shows for the first time that fluoxetine, when given 4 days before MDMA, provides complete protection against the delayed neurotoxic loss of 5-HT nerve terminals induced by this amphetamine derivative and still provides partial protection when given 7 days earlier. Other authors have previously reported that fluoxetine protected against MDMA-induced neurodegeneration, but in their studies the 5-HT uptake inhibitor was co-administered with the MDMA (Schmidt, 1987; Malberg et al., 1996; Shankaran et al., 1999) or given up to 6 h later (Schmidt, 1987; Shankaran et al., 1999). In the current study, protection was observed not only in the attenuation of the MDMA-induced decrease in 5-HT and 5-HIAA concentration in hippocampus, cortex and striatum, but also in the attenuation of the reduction in cortical 5-HT uptake sites labelled with [3H]-paroxetine. The binding of [3H]-paroxetine decreases following administration of compounds shown histologically to produce neuronal damage, including, for example, selective 5-HT neurotoxins such as 5,7-dihydroxytryptamine (Habert et al., 1985) and MDMA (Hewitt & Green, 1994). Therefore, the ability of fluoxetine to attenuate the loss of 5-HT transporter gives confidence in the conclusion that fluoxetine is producing true neuroprotection.
When [3H]-paroxetine binding was used as a marker rather than the 5-HT content, the degree of protection seen when fluoxetine was given concurrently with MDMA was smaller than that observed when fluoxetine had been administered 2 – 4 days earlier (Figure 1). This effect is probably due to the existence of residual drug (fluoxetine or norfluoxetine) in the tissue interfering with the binding parameters 7 days after fluoxetine administration (Figure 3).
The long-lasting neuroprotective effect of fluoxetine appears to be closely related to the presence in the brain of fluoxetine and its active and major metabolite norfluoxetine, whose presence was still detectable for up to 7 days after administration of fluoxetine. Both fluoxetine and norfluoxetine act primarily as 5-HT uptake inhibitors (Bolden-Watson & Richelson, 1993; Wong et al., 1993; Sanchez & Hyttel, 1999) and both have similar efficacy as 5-HT uptake inhibitors (Wong et al., 1993). Since the compounds have similar potency, it was possible to plot the concentration of [fluoxetine+norfluoxetine] in nmol g−1 versus the degree of protection seen over time (using the loss in 5-HT content in cortex). This revealed a simple hyperbolic plot indicating that protection probably relates to tissue drug concentration (Figure 7).
Figure 7.
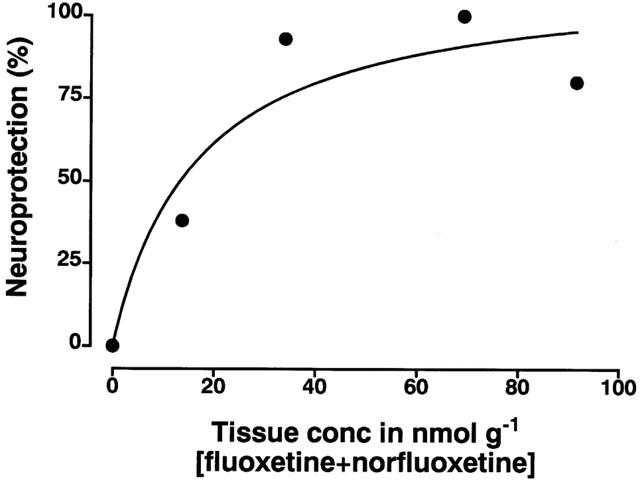
Plot of the mean concentration of fluoxetine plus norfluoxetine in cortical tissue at various times post fluoxetine injection (taken from data presented in Figure 4) and the degree of neuroprotection measured as the per cent of loss of cortical tissue 5-HT seen following fluoxetine administration to MDMA-treated rats versus the loss seen following MDMA alone.
A plot of the changes in KD value versus concentration of [fluoxetine+norfluoxetine] revealed, not unexpectedly, that there is a linear relationship between the combined concentration of these two highly liposoluble drugs and the KD value (r=0.99). There was also a reasonably linear relationship between the KD and Bmax value at every time point studied (r=0.91). It is probable that the Bmax change seen also relates to the presence of the drug interfering with the binding analysis rather than any down regulation of the receptor. This interpretation would be consistent with the failure of others to find down regulation of the uptake site even on chronic administration of 5-HT uptake inhibitor compounds (Cheetham et al., 1993; Dean et al., 1997). However we cannot rule out totally that a down regulation of the uptake site has occurred.
What appears clear is that a defined concentration of the drug must be present in the tissue to inhibit uptake and thereby provide neuroprotection. This view is supported by the study with fluvoxamine, since the rapid clearance of this compound was accompanied by loss of neuroprotection.
Fluoxetine not only provided long-lasting protection against the long-term neurotoxicity induced by MDMA but also produced long-lasting prevention of the acute 5-HT depletion by p-chloroamphetamine. Administration to rats of one dose of fluoxetine (10 mg kg−1, i.p.) between 4 and 24 h before p-chloroamphetamine completely prevented the reduction of the cortical 5-HT concentration induced by p-chloroamphetamine 4 h later. This effect was smaller when the pretreatment interval was 48 h (Fuller et al., 1975).
There are several lines of evidence indicating that the continuous presence of selective 5-HT uptake inhibitors tends to produce changes in the function of the transporter within minutes (Blakely et al., 1998; Benmansour et al., 1999; Ramamoorthy et al., 1998) and that the changes are due to protein kinase C (PKC) activation in a similar way to that occurring for the 5-HT receptors (Qian et al., 1997). PKC activation in turns leads to transporter phosphorylation, transporter redistribution from the cell surface (sequestration) and loss of functional uptake activity. In contrast to 5-HT and amphetamines, antidepressants facilitate the 5-HT transporter phosphorylation and block 5-HT uptake (Ramamoorthy & Blakely, 1999). Since MDMA is a substrate for the 5-HT transporter, an internalization of cell-surface transporter protein and therefore, a decrease in the density of transporter proteins in the cell membrane induced by the presence of fluoxetine or norfluoxetine, could involve a significant and lasting blockade in the uptake of MDMA or most likely of a metabolic product to the presynaptic 5-HT nerve terminals (Esteban et al., 2001).
Two and four days after fluoxetine administration, there is a decrease in brain 5-HIAA concentration whereas 5-HT content remains unaltered. This effect, previously described by others (Baldessarini et al., 1992), is probably due to decreased 5-HT synthesis resulting from compensatory changes initiated by the increase in the synaptic 5-HT concentration (Bymaster & Wong, 1974). However, a reduction in 5-HT synthesis following fluoxetine administration is not the cause of fluoxetine-induced neuroprotection since administration of the tryptophan hydroxylase inhibitor p-chlorophenylalanine (PCPA) does not influence the MDMA-induced decrease in cortical 5-HT uptake sites (Brodkin et al., 1993).
All current evidence suggests that the neurotoxicity of MDMA is not due to the parent compound but to a toxic metabolite probably formed peripherally. Intracerebral administration of MDMA at a dose that produced a concentration similar or higher than that reached following a neurotoxic dose of MDMA (15 mg kg−1) given peripherally, does not produce neurotoxic damage (Esteban et al., 2001). There is evidence that intrastriatal or intracortical administration of glutathione and N-acetylcysteine conjugates of α-methyldopamine (a major metabolite of MDMA) causes significant decreases in striatal and cortical 5-HT concentrations 7 days later (Bai et al., 1999). Nevertheless, the role for these compounds on MDMA neurotoxicity remains to be determined.
It is well known that in Dark Agouti rats MDMA is metabolized by CYP2D1, the rat equivalent to the human CYP2D6, and that fluoxetine and its metabolite, norfluoxetine, are potent inhibitors of CYP2D6 (Hamelin et al., 1996). In addition, fluoxetine and norfluoxetine are detected in the brain several days after fluoxetine injection. Therefore, it could be argued that the protective effect of fluoxetine results from its ability to inhibit MDMA metabolism. This does not appear to be the case since fluoxetine did not alter the concentration of MDMA in the brain. When MDMA was administered to rats pretreated with fluoxetine 2 days earlier (a time when the concentration of both fluoxetine and its metabolite are still high), the cortical MDMA levels observed 60 min later were similar to that found in rats not given fluoxetine. We can state with some degree of confidence that fluoxetine does not prevent MDMA toxicity by inhibiting the metabolism of the drug. It thus seems likely that fluoxetine and its major metabolite are preventing the entry of a neurotoxic metabolite of MDMA into the nerve ending.
What can also be stated unequivocally is that the neuroprotective effect of fluoxetine or fluvoxamine against MDMA toxicity is not related to an effect on body temperature. The hyperthermic response immediately following MDMA was similar in rats receiving saline or either fluoxetine or fluvoxamine. In addition, these data support the notion that the rise in rectal temperature induced by MDMA is not obviously related to its ability to increase 5-HT release (Mechan et al., 2000).
In summary this study shows that the neuroprotective effect of fluoxetine is due to the long term presence of either the parent compound or to its active metabolite, norfluoxetine in the brain. Both compounds block 5-HT transporter selectively, inhibit 5-HT uptake and presumably interfere with the entry of MDMA or a neurotoxic metabolite into the presynaptic nerve terminal.
Acknowledgments
This work was supported by grants from CICYT (SAF98-0074, Ministerio de Education) and Delegacion del Gobierno para el Plan Nacional sobre Drogas (Ministerio del Interior). The authors are grateful to Servicio de Restricción de Estupefacientes, Ministerio de Sanidad, for supplying MDMA and to Lilly, S.A. for supplying fluoxetine. J. Camarero thanks FIS for a fellowship.
Abbreviations
- ANOVA
analysis of variance
- Bmax
maximal number of binding sites
- 2,3-DHBA
dihydroxybenzoic acid
- 3,4-DHMA
dihydroxymethamphetamine
- EDTA
ethylenediaminetetraacetic acid
- 5-HIAA
5-hydroxyindoleacetic acid
- h.p.l.c.
high performance liquid chromatography
- 5-HT
5-hydroxytryptamine
- KD
dissociation constant
- MAO
monoamine oxidase
- MDMA
(±) 3,4-methylenedioxymethamphetamine
References
- AZMITIA E.C., MURPHY R.B., WHITAKER-AZMITIA P.M. MDMA (ecstasy) effects on cultured serotonergic neurons: evidence for Ca2+-dependent toxicity linked to release. Brain Res. 1990;510:97–103. doi: 10.1016/0006-8993(90)90732-q. [DOI] [PubMed] [Google Scholar]
- BALDESSARINI R.J., MARSH E.R., KULA N.S. Interactions of fluoxetine with metabolism of dopamine and serotonin in rat brain regions. Brain Res. 1992;579:152–156. doi: 10.1016/0006-8993(92)90754-w. [DOI] [PubMed] [Google Scholar]
- BAI F., LAU S.S., MONKS T.J. Glutathione and N-acetylcysteine conjugates of α-methyldopamine produce serotonergic neurotoxicity: Possible role in methylenedioxyamphetamine-mediated neurotoxicity. Chem. Res. Toxicol. 1999;12:1150–1157. doi: 10.1021/tx990084t. [DOI] [PubMed] [Google Scholar]
- BENFIELD P., HEEL R.C., LEWIS S.P. Fluoxetine. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy in depressive illness. Drugs. 1986;32:481–508. doi: 10.2165/00003495-198632060-00002. [DOI] [PubMed] [Google Scholar]
- BENMANSOUR S., CECCHI M., MORILAK D.A., GERHARDT G.A., JAVORS M.A., GOULD G.G., FRAZER A. Effects of chronic antidepressant treatments on serotonin transporter function, density and mRNA level. J. Neurosci. 1999;19:10494–10501. doi: 10.1523/JNEUROSCI.19-23-10494.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERGER U.V., GU X.F., AZMITIA E.C. The substituted amphetamines 3,4-methylenedioxymethamphetamine, methamphetamine, p-chloroamphetamine and fenfluramine induce 5-hydroxytryptamine release via a common mechanism blocked by fluoxetine and cocaine. Eur. J. Pharmacol. 1992;215:153–160. doi: 10.1016/0014-2999(92)90023-w. [DOI] [PubMed] [Google Scholar]
- BERGSTROM R.F., PEYTON A.L., LEMBERGER L. Quantification and mechanism of the fluoxetine and tricyclic antidepressant interaction. Clin. Pharmacol. Ther. 1992;51:239–248. doi: 10.1038/clpt.1992.18. [DOI] [PubMed] [Google Scholar]
- BLAKELY R.D., RAMAMOORTHY S., SCHROETER S., QIAN Y., APPARSUNDARAM S., GALLI A., DEFELICE L.J. Regulated phosphorylation and trafficking of antidepressant-sensitive serotonin transporter proteins. Biol. Psychiatry. 1998;44:169–178. doi: 10.1016/s0006-3223(98)00124-3. [DOI] [PubMed] [Google Scholar]
- BOLDEN-WATSON C., RICHELSON E. Blockade of newly-developed antidepressants of biogenic amine uptake into rat brain synaptosomes. Life Sci. 1993;52:1023–1029. doi: 10.1016/0024-3205(93)90194-8. [DOI] [PubMed] [Google Scholar]
- BOSKER F.J., KLOMPMAKERS A.A., WESTENBERG H.G. Effects of single and repeated oral administration of fluvoxamine on extracellular serotonin in the median raphe nucleus and dorsal hippocampus of the rat. Neuropharmacology. 1995;34:501–508. doi: 10.1016/0028-3908(95)00023-y. [DOI] [PubMed] [Google Scholar]
- BRODKIN J., MALYALA A., NASH J.F. Effect of acute monoamine depletion on 3,4-methylenedioxymethamphetamine-induced neurotoxicity. Pharmacol. Biochem. Behav. 1993;45:647–653. doi: 10.1016/0091-3057(93)90520-4. [DOI] [PubMed] [Google Scholar]
- BYMASTER F.P., WONG D.T. Effect of Lilly 110140, 3-(p-trifluoromethylphenoxy)-N-methyl-3-phenylpropanolamine on synthesis of 3H-serotonin from 3H-tryptophan in rat brain. Pharmacologist. 1974;16:244. [Google Scholar]
- CADET J.L., LADENHEIM B., HIRATA H., ROTHMAN R.B., ALI S., CARLSON E., EPSTEIN C., MORAN T.H. Superoxide radicals mediate the biochemical effects of methylenedioxymethamphetamine (MDMA): Evidence from using CuZn-superoxide dismutase transgenic mice. Synapse. 1995;21:169–176. doi: 10.1002/syn.890210210. [DOI] [PubMed] [Google Scholar]
- CHEETHAM S.C., VIGGERS J.A., SLATER N.A., HEAL D.J., BUCKETT W.R. [3H]paroxetine binding in rat frontal cortex strongly correlates with [3H]5-HT uptake; effect of administration of various antidepressant treatments. Neuropharmacology. 1993;32:737–743. doi: 10.1016/0028-3908(93)90181-2. [DOI] [PubMed] [Google Scholar]
- COLADO M.I., GREEN A.R. The spin trap reagent α-phenyl-N-tert-butyl nitrone prevents ‘ecstasy'-induced neurodegeneration of 5-hydroxytryptamine neurones. Eur. J. Pharmacol. 1995;280:343–346. doi: 10.1016/0014-2999(95)00298-y. [DOI] [PubMed] [Google Scholar]
- COLADO M.I., MURRAY T.K., GREEN A.R. 5-HT loss in rat brain following 3,4-methylenedioxymethamphetamine (MDMA), p-chloroamphetamine and fenfluramine administration and effects of chlormethiazole and dizocilpine. Br. J. Pharmacol. 1993;108:583–589. doi: 10.1111/j.1476-5381.1993.tb12846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLADO M.I., O'SHEA E., GRANADOS R., ESTEBAN B., MARTIN A.B., GREEN A.R. Studies on the role of dopamine in the degeneration of 5-HT nerve endings in the brain of Dark Agouti rats following 3,4-methylenedioxymethamphetamine (MDMA or ‘ecstasy') administration. Br. J. Pharmacol. 1999;126:911–924. doi: 10.1038/sj.bjp.0702373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLADO M.I., O'SHEA E., GRANADOS R., MISRA A., MURRAY T.K., GREEN A.R. A study of the neurotoxic effect of MDMA (‘ecstasy') on 5-HT neurones in the brains of mothers and neonates following administration of the drug during pregnancy. Br. J. Pharmacol. 1997a;121:827–833. doi: 10.1038/sj.bjp.0701201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLADO M.I., O'SHEA E., GRANADOS R., MURRAY T.K., GREEN A.R. In vivo evidence for free radical involvement in the degeneration of rat brain 5-HT following administration of MDMA (‘ecstasy') and p-chloroamphetamine but not the degeneration following fenfluramine. Br. J. Pharmacol. 1997b;121:889–900. doi: 10.1038/sj.bjp.0701213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLADO M.I., WILLIAMS J.L., GREEN A.R. The hyperthermic and neurotoxic effects of ‘Ecstasy' (MDMA) and 3,4-methylenedioxyamphetamine (MDA) in the Dark Agouti (DA) rat, a model of the CYP2D6 poor metabolizer phenotype. Br. J. Pharmacol. 1995;115:1281–1289. doi: 10.1111/j.1476-5381.1995.tb15037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CRESPI D., MENNINI T., GOBBI M. Carrier-dependent and Ca2+-dependent 5-HT and dopamine release induced by (+)-amphetamine, 3,4-methylendioxymethamphetamine, p-chloroamphetamine and (+)-fenfluramine. Br. J. Pharmacol. 1997;121:1735–1743. doi: 10.1038/sj.bjp.0701325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEAN B., PEREIRA A., PAVEY G., SINGH B. Repeated antidepressant drug treatment, time of death and frequency of handling do not affect [3H]paroxetine binding in rat cortex. Psychiat. Res. 1997;73:173–179. doi: 10.1016/s0165-1781(97)00125-x. [DOI] [PubMed] [Google Scholar]
- ESTEBAN B., O'SHEA E., CAMARERO J., SANCHEZ V., GREEN A.R., COLADO M.I. 3,4-Methylenedioxymethamphetamine induces monoamine release, but not toxicity, when administered centrally at a concentration occurring following a peripherally injected neurotoxic dose. Psychopharmacology. 2001;154:251–260. doi: 10.1007/s002130000645. [DOI] [PubMed] [Google Scholar]
- FITZGERALD J.L., REID J.J. Interactions of methylenedioxymethamphetamine with monoamine transmitter release mechanisms in rat brain slices. Naunyn Schmiedeberg's Arch. Pharmacol. 1993;347:313–323. doi: 10.1007/BF00167451. [DOI] [PubMed] [Google Scholar]
- FULLER R.W., KENNETH W.P., MOLLOY B.B. Effect of 3-(p-trifluoromethylphenoxy)-N-methyl-3-phenylpropylamine on the depletion of brain serotonin by 4-chloroamphetamine. J. Pharmacol. Exp. Ther. 1975;193:796–803. [PubMed] [Google Scholar]
- FULLER R.W., SNODDY H.D., PERRY K.W., BYMASTER F.P., WONG D.T. Importance of duration of drug action in the antagonism of p-chloroamphetamine depletion of brain serotonin-comparison of fluoxetine and chlorimipramine. Biochem. Pharmacol. 1978;27:193–198. doi: 10.1016/0006-2952(78)90300-3. [DOI] [PubMed] [Google Scholar]
- GARDIER A.M., LEPOUL E., TROUVIN J.H., CHANUT E., DESSALLES M.C., JACQUOT C. Changes in dopamine metabolism in rat forebrain regions after cessation of long-term fluoxetine treatment: Relationship with brain concentrations of fluoxetine and norfluoxetine. Life Sci. 1994;54:51–56. doi: 10.1016/0024-3205(94)00821-3. [DOI] [PubMed] [Google Scholar]
- GOUGH B., ALI S.F., SLIKKER W., HOLSON R.R. Acute effects of 3,4-methylenedioxymethamphetamine (MDMA) on monoamines in rat caudate. Pharmacol. Biochem. Behav. 1991;39:619–623. doi: 10.1016/0091-3057(91)90137-q. [DOI] [PubMed] [Google Scholar]
- GU X.F., AZMITIA E.C. Integrative transporter-mediated release from cytoplasmic and vesicular 5-hydroxytryptamine stores in cultured neurons. Eur. J. Pharmacol. 1993;235:51–57. doi: 10.1016/0014-2999(93)90819-4. [DOI] [PubMed] [Google Scholar]
- GUDELSKY G.A., NASH J.F. Carrier-mediated release of serotonin by 3,4-methylenedioxymethamphetamine: implications for serotonin-dopamine interactions. J. Neurochem. 1996;66:243–249. doi: 10.1046/j.1471-4159.1996.66010243.x. [DOI] [PubMed] [Google Scholar]
- HABERT E., GRAHAM D., TAHRAOUI K., CLAUSTRE Y., LANGER S.Z. Characterization of [3H]paroxetine binding to rat cortical membranes. Eur. J. Pharmacol. 1985;118:107–114. doi: 10.1016/0014-2999(85)90668-5. [DOI] [PubMed] [Google Scholar]
- HAMELIN B.A., TURGEON J., VALLEE F., BELANGER P.M., PAQUET F., LEBEL M. The disposition of fluoxetine but not sertraline is altered in poor metabolizers of debrisoquin. Clin. Pharmacol. Ther. 1996;60:512–521. doi: 10.1016/S0009-9236(96)90147-2. [DOI] [PubMed] [Google Scholar]
- HEKMATPANAH C.R., PEROUTKA S.J. 5-Hydroxytryptamine uptake blockers attenuate the 5-hydroxytryptamine-releasing effect of 3,4-methylenedioxymethamphetamine and related agents. Eur. J. Pharmacol. 1990;177:95–98. doi: 10.1016/0014-2999(90)90555-k. [DOI] [PubMed] [Google Scholar]
- HEWITT K.E., GREEN A.R. Chlormethiazole, dizocilpine and haloperidol prevent the degeneration of serotonergic nerve terminals induced by administration of MDMA (‘ecstasy') to rats. Neuropharmacology. 1994;33:1589–1595. doi: 10.1016/0028-3908(94)90134-1. [DOI] [PubMed] [Google Scholar]
- HOLLADAY J.W., DEWEY M.J., YOO S.D. Pharmacokinetics and antidepressant activity of fluoxetine in transgenic mice with elevated serum alpha-1-acid glycoprotein levels. Drug Metab. Dispos. 1998;26:20–24. [PubMed] [Google Scholar]
- HORNG J.S., WONG D.T. Effects of serotonin uptake inhibitor, Lilly 110140, on transport of serotonin in rat and human blood platelets. Biochem. Pharmacol. 1976;25:865–867. doi: 10.1016/0006-2952(76)90162-3. [DOI] [PubMed] [Google Scholar]
- JOHNSON M.P., CONARTY P.F., NICHOLS D.E. [3H]monoamine releasing and uptake inhibition properties of 3,4-methylenedioxymethamphetamine and p-chloroamphetamine analogues. Eur. J. Pharmacol. 1991;200:9–16. doi: 10.1016/0014-2999(91)90659-e. [DOI] [PubMed] [Google Scholar]
- JOHNSON M.P., HOFFMAN A.J., NICHOLS D.E. Effects of the enantiomers of MDA, MDMA and related analogues on [3H]serotonin and [3H]dopamine release from superfused rat brain slices. Eur. J. Pharmacol. 1986;132:269–276. doi: 10.1016/0014-2999(86)90615-1. [DOI] [PubMed] [Google Scholar]
- LEFEBVRE M., MARCHAND M., HOROWITZ J.M., TORRES G. Detection of fluoxetine in brain, blood, liver and hair of rats using gas chromatography-mass spectrometry. Life Sci. 1999;64:805–811. doi: 10.1016/s0024-3205(98)00622-5. [DOI] [PubMed] [Google Scholar]
- LEMBERGER L., BERGSTROM R.F., WOLEN R.L., FARID N.A., ENAS G.G., ARONOFF G.R. Fluoxetine: clinical pharmacology and physiologic disposition. J. Clin. Psychiatry. 1985;46:14–19. [PubMed] [Google Scholar]
- LEONARDI E.T., AZMITIA E.C. MDMA (ecstasy) inhibition of MAO type A and type B: comparisons with fenfluramine and fluoxetine (Prozac) Neuropsychopharmacology. 1994;10:231–238. doi: 10.1038/npp.1994.26. [DOI] [PubMed] [Google Scholar]
- LOWRY O.H., ROSEBROUGH N.J., FARR A.L., RANDALL R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- MALBERG J.E., SABOL K.E., SEIDEN L.S. Co-administration of MDMA with drugs that protect against MDMA neurotoxicity produces different effects on body temperature in the rat. J. Pharmacol. Exp. Ther. 1996;278:258–267. [PubMed] [Google Scholar]
- MCKENNA D.J., GUAN X.M., SHULGIN A.T. 3,4-Methylenedioxyamphetamine (MDA) analogues exhibit differential effects on synaptosomal release of 3H-dopamine and 3H-5-hydroxytryptamine. Pharmacol. Biochem. Behav. 1991;38:505–512. doi: 10.1016/0091-3057(91)90005-m. [DOI] [PubMed] [Google Scholar]
- MECHAN A.O., COLADO M.I., ESTEBAN B., ELLIOTT J.M., GREEN A.R. Evidence against MDMA-induced hyperthermia being mediated by 5-HT release in the brain. Br. J. Pharmacol. 2000;131:153P. [Google Scholar]
- MOLLIVER M.E., BERGER U.V., MAMOUNAS L.A., MOLLIVER D.C., O'HEARN E., WILSON M.A. Neurotoxicity of MDMA and related compounds: anatomic studies. Ann. N. Y. Acad. Sci. 1990;600:649–661. doi: 10.1111/j.1749-6632.1990.tb16916.x. [DOI] [PubMed] [Google Scholar]
- NICHOLS D.E., LLOYD D.H., HOFFMAN A.J., NICHOLS M.B., YIM G.K. Effects of certain hallucinogenic amphetamine analogues on the release of [3H]serotonin from rat brain synaptosomes. J. Med. Chem. 1982;25:530–535. doi: 10.1021/jm00347a010. [DOI] [PubMed] [Google Scholar]
- O'HEARN E., BATTAGLIA G., DE SOUZA E.B., KUHAR M.J., MOLLIVER M.E. Methylenedioxyamphetamine (MDA) and methylenedioxymethamphetamine (MDMA) cause selective ablation of serotonergic axon terminals in forebrain: immunocytochemical evidence for neurotoxicity. J. Neurosci. 1988;8:2788–2803. doi: 10.1523/JNEUROSCI.08-08-02788.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'SHEA E., GRANADOS R., ESTEBAN E., COLADO M.I., GREEN A.R. The relationship between the degree of neurodegeneration of rat brain 5-HT nerve terminals and the dose and frequency of administration of MDMA (‘ecstasy') Neuropharmacology. 1998;37:919–926. doi: 10.1016/s0028-3908(98)00029-x. [DOI] [PubMed] [Google Scholar]
- POBLETE J.C., WHITAKER-AZMITIA P.M., AZMITIA E.C. The effects of drugs of abuse and selected serotonin agonists on the high affinity serotonin transporter displacement of [3H]-paroxetine. Soc. Neurosci. Abstr. 1989;15:418. [Google Scholar]
- PRESKORN S.H. Clinically relevant pharmacology of selective serotonin reuptake inhibitors. An overview with emphasis on pharmacokinetics and effects on oxidative drug metabolism. Clin. Pharmacokinet. 1997;32:1–21. doi: 10.2165/00003088-199700321-00003. [DOI] [PubMed] [Google Scholar]
- QIAN Y., GALLI A., RAMAMOORTHY S., RISSO S., DEFELICE L.J., BLAKELY R.D. Protein kinase C activation regulates human serotonin transporters in HEK-293 cells via altered cell surface expression. J. Neurosci. 1997;17:45–57. doi: 10.1523/JNEUROSCI.17-01-00045.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAMAMOORTHY S., BLAKELY R.D. Phosphorylation and sequestration of serotonin transporters differentially modulated by psychostimulants. Science. 1999;285:763–766. doi: 10.1126/science.285.5428.763. [DOI] [PubMed] [Google Scholar]
- RAMAMOORTHY S., GIOVANETTI E., QIAN Y., BLAKELY R.D. Phosphorylation and regulation of antidepressant-sensitive serotonin transporters. J. Biol. Chem. 1998;273:2458–2466. doi: 10.1074/jbc.273.4.2458. [DOI] [PubMed] [Google Scholar]
- RUDNICK G., WALL S.C. The molecular mechanism of ‘ecstasy' [3,4-methylenedioxymethamphetamine (MDMA)]: Serotonin transporters are targets for MDMA-induced serotonin release. Proc. Natl. Acad. Sci. U.S.A. 1992;89:1817–1821. doi: 10.1073/pnas.89.5.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SANCHEZ C., HYTTEL J. Comparison of the effects of antidepressants and their metabolites on reuptake of biogenic amines and on receptor binding. Cell. Mol. Neurobiol. 1999;19:467–489. doi: 10.1023/a:1006986824213. [DOI] [PubMed] [Google Scholar]
- SCHMIDT C.J. Neurotoxicity of the psychedelic amphetamine, methylenedioxymethamphetamine. J. Pharmacol. Exp. Ther. 1987;240:1–7. [PubMed] [Google Scholar]
- SCHMIDT C.J., BLACK C.K., TAYLOR V.L. Antagonism of the neurotoxicity due to a single administration of methylenedioxymethamphetamine. Eur. J. Pharmacol. 1990;181:59–70. doi: 10.1016/0014-2999(90)90245-2. [DOI] [PubMed] [Google Scholar]
- SCHMIDT C.J., BLACK C.K., TAYLOR V.L. L-DOPA potentiation of the serotonergic deficits due to a single administration of 3,4-methylenedioxymethamphetamine to rats. Eur. J. Pharmacol. 1991;203:41–49. doi: 10.1016/0014-2999(91)90788-r. [DOI] [PubMed] [Google Scholar]
- SCHMIDT C.J., LEVIN J.A., LOVENBERG W. In vitro and in vivo neurochemical effects of methylenedioxymethamphetamine on striatal monoaminergic systems in the rat brain. Biochem. Pharmacol. 1987;36:747–755. doi: 10.1016/0006-2952(87)90729-5. [DOI] [PubMed] [Google Scholar]
- SHANKARAN M., YAMAMOTO B.K., GUDESLKY G.A. Involvement of the serotonin transporter in the formation of hydroxyl radicals induced by 3,4-methylenedioxymethamphetamine. Eur. J. Pharmacol. 1999;385:103–110. doi: 10.1016/s0014-2999(99)00728-1. [DOI] [PubMed] [Google Scholar]
- SHARKEY J., MCBEAN D.E., KELLY P.A. Alterations in hippocampal function following repeated exposure to the amphetamine derivative methylenedioxymethamphetamine (‘ecstasy') Psychopharmacology. 1991;105:113–118. doi: 10.1007/BF02316872. [DOI] [PubMed] [Google Scholar]
- SLIKKER W., HOLSON R.R., ALI S.F., KOLTA M.G., PAULE M.G., SCALLET A.C., MCMILLAN D.E., BAILEY J.R., HONG J.S., SCALZO F.M. Behavioral and neurochemical effects of orally administered MDMA in the rodent and nonhuman primate. Neurotoxicology. 1989;10:529–542. [PubMed] [Google Scholar]
- SPRAGUE J.E., NICHOLS D.E. The monoamine oxidase-B inhibitor L-deprenyl protects against 3,4-methylenedioxymethamphetamine-induced lipid peroxidation and long-term serotonergic deficits. J. Pharmacol. Exp. Ther. 1995;273:667–673. [PubMed] [Google Scholar]
- SPROUSE J.S., BRADBERRY C.W., ROTH R.H., AGHAJANIAN G.K. MDMA (3,4-methylenedioxymethamphetamine) inhibits the firing of dorsal raphe neurons in brain slices via release of serotonin. Eur. J. Pharmacol. 1989;167:375–383. doi: 10.1016/0014-2999(89)90446-9. [DOI] [PubMed] [Google Scholar]
- STANFORD S.C. Prozac: panacea or puzzle. Trends Pharmacol. Sci. 1996;17:150–154. doi: 10.1016/0165-6147(96)81591-4. [DOI] [PubMed] [Google Scholar]
- STEELE T.D., NICHOLS D.F., YIM G.K. Stereochemical effects of 3,4-methylenedioxymethamphetamine (MDMA) and related amphetamine derivatives on inhibition of uptake of [3H]monoamines into synaptosomes from different regions of rat brain. Biochem. Pharmacol. 1987;36:2297–2303. doi: 10.1016/0006-2952(87)90594-6. [DOI] [PubMed] [Google Scholar]
- STONE D.M., JOHNSON M., HANSON G.R., GIBB J.W. Role of endogenous dopamine in the central serotonergic deficits induced by 3,4-methylenedioxymethamphetamine. J. Pharmacol. Exp. Ther. 1988;247:79–87. [PubMed] [Google Scholar]
- THOMAS C.E., HUBER E.W., OHLWEILER D.F. Hydroxyl and peroxyl radical trapping by the monoamine oxidase-B inhibitors deprenyl and MDL 72,974A: implications for protection of biological substrates. Free Radic. Biol. Med. 1997;22:733–737. doi: 10.1016/s0891-5849(96)00402-9. [DOI] [PubMed] [Google Scholar]
- VAN HARTEN J. Overview of the pharmacokinetics of fluvoxamine. Clin. Pharmacokinet. 1995;29:1–9. doi: 10.2165/00003088-199500291-00003. [DOI] [PubMed] [Google Scholar]
- WONG D.T., BYMASTER F.P., REID L.R., THRELKELD P.G. Fluoxetine and two other serotonin uptake inhibitors without affinity for neuronal receptors. Biochem. Pharmacol. 1983;32:1287–1293. doi: 10.1016/0006-2952(83)90284-8. [DOI] [PubMed] [Google Scholar]
- WONG D.T., BYMASTER F.P., REID L.R., MAYLE D.A., KRUSHINSKI J.H., ROBERTSON D.W. Norfluoxetine enantiomers as inhibitors of serotonin uptake in the rat brain. Neuropsychopharmacology. 1993;8:337–344. doi: 10.1038/npp.1993.33. [DOI] [PubMed] [Google Scholar]
- YEH S.Y. N-tert-butyl-alpha-phenylnitrone protects against 3,4-methylenedioxymethamphetamine-induced depletion of serotonin in rats. Synapse. 1999;31:169–177. doi: 10.1002/(SICI)1098-2396(19990301)31:3<169::AID-SYN1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]