

Abstract
Granulocyte/macrophage colony-stimulating factor (GM-CSF) is a pro-inflammatory cytokine secreted by cells of the monocyte/macrophage lineage and has been implicated in the pathogenesis of bronchitis and asthma.
In the present study we have evaluated the effect of several cyclic AMP-elevating agents on lipopolysaccharide (LPS)-induced GM-CSF release from human monocytes and the extent to which the anti-inflammatory cytokine, interleukin (IL)-10, is involved.
LPS evoked a concentration-dependent generation of GM-CSF from human monocytes that was inhibited, at the mRNA and protein level, by 8-Br-cyclic AMP, cholera toxin, prostaglandin E2 (PGE2) and a number of structurally dissimilar phosphodiesterase (PDE) 4 inhibitors.
Pre-treatment of monocytes with a concentration of an anti-IL-10 monoclonal antibody that abolished the inhibitory action of a maximally effective concentration of exogenous human recombinant IL-10, significantly augmented LPS-induced GM-CSF generation. This effect was associated with a parallel upwards displacement of the concentration-response curves that described the inhibition of GM-CSF by PGE2, 8-Br-cyclic AMP and the PDE4 inhibitor, rolipram, without significantly changing the potency of any drug. Consequently, the maximum percentage inhibition of GM-CSF release was reduced. Further experiments established that the reduction in the maximum inhibition of GM-CSF release seen in anti-IL-10-treated cells was not due to functional antagonism as rolipram, PGE2 and 8-Br-cyclic AMP were equi-effective at all concentrations of LPS studied.
These data indicate that cyclic AMP-elevating drugs attenuate the elaboration of GM-CSF from LPS-stimulated human monocytes by a mechanism that is not mediated via IL-10. Suppression of GM-CSF from monocytes may explain, at least in part, the efficacy of PDE4 inhibitors in clinical trials of chronic obstructive pulmonary disease.
Keywords: Granulocyte/macrophage colony-stimulating factor; human monocytes; phosphodiesterase 4; cyclic adenosine-3′,5′-monophosphate, interleukin-10
Introduction
Granulocyte/macrophage colony-stimulating factor (GM-CSF) is a 14 – 35 kDa acidic glycoprotein that is secreted by many cells including those of the human monocyte/macrophage lineage (Blanchard et al., 1991; Meja et al., 2000; Sallerfors & Olofsson, 1992). Many studies, in vitro and in vivo, have shown that GM-CSF primes and activates a wide variety of inflammatory and immune cells and is implicated in the pathogenesis of a number of respiratory diseases including chronic obstructive pulmonary disease (COPD) and asthma. For example, increased expression of GM-CSF has been detected in the bronchoalveolar lavage (BAL) fluid of patients with asthma and in bronchitic subjects during exacerbations (Balbi et al., 1997). In addition, the number of GM-CSF+ T-lymphocytes, eosinophils and monocytes in the BAL fluid of asthmatic subjects after allergen provocation is increased when compared to normal individuals (Broide et al., 1991; 1992; Davies et al., 1995; Hallsworth et al., 1994; Sousa et al., 1993; Woolley et al., 1995), which is consistent with the elevated level of circulating GM-CSF in people with acute severe asthma (Brown et al., 1991). Furthermore, intrapulmonary transfer of the GM-CSF gene to rats leads to eosinophilia and monocytosis that is associated with irreversible fibrosis and airway remodelling (Xing et al., 1996). In mice, transfer of the same gene contributes to airways inflammation by prolonging interleukin (IL)-4- and IL-5-driven leukocyte infiltration (Lei et al., 1998).
The possible contribution of GM-CSF to the pathogenesis of airway inflammatory diseases provides a compelling rationale for targeting the generation and/or release of this cytokine with small molecule inhibitors. Novel compounds that show promise in the treatment of asthma and COPD include inhibitors of the cyclic AMP-specific, or type 4, family of cyclic nucleotide phosphodiesterases (PDE), some of which are in phase III clinical trials (Giembycz, 2000; 2001; Norman, 2000; Torphy, et al., 1999). However, although cyclic AMP-elevating agents generally suppress the production of pro-inflammatory cytokines there is good evidence that cytokine gene expression can also be up-regulated (Zidek, 1999). Indeed, the release of GM-CSF from human bone marrow stromal cells and T-lymphocytes can be attenuated and augmented under certain conditions (Borger et al., 1996; Bug et al., 1998; Kambayashi et al., 1995; Quill et al., 1989).
It has been proposed that the ability of PDE4 inhibitors and PGE2 to suppress the generation of pro-inflammatory cytokines (e.g. tumour necrosis factor-α [TNFα], IL-6) from murine macrophages requires the formation of IL-10 (Kambayashi et al., 1995; Strassmann et al., 1994). A similar conclusion was reached from in vivo studies of endotoxin shock with cell permeant cyclic AMP analogues (Arai et al., 1995), iloprost (Grundmann et al., 1992), pentoxifylline and a novel alkylxanthine PDE inhibitor, A802715 (Jilg et al., 1996). In the former investigation an anti-IL-10 neutralizing antibody inhibited the ability of dibutyryl cyclic AMP to protect against endotoxin-induced liver injury in mice rendered hyper-sensitive to LPS by the intravenous administration of Propionibacterium acnes. Collectively, these results led to a proposal that cyclic AMP augments IL-10 secretion in LPS-sensitive cells, which acts in an autocrine manner to suppress the elaboration of pro-inflammatory cytokines (Arai et al., 1995; Jilg et al., 1996; Kambayashi et al., 1995). These results are potentially important given that IL-10 is known to suppress certain Th1- and Th2-driven indices of inflammation (Pretolani & Goldman, 1997; Schreiber et al., 1995). Moreover, from a molecular perspective these data are consistent with the description of a cyclic AMP-response element (CRE) within the promoter region of the murine IL-10 gene (Kim et al., 1992), and the fact that cyclic AMP-elevating agents increase the number of IL-10 mRNA transcripts and augment IL-10 release (Kambayashi et al., 1995).
In the present study, we have evaluated the effect of a variety of cyclic AMP-elevating agents on LPS-induced GM-CSF release from human monocytes and, as the human IL-10 gene also features a putative CRE (Platzer et al., 1994; 1995), the extent to which IL-10 is involved.
Methods
Isolation and purification of human mononuclear cells
Blood was collected from normal healthy individuals by ante-cubital venepuncture into acid citrate dextrose (in mM): disodium citrate 160, glucose 110 – pH 7.4 and mixed with 6% Hespan (hydroxymethyl starch) to sediment erythrocytes. After standing at room temperature for 90 min, the leukocyte-rich plasma was removed and centrifuged at 312×g for 7 min. The resulting cell pellet was resuspended gently in approximately 7 ml of buffer A (in mM: KH2PO4 5, K2HPO4 5, NaCl 110 – pH 7.4) made 50% with Percoll and layered over a discontinuous Percoll density gradient (63% and 73%) in buffer A. Mononuclear cells were subsequently separated from polymorphonuclear cells by centrifugation at 1200×g for 25 min at 18°C. Using this procedure, mononuclear cells were recovered from the 50%/63% Percoll interface.
Mononuclear cells were washed three times in Ca2+/Mg2+-free Hanks' balanced salt solution (HBSS) to remove Percoll and finally suspended in Ca2+/Mg2+-free HBSS at a concentration of 106 ml−1. Cells (5×105) were added to 24-well culture plates (Greiner Labortecnik Ltd, Dursley, Gloucestershire) containing 500 μl Dutch-modified RPMI 1640 (RPMI 1640 supplemented with 10% foetal calf serum (FCS), 2 mM L-glutamine, 100 units ml−1 penicillin and 100 μg ml−1 streptomycin) and allowed to adhere to the plastic for 90 min at 37°C in a humidified incubator under an atmosphere of 5% CO2. The purity of the adherent cell population was routinely >94% monocytes. Plates were agitated, non-adherent cells decanted and the resulting monocytes were cultured for various times (see text and figure legends for details) in 1 ml supplemented Dutch-modified RMPI 1640 in the absence and presence of the drugs under investigation. The GM-CSF released into the culture supernatant was subsequently measured by an amplified sandwich ELISA.
Measurement of GM-CSF
Ninety-six well round-bottom plates were coated with 50 μl of a rat, anti-human GM-CSF monoclonal antibody diluted 1 : 250 in buffer B (in mM: NaHCO3 100, NaN3 15, pH 8.2) and left overnight at 4°C. Plates were subsequently washed in buffer B and immediately blocked with 200 μl FCS (10% in buffer B) for 2 h at room temperature. After an additional wash with buffer B, 100 μl GM-CSF standards, quality controls and unknown samples, in supplemented Dutch-modified RPMI 1640, were added to the plates and left for 18 h at 4°C. Plates were washed in buffer B, incubated for 45 min at room temperature with 100 μl of a biotinylated rat, anti-human monoclonal GM-CSF antibody diluted 1 : 500 in buffer B supplemented with 10% FCS, washed again, and then incubated for an additional 30 min at room temperature with 100 μl of avidin-peroxidase diluted 1 : 400 in buffer B (supplemented with 10% FCS). Plates were washed again and developed with 100 μl ABTS (2,2′-azino-bis-(3-ethylbenzothiazoline-6-sulphonic acid) substrate solution (0.55 mM ABTS; 0.1 M citric acid, pH 4.35; 0.1% (30%) H2O2). GM-CSF was measured colorimetrically at 405 nm and quantified by interpolation from a standard curve constructed to known concentrations of human recombinant GM-CSF. The detection limit of this assay is 16 pg ml−1.
A previous study has established that exposure of human monocytes to bacterial LPS evokes a time- and concentration-dependent elaboration of GM-CSF with a t1/2 and EC50 of 14.3 h and 390 pg ml−1 respectively (Meja et al., 2000). Unless stated otherwise, LPS was used in all experiments at 3 ng ml−1 (EC90) and GM-CSF was measured in the culture supernatant 18 h after the addition of LPS (see Meja et al., 2000 for further details).
Semi-quantitative RT-PCR
Total RNA was extracted from 4×106 adherent monocytes using the Qiagen RNeasy mini kit according to the manufacturer's instructions and up to 1 μg was treated with DNaseI to remove contaminating genomic DNA. Five hundred nanograms of RNA were reverse transcribed in a total volume of 20 μl in 50 mM Tris-HCl (pH 8.3; 25°C) containing 10 mM MgCl2, 500 μM spermidine, 10 mM DTT, 9 u AMV reverse transcriptase, 40 u RNase inhibitor, 0.5 μg random hexamers (Pharmacia, Uppsala, Sweden) and 1 mM deoxynucleotides. A RT-generated cDNA encoding the GM-CSF gene was amplified by PCR using specific primers designed from the reported primary sequences (Table 1) deposited with the GenBank data base. To confirm the integrity of RNA and equal loading of sample, RT-PCR analysis of the GAPDH gene was routinely performed using primers synthesized from the sequence described in Maier et al. (1990). PCR amplification (25 and 28 cycles for GAPDH and GM-CSF respectively) was conducted in a reaction volume of 25 μl containing 0.5 u Taq polymerase at a denaturing temperature of 94°C for 30 s, specific annealing temperature (Table 1) and an extension temperature of 72°C for 30 s. The cycle number, which achieved exponential amplification where product was proportional to starting cDNA, was determined empirically by performing PCR on an ‘average' cDNA sample by combining cDNA from all samples within one experiment. PCR products were subsequently size-fractionated on 2% agarose/TAE gels, stained with ethidium bromide and visualized under UV light. To confirm identity with the published cDNA-sequences, the GM-CSF and GAPDH amplification products were cloned in to pGEM5z®-vectors (Promega, Southampton, U.K.) and double-stranded sequencing was performed using the T7 Sequenase 2.0 system (Amersham, Buckinghamshire, U.K.).
Table 1.
Primers and conditions used in RT-PCR experiments

After agarose gel electrophoresis, Southern blotting and hybridization were performed to confirm the identity of PCR products and to check for possible genomic contamination. To quantify product formation, aliquots (5 μl) of the PCR-product were ‘dot-blotted' and hybridized with the appropriate radiolabelled cloned cDNA. After washing at high stringency the radioactivity associated with each ‘dot-blot' was determined by Cérenkov counting. GM-CSF transcripts are expressed as ratio to GAPDH and relative values are plotted as means±s.e.mean.
We have previously established that exposure of human monocytes to bacterial LPS (3 ng ml−1) evokes a time-dependent accumulation of GM-CSF mRNA transcripts with a t1/2 of 0.8 h (Meja et al., 2000). In all experiments LPS was used at 3 ng ml−1 and GM-CSF mRNA was measured 3 h after stimulation (see Meja et al., 2000 for further details).
Drugs and analytical reagents
Percoll was obtained from Pharmacia/LKB (Milton Keynes, Buckinghamshire, U.K.), and LPS (from Salmonella enteritidis), 8-Br-cyclic AMP, salbutamol, PGE2, avidin-peroxidase (code A3151), FCS, ABTS and HBSS were from the Sigma Chemical Company (Poole, Dorset, U.K.). GM-CSF standards (code RH-CSF-C) and quality controls (code 88/646) were obtained from Genzyme and the National Institute for Biological Standards and Controls respectively. Rat, anti-human GM-CSF and biotinylated rat, anti-human GM-CSF monoclonal antibodies (codes 118551D and 18526D respectively) were purchased from Pharmingen/Cambridge Bioscience (Cambridge, U.K.). Fluorescein-conjugated streptavidin (code F0422) and biotinylated swine, anti-rabbit IgG (code E0353) were from Dako (Cambridge, U.K.). Human recombinant IL-10 (code 217-IL-005), IL-10 standards (code 217-IL-005) and PCR primers for GM-CSF and GAPDH genes were obtained from R & D systems Europe Ltd. (Abingdon, Oxfordshire, U.K.). Alkaline phosphatase-labelled sheep, anti-rabbit IgG antibodies were purchased from Stratech Scientific Ltd (Luton, Bedfordshire, U.K.). Taq DNA-polymerase was from Bioline (Finchley, London, U.K.), L-glutamine and RPMI 1640 were from Gibco (Rickmansworth, Hertfordshire), and AMV reverse transcriptase, RNasin®-ribonuclease-inhibitor and deoxynucleotides were purchased from Promega (Southampton, Hampshire, U.K.). Multiprime® DNA-labelling system and nitrocellulose membranes were supplied by Amersham International (Little Chalfont, Buckinghamshire, U.K.). Rolipram, Ro 20 – 1724, benafentrine, piclamilast, and SK&F 95654 and denbufylline were provided by Schering (Berlin, Germany), Calbiochem (Nottingham, U.K.), Byk-Gulden (Konstanz, Germany), Rhone-Poulenc Rorer (Dagenham, Essex, U.K.) and GlaxoSmithKline (King of Prussia, U.S.A.) respectively. All other reagents were from BDH (Poole, Dorset, U.K.).
Data and statistical analyses
Data points, and values in the text and figure legends, represent the mean±s.e.mean of n independent determinations. Concentration-response curves were analysed by least-squares, non-linear iterative regression with the ‘PRISM' curve fitting program (GraphPad, San Diego, California, U.S.A.) and EC50/IC50 values were interpolated from curves of best-fit. When statistical evaluation was required, data were analysed by Student's t-test for paired data or by one-way ANOVA/Newman-Keuls multiple comparison test. The null hypothesis was rejected when P<0.05.
Results
Effect of rolipram and other isoenzyme-selective PDE inhibitors on LPS-induced GM-CSF release
Pre-treatment (20 min) of monocytes with the PDE4 inhibitor, rolipram (0.2, 2 and 20 μM) produced in a non-competitive inhibition of LPS (100 pg ml−1 to 1 μg ml−1)-induced GM-CSF generation with respect to vehicle-treated cells (Figure 1a). Thus, there was a progressive, concentration-related reduction in the maximum response without any change in the potency of LPS (EC50 values=0.28, 0.31, 0.17 and 0.5 ng ml−1 for vehicle and 0.2, 2 and 20 μM rolipram respectively). At a fixed concentration of LPS (3 ng ml−1), rolipram inhibited GM-CSF release in a concentration-dependent manner (EC50 ∼1.8 μM) reducing output by ∼80% at the maximally effective concentration (Figure 1b; Table 2).
Figure 1.
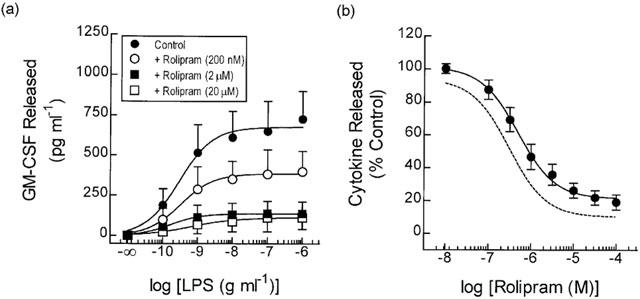
Effect of rolipram on LPS-induced GM-CSF release. Adherent monocytes were treated for 20 min with rolipram and then exposed to LPS. Cells were maintained at 37°C in a thermostatically-controlled incubator under a 5% CO2 atmosphere and the amount of GM-CSF released into the culture supernatant was measured at 18 h by ELISA. The effect of rolipram on the LPS concentration-response relationship and at a fixed submaximal concentration of LPS (3 ng ml−1) is shown in (a) and (b) respectively. The dotted line in (b) shows the position of the concentration-response curve that describes the inhibition of TNFα generation under identical experimental conditions (data taken from Seldon et al., 1995; 1998b). Each data point represents the mean±s.e.mean of nine independent determinations.
Table 2.
Effect of cyclic AMP PDE inhibitors and other drugs on LPS-induced GM-CSF and TNFα generation from human peripheral blood monocytes

Pre-treatment (20 min) of monocytes with the PDE inhibitors Ro 20 – 1724, denbufylline, piclamilast (all PDE4-selective) and benafentrine (PDE3/4-selective) inhibited LPS-induced GM-CSF release in a concentration-dependent manner with EC50 values of 41, 3.5, 0.009 and 22 μM respectively (Figure 2; Table 2). A selective inhibitor of PDE3 (SK&F 95654) did not affect LPS-induced GM-CSF generation at concentrations where isoenzyme-selectivity is preserved (data not shown).
Figure 2.

Effect of PDE4 inhibitors on LPS-induced GM-CSF release. Monocytes were treated with Ro 20-1724 (a), denbufylline (b), benafentrine (c) and piclamilast (d), exposed to LPS and the amount of GM-CSF released into the culture supernatant was measured by ELISA. The dotted line in each panel shows the position of the concentration-response curve that describes the inhibition of TNFα generation (data taken from Seldon et al., 1995; 1998b). Each data point represents the mean±s.e.mean of three to six independent determinations.
Effect of 8-Br-cyclic AMP and cyclic AMP-elevating drugs on LPS-induced GM-CSF release
Exposure (30 min) of human monocytes to 8-Br-cyclic AMP suppressed LPS-induced GM-CSF release in a concentration-dependent manner with an IC50 of 271 μM (Figure 3a; Table 2). Maximum inhibition was achieved at 10 mM 8-Br-cyclic AMP, a concentration that inhibited cytokine generation by >90%. Similarly, incubation (2 h) of monocytes with cholera toxin (CTX) prior to the addition of LPS reduced the release of GM-CSF by 54% at the maximally effective concentration (Figure 3b).
Figure 3.
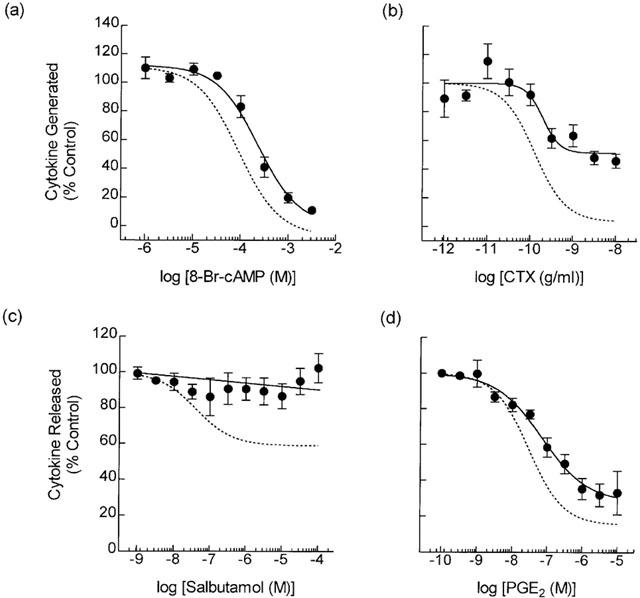
Effect of other cAMP-elevating drugs on LPS-induced GM-CSF release. Monocytes were treated with 8-Br-cyclic AMP (30 min; panel a), CTX (2 h; b), salbutamol (5 min; c), or PGE2 (5 min; d) exposed to LPS and the amount of GM-CSF released into the culture supernatant was measured by ELISA. The dotted line in each panel shows the position of the concentration-response curve that describes the inhibition of TNFα generation (data taken from Seldon et al., 1995; 1998b). Each data point represents the mean±s.e.mean of three to five independent determinations.
Figures 3c,d show the effect of salbutamol and PGE2 on LPS-induced GM-CSF generation. Contrary to its inhibitory effect on TNFα secretion (Figure 3c – dotted line; Seldon et al., 1995) salbutamol (5 min pre-treatment) did not affect the elaboration of GM-CSF at any concentration examined. In contrast PGE2, which acts through an inhibitory EP2-like receptor on monocytes (Meja et al., 1997), reduced GM-CSF output in a concentration-dependent manner with an IC50 of 307 nM (Figure 3d).
Reduced sensitivity of LPS-induced GM-CSF release to cyclic AMP
A consistent finding of these studies was that the LPS-driven release of GM-CSF was significantly less sensitive (four- to 13-times) to all of the PDE inhibitors studied when compared to the elaboration of TNFα (dotted lines) under identical experimental conditions (Figures 1b, 2) (Seldon et al., 1995). Moreover, GM-CSF release was never abolished (cf. TNFα). Indeed, a maximally effective concentration of piclamilast (100 nM), denbufylline (100 μM) and benafentrine (100 μM) suppressed GM-CSF by only ∼50, 70 and 80% respectively (Figure 2d). CTX, PGE2 and 8-Br-cyclic AMP were also less potent and less effective at inhibiting the release of GM-CSF than of TNFα, while salbutamol was inactive (Figure 3, dotted line; see Table 2).
Effect of rolipram, 8-Br-cyclic AMP, salbutamol and PGE2 on GM-CSF mRNA expression
In unstimulated adherent monocytes GM-CSF mRNA transcripts were present in a very low copy number as assessed by RT-PCR (28 cycles of amplification). However, 3 h after stimulation of monocytes with LPS (3 ng ml−1) GM-CSF mRNA expression was increased 51 fold relative to the ‘house-keeping' gene GAPDH (Figure 4). Pre-treatment of monocytes with PGE2 (1 μM; 5 min), 8-Br-cyclic AMP (1 mM; 30 min) and rolipram (10 μM; 20 min) significantly attenuated this response by 44, 58 and 57% respectively (Figure 4), whereas salbutamol (1 μM; 5 min) was without effect on GM-CSF mRNA expression (Figure 4).
Figure 4.

Effect of cyclic AMP-elevating drugs on LPS-induced GM-CSF mRNA expression. Adherent monocytes were pre-treated with salbutamol (5 min; 1 μM), PGE2 (5 min; 1 μM), 8-Br-cyclic AMP (30 min; 1 mM), rolipram (20 min; 10 μM) or vehicle and exposed to LPS (3 ng ml−1). After 3 h, RNA was extracted and 0.5 μg was reversed transcribed to generate cDNAs for GM-CSF and GAPDH using the primer pairs shown in Table 1. PCR was performed with reverse transcribed cDNA, the products subjected to electrophoresis on 1.5% agarose gels and DNA subsequently visualized after staining with ethidium bromide. PCR products were quantified by Southern blotting and standardized against GAPDH. RT-PCR product sizes for GM-CSF and GAPDH were 500 bp (28 cycles) and 598 bp (24 cycles) respectively. (a) and (b) show the mean data of four independent experiments and a representative gel (prior to Southern hybridization) respectively. 1, Control; 2, LPS+Rolipram; 3, LPS+8-Br-cyclic AMP; 4, LPS; 5, Control; 6, LPS+Salbutamol; 7, LPS+PGE2; 8, LPS. *P<0.05, significant inhibition of LPS-induced GM-CSF mRNA expression.
Effect of IL-10 on LPS-induced GM-CSF generation and reversal by an anti-IL-10 antibody
Pre-treatment of monocytes with human recombinant (hr) IL-10 suppressed LPS-induced GM-CSF release in a concentration-dependent manner (IC50=71.2±19.1 pg ml−1) with complete inhibition achieved between 3 – 10 ng ml−1 (Figure 5). The introduction of an anti-IL-10 neutralizing antibody to monocyte cultures attenuated the inhibitory effect of hrIL-10 (1 ng ml−1) in a concentration-dependent manner (EC50=9.5±0.9 ng ml−1) with complete reversal at 0.1 – 1 μg ml−1 (Figure 5). An isotype-matched control antibody (rat IgG1k) failed to block the inhibitory effect of hrIL-10 (data not shown).
Figure 5.
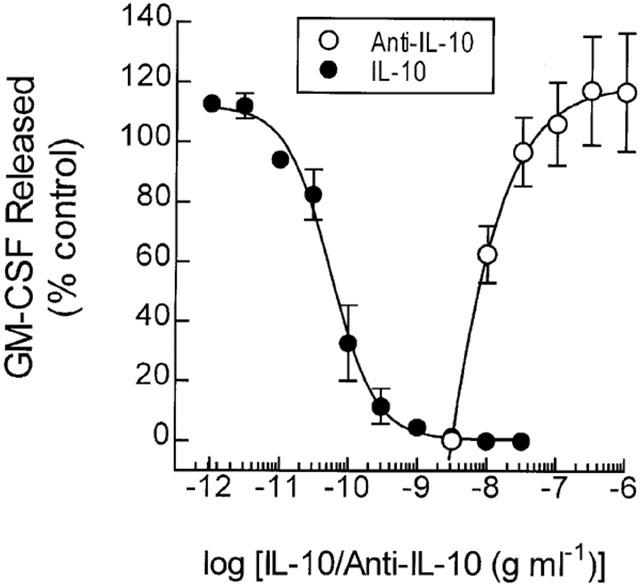
Inhibition of LPS-induced GM-CSF release by hrIL-10 and reversal by anti-IL-10. Adherent monocytes were pre-treated (20 min) with hrIL-10, or hrIL-10 in the presence of anti-IL-10 before being exposed to LPS (3 ng ml−1). The amount of GM-CSF released in to the culture supernatant was then measured by ELISA at 18 h. Each data point represents the mean±s.e.mean of four independent determinations.
Effect of an anti-IL-10 antibody on LPS-induced GM-CSF generation
Pre-treatment (20 min) of monocytes with an anti-IL-10 antibody at 3, 30 and 300 ng ml−1 had no effect on the potency of LPS for GM-CSF generation (EC50's=0.24±0.07, 0.27±0.07 and 0.33±0.1 ng ml−1 respectively) when compared to untreated cells (EC50=0.23±0.07; Figure 6). However, the anti-IL-10 antibody, but not rat IgG1k, significantly augmented the maximum amount of GM-CSF released confirming previous reports that IL-10 is generated in response to LPS (Seldon et al., 1998b) and acts in a negative autocrine factor to temper the production of GM-CSF. This effect was concentration-related such that LPS (100 ng ml−1)-induced GM-CSF release was augmented 1.4 fold from 547±89 to 769±117 pg ml−1 (P<0.05) in the presence of 300 ng ml−1 of the anti-IL-10 antibody (Figure 6). The potentiation of GM-CSF release by the anti-IL-10 antibody varied markedly (14 – 195%) between subjects (mean=69±22%, n=18), which presumably reflects the significant variation in monocyte IL-10 production between donors (Platzer et al., 1995; Seldon et al., 1998b).
Figure 6.
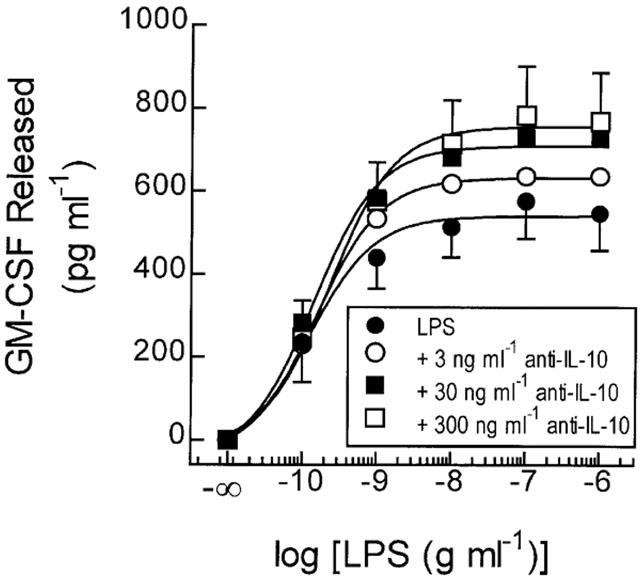
Effect of an anti-IL-10 antibody on LPS-induced GM-CSF release. Adherent monocytes were pre-treated (20 min) with anti-IL-10 (as indicated) before being exposed to LPS. The amount of GM-CSF released in to the culture supernatant was then measured by ELISA at 18 h. Each data point represents the mean±s.e.mean of four independent determinations.
Inhibition of LPS-induced GM-CSF generation by rolipram, PGE2 and 8-Br-cyclic AMP: effect of an anti-IL-10 antibody
Pre-treatment of monocytes with rolipram, PGE2 and 8-Br-cyclic AMP inhibited LPS-induced GM-CSF release in a concentration-dependent manner (Figure 7). In the presence of a concentration (100 ng ml−1) of the anti-IL-10 antibody that abolished the inhibitory action of a maximally effective concentration of exogenous hrIL-10, there was an upwards shift of the concentration-response curves that described the inhibition of GM-CSF by each drug without a significant change in their potency. As a consequence, the maximum percentage inhibition of GM-CSF release was significantly attenuated (Figure 7; Table 3). Since the anti-IL-10 antibody significantly augmented LPS-induced GM-CSF release it was reasoned that the reduced ability of cyclic AMP to antagonize inhibitory effect of LPS might be due to functional antagonism. To address this possibility, experiments were conducted at different concentrations of LPS in the absence and presence of anti-IL-10 (100 ng ml−1). As shown in Table 4, the maximum inhibition of GM-CSF release effected by rolipram, PGE2 and 8-Br-cyclic AMP was identical at all concentrations of LPS studied.
Figure 7.

Effect of an anti-IL-10 antibody on the inhibitory effect of cyclic AMP-elevating drugs on LPS-induced GM-CSF release. Monocytes were treated with rolipram (a), PGE2 (b), 8-Br-cyclic AMP (c) and salbutamol (d) in the absence and presence of anti-10 (100 ng ml−1), exposed to LPS (3 ng ml−1) and the amount of GM-CSF released into the culture supernatant was measured by ELISA. Each data point represents the mean±s.e.mean of three to six independent determinations.
Table 3.
Effect of an anti-IL-10 antibody on the inhibition of LPS-induced GM-CSF generation by rolipram, 8-Br-cyclic AMP and PGE2

Table 4.
Effect of an anti-IL-10 antibody on the inhibitory effect of rolipram, 8-Br-cyclic AMP and PGE2 on GM-CSF release at different concentrations of LPS

Discussion
In the present study we have confirmed previous investigations that human peripheral blood monocytes release the haematopoietic pro-inflammatory cytokine, GM-CSF in response to bacterial LPS (Blanchard et al., 1991; de Waal Malefyt et al., 1991; Meja et al., 2000). In addition, we have extended this finding by providing evidence that cyclic AMP-elevating drugs act at both the transcriptional and post-transcriptional level to suppress GM-CSF output by a mechanism that is not mediated by IL-10.
Effect of cyclic AMP-elevating drugs on LPS-induced GM-CSF mRNA expression and protein release
Pre-treatment of monocytes with the PDE inhibitors rolipram, Ro 20-1724, denbufylline, piclamilast and benafentrine inhibited LPS-induced GM-CSF release with a rank order of potency similar to that reportedly previously for the inhibition of PDE4 in monocyte lysates (Seldon et al., 1995). These results are consistent with PDE4 being the major cyclic AMP hydrolyzing activity in monocytes (Gantner et al., 1997; Seldon et al., 1995) and the ability of PDE4 inhibitors to attenuate GM-CSF release from human bone marrow stromal cells and T-lymphocytes (Borger et al., 1996; Bug et al., 1998). In contrast, a selective inhibitor of PDE3 (SK&F 95654) did not affect LPS-induced GM-CSF generation at concentrations where isoenzyme-selectivity is preserved. Although appreciable PDE3 is present in the particulate fraction of human monocytes (Gantner et al., 1997), these data show that inhibition of this enzyme family does not regulate the release of GM-CSF, which is in agreement with the inability of PDE3 inhibitors to suppress the elaboration of TNFα and IL-2 from monocytes (Seldon et al., 1995) and mitogen-stimulated T-lymphocytes respectively (Giembycz et al., 1996).
Further studies were performed to assess whether agonists (PGE2 and salbutamol) that can act via Gs-coupled receptors could also suppress GM-CSF generation. Unexpectedly, salbutamol did not affect the elaboration of GM-CSF at any concentration examined under conditions where PGE2, which acts through an inhibitory ‘EP2-like' receptor on monocytes (Meja et al., 1997), reduced GM-CSF output. The reason for this discrepancy is unclear given that other cyclic AMP-elevating drugs were active in this system. As LPS-induced TNFα release is suppressed by salbutamol an absence of functional β2-adrenoceptors cannot account for this difference (Seldon et al., 1995; 1998a). One possibility is that the EP2-like receptor is coupled more efficiently to Gs/adenylyl cyclase on human monocytes than is the β2-adrenoceptor. Equally, the density of EP2-like receptors might be significantly greater than the number of β2-adrenoceptors such that the cyclic AMP signal generated by salbutamol is relatively modest when compared to that effected by PGE2. Both these ideas are supported by the finding that PGE2 activates cyclic AMP-dependent protein kinase (PKA) to a significantly greater extent in human monocytes than salbutamol under identical experimental conditions (Seldon et al., 1995). Similarly, PGE2-induced cyclic AMP accumulation and PKA activation are markedly potentiated by the PDE4 inhibitor, rolipram, when compared to the same responses elicited by salbutamol (Seldon et al., 1995). Taken together, these data suggest that the extent to which cyclic AMP is elevated and PKA activated might be primary determinants for inhibition of LPS-induced GM-CSF release from human monocytes.
A consistent finding of these studies was that the LPS-driven release of GM-CSF was significantly less sensitive (four- to 13-times) to all of the PDE inhibitors studied and the inhibition produced less when compared to the elaboration of TNFα (Seldon et al., 1995). Indeed, at maximally effective concentrations piclamilast, denbufylline and benafentrine suppressed the elaboration of GM-CSF by only ∼50, 70 and 80% respectively. This reduction in potency and activity held for other cyclic AMP-elevating drugs including PGE2, CTX, and 8-Br-cyclic AMP suggesting that LPS regulates the expression of GM-CSF and TNFα by distinct mechanisms that display different sensitivities to cyclic AMP.
We have previously reported that LPS evokes a time-dependent and transient accumulation of GM-CSF mRNA transcripts in human monocytes that is abolished by actinomycin D and cycloheximide indicative of de novo transcription and translation of the GM-CSF gene (Meja et al., 2000). In unstimulated adherent monocytes GM-CSF mRNA is barely detectable but increases more than 50 fold after 3 h exposure of cells to LPS. In the present manuscript we have shown that pre-treatment of monocytes with PGE2, 8-Br-cyclic AMP and rolipram prior to LPS significantly attenuated GM-CSF mRNA expression at 3 h by 40 to 60%. Other investigators have established that cyclic AMP-elevating drugs attenuate GM-CSF release in T-lymphocytes by reducing both the transcription rate of the GM-CSF gene and GM-CSF mRNA stability (Borger et al., 1996). Although either of these mechanism could equally account for the reduction in GM-CSF output from human monocytes, the finding that significant GM-CSF mRNA was present in 8-Br-cyclic AMP-, PGE2- and rolipram-treated cells under conditions where GM-CSF release was suppressed by 80 to 90% indicates that cyclic AMP also impairs translation of GM-CSF mRNA to protein.
Pre-treatment of monocytes with salbutamol had no effect on GM-CSF mRNA expression despite clear activation of the cyclic AMP/PKA cascade in these cells (Seldon et al., 1995). This result is consistent with the lack of effect of salbutamol on GM-CSF output and endorses the contention that the magnitude of the cyclic AMP/PKA response may be critical in determining whether cytokine expression is repressed.
Role of IL-10 in GM-CSF expression
As described in the introduction there is evidence from both in vitro and in vivo studies that the generation of IL-10 by LPS-sensitive cells is enhanced by cyclic AMP, which acts in an autocrine manner to inhibit pro-inflammatory cytokine production (Arai et al., 1995; Jilg et al., 1996; Kambayashi et al., 1995; Strassmann et al., 1994). To establish whether such a mechanism could account for the inhibitory effect of cyclic AMP on GM-CSF production in human monocytes the effect of neutralising IL-10 was studied. As shown in Figure 7 an anti-IL-10 antibody, at a concentration that abolished the inhibitory action of a maximally effective concentration of exogenous hrIL-10, produced a parallel upwards shift of the concentration-response curves that described the inhibition of GM-CSF release by rolipram, PGE2 and 8-Br-cyclic AMP without changing their potency. Although the maximum percentage inhibition of GM-CSF release was reduced this was not due to inhibition of the effect of rolipram, PGE2 and 8-Br-cyclic AMP but merely a reflection of the higher starting baseline. Curiously, these results are contrary to comparable studies performed with murine macrophages (Kambayashi et al., 1995; Strassmann et al., 1994), Kuppfer cells (Arai et al., 1995) and in vivo murine models of endotoxin shock (Arai et al., 1995; Jilg et al., 1996). In each of these experimental settings an anti-IL-10 antibody was reported to abolish or markedly attenuate the protection of LPS-evoked TNFα and IL-6 production, liver injury and lethality afforded by agents that elevate cyclic AMP.
Currently, it is unclear why anti-IL-10 reverses the inhibitory effect of cyclic AMP-elevating drugs on cytokine production from LPS-sensitive cells of murine origin but not from human monocytes. We have previously reported that this disparity is not due to a failure of monocytes to release IL-10 or the ability of cyclic AMP-elevating agents to augment this response (Seldon et al., 1998). Dissimilar kinetics of IL-10 versus GM-CSF release also cannot explain this difference (Meja et al., 2000; Seldon et al., 1998b). However, the possibility that transcriptional/post-transcriptional control of certain pro-inflammatory cytokine genes varies between humans and mice, and/or monocytes and macrophages is worthy of consideration and is currently being assessed in the authors' laboratory.
The upwards shift of the concentration-response curves that described the suppression of LPS-induced GM-CSF generation by 8-Br-cyclic AMP, PGE2 and rolipram in anti-IL-10-treated monocytes was parallel such that the maximum response produce by these drugs was significantly reduced (Table 3). It was reasoned that one explanation for the reduced effect was functional antagonism and this possibility was addressed by performing experiments at different concentrations of LPS in the absence and presence of a neutralizing anti-IL-10 antibody. However, as shown in Table 4 the maximum inhibition of GM-CSF release effected by rolipram, PGE2 and 8-Br-cyclic AMP was identical at all concentrations of LPS studied indicating that in anti-IL-10-treated monocytes only about 50% of the GM-CSF released by LPS is sensitive to inhibition by cyclic AMP.
In conclusion, the results of the present study demonstrate unequivocally that cyclic AMP-elevating drugs attenuate the elaboration of GM-CSF from LPS-stimulated human monocytes by a mechanism that is not mediated via the inhibitory cytokine, IL-10. As GM-CSF primes eosinophils and neutrophils and enhances their survival, suppression of this cytokine from monocytes/macrophages in the airways could explain, at least in part, the reported efficacy of PDE4 inhibitors clinical trials of COPD (Giembycz, 2001; Torphy et al., 1999).
Acknowledgments
The authors thank the National Asthma Campaign (U.K.) and GlaxoSmithKline for financial support.
Abbreviations
- BAL
bronchoalveolar lavage
- 8-Br-cyclic AMP
8-bromo-cyclic adenosine-3′,5′-monophosphate
- cyclic AMP
cyclic adenosine-3′,5′-monophosphate
- CTX
cholera toxin
- FCS
foetal calf serum
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GM-CSF
granulocyte/macrophage colony-stimulating factor
- HBSS
Hanks' balanced salt solution
- IL
interleukin
- LPS
lipopolysaccharide
- PDE
phosphodiesterase
- PGE2
prostaglandin E2
- TNFα
tumour necrosis factor-α
References
- ARAI T., HIROMATSU K., KOBAYASHI N., TAKANO M., ISHIDA H., NIMURA Y., YOSHIKAI Y. IL-10 is involved in the protective effect of dibutyryl cyclic adenosine monophosphate on endotoxin-induced inflammatory liver injury. J. Immunol. 1995;155:5743–5749. [PubMed] [Google Scholar]
- BALBI B., BASON C., BALLEARI E., FIASELLA F., PESCI A., GHIO R., FABIANO F. Increased bronchoalveolar granulocytes and granulocyte/macrophage colony-stimulating factor during exacerbations of chronic bronchitis. Eur. Respir. J. 1997;10:846–850. [PubMed] [Google Scholar]
- BLANCHARD D.K., MICHELINI-NORRIS M.B., PEARSON C.A., MCMILLEN S., DJEU J.Y. Production of granulocyte-macrophage colony-stimulating factor (GM-CSF) by monocytes and large granular lymphocytes stimulated with Mycobacterium avium - M. intracellulare: activation of bactericidal activity by GM-CSF. Infect. Immun. 1991;59:2396–2402. doi: 10.1128/iai.59.7.2396-2402.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BORGER P., KAUFFMAN H.F., VIJGEN J.L., POSTMA D.S., VALLENGA E. Activation of the cAMP-dependent signalling pathway downregulates the expression of interleukin-3 and granulocyte/macrophage colony-stimulating factor in activated human T-lymphocytes. Exp. Hematol. 1996;24:108–115. [PubMed] [Google Scholar]
- BROIDE D.H., GLEICH G.J., CUOMO A.J., COBURN D.A., FEDERMAN E.C., SCHWARTZ L.B., WASSERMAN S.I. Evidence of ongoing mast cell and eosinophil degranulation in symptomatic asthma airway. J. Allergy Clin. Immunol. 1991;88:637–648. doi: 10.1016/0091-6749(91)90158-k. [DOI] [PubMed] [Google Scholar]
- BROIDE D.H., PAINE M.M., FIRESTEIN G.S. Eosinophils express interleukin 5 and granulocyte macrophage-colony-stimulating factor mRNA at sites of allergic inflammation in asthmatics. J. Clin. Invest. 1992;90:1414–1424. doi: 10.1172/JCI116008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BROWN P.H., CROMPTON G.K., GREENING A.P. Proinflammatory cytokines in acute asthma. Lancet. 1991;338:590–593. doi: 10.1016/0140-6736(91)90605-o. [DOI] [PubMed] [Google Scholar]
- BUG G., AMAN J., HUBER C., PESCHEL C., DERIGS H.G. cAMP analogues downregulate the expression of granulocyte/macrophage colony-stimulating factor (GM-CSF) in human bone marrow stromal cells in vitro. Mediators Inflamm. 1998;7:195–199. doi: 10.1080/09629359891135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVIES R.J., WANG J.H., TRIGG C.J., DEVALIA J.L. Expression of granulocyte/macrophage-colony-stimulating factor, interleukin-8 and RANTES in the bronchial epithelium of mild asthmatics is down-regulated by inhaled beclomethasone dipropionate. Int. Arch. Allergy Immunol. 1995;107:428–429. doi: 10.1159/000237068. [DOI] [PubMed] [Google Scholar]
- DE WAAL MALEFYT R., ABRAMS J., BENNETT B., FIGDOR C.G., DE VRIES J.E. Interleukin 10 (IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J. Exp. Med. 1991;174:1209–1220. doi: 10.1084/jem.174.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GANTNER F., KUPFERSCHMIDT R., SCHUDT C., WENDEL A., HATZELMANN A. In vitro differentiation of human monocytes to macrophages: change of PDE profile and its relationship to suppression of tumour necrosis factor-alpha release by PDE inhibitors. Br. J. Pharmacol. 1997;121:221–231. doi: 10.1038/sj.bjp.0701124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GIEMBYCZ M.A. Phosphodiesterase 4 inhibitors and the treatment of asthma: where are we now and where do we go from here. Drugs. 2000;59:193–212. doi: 10.2165/00003495-200059020-00004. [DOI] [PubMed] [Google Scholar]
- GIEMBYCZ M.A. Cilomilast: a selective phosphodiesterase 4 inhibitor for the treatment of asthma and chronic obstructive pulmonary disease. Exp. Opin. Invest. Drugs. 2001;10:1361–1379. doi: 10.1517/13543784.10.7.1361. [DOI] [PubMed] [Google Scholar]
- GIEMBYCZ M.A., CORRIGAN C.J., SEYBOLD J., NEWTON R., BARNES P.J. Identification of cyclic AMP phosphodiesterases 3, 4 and 7 in human CD4+ and CD8+ T-lymphocytes: role in regulating proliferation and the biosynthesis of interleukin-2. Br. J. Pharmacol. 1996;118:1945–1958. doi: 10.1111/j.1476-5381.1996.tb15629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRUNDMANN H.J., HAHNLE U., HEGENSCHEID B., SAHLMULLER G., BIENZLE U., BLITSTEIN WILLINGER E. Inhibition of endotoxin-induced macrophage tumor necrosis factor expression by a prostacyclin analogue and its beneficial effect in experimental lipopolysaccharide intoxication. J. Infect. Dis. 1992;165:501–505. doi: 10.1093/infdis/165.3.501. [DOI] [PubMed] [Google Scholar]
- HALLSWORTH M.P., SOH C.P., LANE S.J., ARM J.P., LEE T.H. Selective enhancement of GM-CSF, TNFα, IL-1β and IL-8 production by monocytes and macrophages of asthmatic subjects. Eur. Respir. J. 1994;7:1096–1102. [PubMed] [Google Scholar]
- JILG S., BARSIG J., LEIST M., KUSTERS S., VOLK H.D., WENDEL A. Enhanced release of interleukin-10 and soluble tumor necrosis factor receptors as novel principles of methylxanthine action in murine models of endotoxic shock. J. Pharmacol. Exp. Ther. 1996;278:421–431. [PubMed] [Google Scholar]
- KAMBAYASHI T., JACOB C.O., ZHOU D., MAZUREK N., FONG M., STRASSMANN G. Cyclic nucleotide phosphodiesterase type IV participates in the regulation of IL-10 and in the subsequent inhibition of TNFα and IL-6 release by endotoxin-stimulated macrophages. J. Immunol. 1995;155:4909–4916. [PubMed] [Google Scholar]
- KIM J.M., BRANNAN C.I., COPELAND N.G., JENKINS N.A., KHAN T.A., MOORE K.W. Structure of the mouse IL-10 gene and chromosomal localization of the mouse and human genes. J. Immunol. 1992;148:3618–3623. [PubMed] [Google Scholar]
- LEI X.F., OHKAWARA Y., STAMPFLI M.R., GAULDIE J., CROITORU K., JORDANA M., XING Z. Compartmentalized transgene expression of granulocyte-macrophage colony- stimulating factor (GM-CSF) in mouse lung enhances allergic airways inflammation. Clin. Exp. Immunol. 1998;113:157–165. doi: 10.1046/j.1365-2249.1998.00652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAIER J.A., HLA T., MCCAIG T. Cyclo-oxygenase is an immediate early gene induced by interleukin-1 in human endothelial cells. J. Biol. Chem. 1990;265:10805–10808. [PubMed] [Google Scholar]
- MEJA K.K., BARNES P.J., GIEMBYCZ M.A. Characterization of the prostanoid receptors on human blood monocytes at which prostaglandin E2 inhibits lipopolysacchide-induced tumour necrosis factor-α generation. Br. J. Pharmacol. 1997;122:147–157. doi: 10.1038/sj.bjp.0701360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEJA K.K., SELDON P.M., NASUHARA Y., ITO K., BARNES P.J., LINDSAY M.A., GIEMBYCZ M.A. p38 MAP kinase and MKK-1 co-operate in the generation of GM-CSF from LPS-stimulated human monocytes by an NFκB-independent mechanism. Br. J. Pharmacol. 2000;131:1143–1153. doi: 10.1038/sj.bjp.0703684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NORMAN P. PDE Inhibitors: sustained patenting activity as leading drugs near the market. Exp. Opin. Ther. Patents. 2000;10:1415–1427. [Google Scholar]
- PLATZER C., MEISEL C., VOGT K., PLATZER M., VOLK H.D. Up-regulation of monocytic IL-10 by tumor necrosis factor-α and cAMP-elevating drugs. Int. Immunol. 1995;7:517–523. doi: 10.1093/intimm/7.4.517. [DOI] [PubMed] [Google Scholar]
- PLATZER C., VOLK H.D., PLATZER M. 5′ Noncoding sequence of human IL-10 gene obtained by oligo-cassette PCR walking. DNA Seq. 1994;4:399–401. doi: 10.3109/10425179409010188. [DOI] [PubMed] [Google Scholar]
- PRETOLANI M., GOLDMAN M. IL-10: a potential therapy for allergic inflammation. Immunol. Today. 1997;18:277–280. doi: 10.1016/s0167-5699(97)80023-0. [DOI] [PubMed] [Google Scholar]
- QUILL H., GAUR A., PHIPPS R.P. Prostaglandin E2-dependent induction of granulocyte/macrophage colony-stimulating factor secretion by cloned murine helper T-cells. J. Immunol. 1989;142:813–818. [PubMed] [Google Scholar]
- SALLERFORS B., OLOFSSON T. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and granulocyte colony-stimulating factor (G-CSF) secretion by adherent monocytes measured by quantitative immunoassays. Eur. J. Haematol. 1992;49:199–207. doi: 10.1111/j.1600-0609.1992.tb00047.x. [DOI] [PubMed] [Google Scholar]
- SCHREIBER S., HEINIG T., THIELE H.G., RAEDLER A. Immunoregulatory role of interleukin 10 in patients with inflammatory bowel disease. Gastroenterology. 1995;108:1434–1444. doi: 10.1016/0016-5085(95)90692-4. [DOI] [PubMed] [Google Scholar]
- SELDON P.M., BARNES P.J., GIEMBYCZ M.A. Interleukin-10 does not mediate the inhibitory effect of PDE4 inhibitors and other cAMP-elevating drugs on lipopolysaccharide-induced tumor necrosis factor-α generation from human peripheral blood monocytes. Cell Biochem. Biophys. 1998b;29:179–201. doi: 10.1007/BF02737835. [DOI] [PubMed] [Google Scholar]
- SELDON P.M., BARNES P.J., MEJA K., GIEMBYCZ M.A. Suppression of lipopolysaccharide-induced tumor necrosis factor-α generation from human peripheral blood monocytes by inhibitors of phosphodiesterase 4: interaction with stimulants of adenylyl cyclase. Mol. Pharmacol. 1995;48:747–757. [PubMed] [Google Scholar]
- SELDON P.M., STEVENS D.A., ADCOCK I.M., O'CONNOR B.J., BARNES P.J., GIEMBYCZ M.A. Albuterol does not antagonize the inhibitory effect of dexamethasone on monocyte cytokine release. Am. J. Respir. Crit. Care. Med. 1998a;157:803–809. doi: 10.1164/ajrccm.157.3.9707116. [DOI] [PubMed] [Google Scholar]
- SOUSA A.R., POSTON R.N., LANE S.J., NAKHOSTEEN J.A., LEE T.H. Detection of GM-CSF in asthmatic bronchial epithelium and decrease by inhaled corticosteroids. Am. Rev. Respir. Dis. 1993;147:1557–1561. doi: 10.1164/ajrccm/147.6_Pt_1.1557. [DOI] [PubMed] [Google Scholar]
- STRASSMANN G., PATIL KOOTA V., FINKELMAN F., FONG M., KAMBAYASHI T. Evidence for the involvement of interleukin-10 in the differential deactivation of murine peritoneal macrophages by prostaglandin E2. J. Exp. Med. 1994;180:2365–2370. doi: 10.1084/jem.180.6.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TORPHY T.J., BARNETTE M.S., UNDERWOOD D.C., GRISWOLD D.E., CHRISTENSEN S.B., MURDOCH R.D., NIEMAN R.B., COMPTON C.H. Ariflo™ (SB 207499), a second generation phosphodiesterase 4 inhibitor for the treatment of asthma and COPD: from concept to clinic. Pulm. Pharmacol. Ther. 1999;12:131–135. doi: 10.1006/pupt.1999.0181. [DOI] [PubMed] [Google Scholar]
- WOOLLEY K.L., ADELROTH E., WOOLLEY M.J., ELLIS R., JORDANA M., O'BYRNE P.M. Effects of allergen challenge on eosinophils, eosinophil cationic protein, and granulocyte-macrophage colony-stimulating factor in mild asthma. Am. J. Respir. Crit. Care Med. 1995;151:1915–1924. doi: 10.1164/ajrccm.151.6.7767540. [DOI] [PubMed] [Google Scholar]
- XING Z., OHKAWARA Y., JORDANA M., GRAHAM F., GAULDIE J. Transfer of granulocyte-macrophage colony-stimulating factor gene to rat lung induces eosinophilia, monocytosis, and fibrotic reactions. J. Clin. Invest. 1996;97:1102–1110. doi: 10.1172/JCI118503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZIDEK Z. Adenosine - cyclic AMP pathways and cytokine expression. Eur. Cyt. Net. 1999;10:319–328. [PubMed] [Google Scholar]