

Abstract
To elucidate the mechanism of the relaxation mediated by endothelium-derived hyperpolarizing factors (EDHFs), the effect of brefeldin A, a membrane transport blocker, on cytosolic Ca2+ concentration ([Ca2+]i) and tension was determined in the porcine coronary arterial strips. We also examined the effect of brefeldin A on [Ca2+]i in the endothelial cells of the porcine aortic valve.
In the presence of 10 μM indomethacin and 30 μM NG-nitro-L-arginine (L-NOARG), both bradykinin and substance P induced a transient decrease in [Ca2+]i and tension in arterial strips contracted with 100 nM U46619 (thromboxane A2 analogue). A 6 h pre-treatment with 20 μg ml−1 brefeldin A abolished the bradykinin-induced relaxation, while it had no effect on the substance P-induced relaxation.
In the absence of indomethacin and L-NOARG, brefeldin A had no effect on the bradykinin-induced relaxation during the contraction induced by U46619 or 118 mM K+.
The indomethacin/L-NOARG-resistant relaxation induced by bradykinin was completely inhibited by 3 mM tetrabutylammonium (non-specific Ca2+-activated K+ channel blocker), while that induced by substance P was not inhibited by 3 mM tetrabutylammonium or 1 mM 4-aminopyridine (voltage-dependent K+ channels blocker) alone, but completely inhibited by their combination.
Brefeldin A had no effect on the [Ca2+]i elevation in endothelial cells induced by bradykinin or substance P.
In conclusion, bradykinin produce EDHF in a brefeldin A-sensitive mechanism in the porcine coronary artery. However, this mechanism is not active in a substance P-induced production of EDHF, which thus suggests EDHF to be more than a single entity.
Keywords: Brefeldin A, endothelium-derived hyperpolarizing factor, bradykinin, substance P
Introduction
It is widely accepted that the endothelium-dependent vasorelaxation is mediated by at least three factors; prostaglandin I2 (PGI2), nitric oxide (NO) and endothelium-derived hyperpolarizing factor (EDHF). EDHF is suggested to be involved in the NO/PGI2-independent component of the endothelium-dependent relaxation (Vanhoutte & Shimokawa, 1989). However, it is still controversial as to whether the endothelium-dependent hyperpolarization is mediated by a soluble factor, i.e., EDHF or by direct electrical transmission between endothelial cells and smooth muscle cells (Kuhberger et al., 1994). Recent studies have suggested that the cytochrome P450-derived arachidonic acid metabolites, epoxyeicosatrienoic acids (EETs), are EDHF (Feletou & Vanhoutte, 1999). Cytochrome P450 mono-oxygenase is mainly located on endothelial microsomal membranes and synthesizes EETs from arachidonic acid in a nicotinamide adenine dinucleotide phosphate-dependent pathway (Feletou & Vanhoutte, 1996). Moreover, anandamide, one of cannabinoides, and K+ have also been suggested to be the candidates of EDHF (Jarai et al., 1999; Edwards et al., 1998). Controversy also remains regarding the chemical nature of EDHF, and EDHF could be more than a single entity (Feletou & Vanhoutte, 1999).
In eukaryotic cells, the intracellular membrane transport plays a critical role in targeting membrane-bound proteins to the appropriate organelle (Klionsky & Ohsumi, 1999). Brefeldin A, a fungal metabolite, blocks the forward transport between the endoplasmic reticulum and Golgi apparatus, and causes an impaired distribution of the membrane proteins (Klausner et al., 1992). This compound could thus serve as a useful tool to investigate the involvement of cytochrome P450 in the production of EDHF in endothelial cells.
In the present study, we investigated the involvement of brefeldin A-sensitive membrane transport in the EDHF-mediated endothelium-dependent relaxation. For this purpose, we utilized surface fura-2-fluorimetry (Kanaide, 1999) to simultaneously determine the effects of brefeldin A on the cytosolic Ca2+ concentrations ([Ca2+]i) and tension of smooth muscle cells in strips of the porcine coronary artery with an intact endothelium. The endothelium-dependent relaxation was induced by two agonists, bradykinin and substance P. To evaluate the effect of brefeldin A on the EDHF-mediated relaxation, the endothelium-dependent relaxation was examined in the presence of indomethacin and NG-nitro-L-arginine (L-NOARG), which inhibit production of PGI2 and NO, respectively. We further investigated the effect of brefeldin A on the Ca2+ signalling in in situ endothelial cells, using strips of the porcine aortic valve.
Methods
Tissue preparation
Coronary arterial strips: Immediately after the animals, either sex, had been slaughtered by an electric shock and subsequent exsanguination, the left circumflex arteries were excised from the porcine hearts, and brought to the laboratory in ice-cold normal physiological salt solution (PSS). The arterial segments 2 – 3 cm from the origin were used for the experiments. After removing the adventitia, the segments were opened longitudinally, and then cut into circular strips (approximately 1 mm wide, 5 mm long, and 0.1 mm thick). Special care was taken to avoid damaging the endothelium.
Aortic valvular strips: Pocine aortic valves were immediately isolated after slaughter and were brought to the laboratory in ice-cold PSS. The valve leaflets were then cut into strips in an axial direction (approximately 2 mm wide, 5 mm long, and 0.18 mm thick) and used to monitor the [Ca2+]i in the endothelial cells in situ. The centre of each leaflet, corpus arantii, was not used. Special care was taken so as not to touch their surface during the whole procedure of preparation.
Fura-2 loading
The coronary arterial strips were loaded with Ca2+ indicator dye fura-2, by incubation in oxygenated (a mixture of 95% O2 and 5% CO2) Dulbecco's modified Eagle's medium (DMEM) containing 25 μM fura-2/AM (an acetoxymethyl ester form of fura-2) and 5% foetal bovine serum for 4 h at 37°C as previously described (Hirano et al., 1990). The valvular strips were loaded with fura-2 by incubation in oxygenated DMEM containing 50 μM fura-2/AM, 1 mM probenecid (di virgilio et al., 1989) and 5% foetal bovine serum for 90 min at 37°C (Aoki et al., 1994). After loading with fura-2, both vascular and valvular strips were washed to remove any dye in the extracellular space, and equilibrated in normal PSS for at least 1 h before any measurements were begun.
Front-surface fluorimetry of arterial and valvular strips
Experiments on the arterial strips were carried out at 37°C as previously described (Hirano et al., 1990). The fura-2-loaded vascular strips were mounted vertically in a quartz organ bath. Changes in the fluorescence intensity of the fura-2-Ca2+ complex in smooth muscle cells were monitored with a front-surface fluorimeter specifically designed for fura-2 fluorometry (CAM-OF2, Japan Spectroscopic Co., Tokyo, Japan). The ratio of the fluorescence (500 nm) intensities at 340 nm excitation to those at 380 nm excitation (ratio) was monitored as an indication of [Ca2+]i. Under the present experimental conditions (37°C and without probenecid), the fura-2 signals derived from the endothelial cells of the porcine coronary artery were negligible and the signal was exclusively derived from the smooth muscle cells (Kuroiwa et al., 1993). The fluorescence ratio was expressed as a percentage, assigning the value at rest and that at a sustained phase of the U46619-induced pre-contraction to be 0% and 100%, respectively, unless otherwise specified.
The fura-2 fluorimetry of the valvular strips was performed similarly. However, experiments with the valvular strips were carried out at 25°C in order to prevent any leakage of the fluorescent dye (Aoki et al., 1994). Fluorescence-microscopic observations revealed only the endothelial lining to be positive for fura-2 fluorescence (Kuroiwa et al., 1993). The fluorescence ratio was expressed as a percentage, while assigning the value at rest and that at the peak level of [Ca2+]i increase induced by 10 μM ATP to be 0% and 100%, respectively.
Measurement of tension development
The arterial strips, mounted vertically in a quartz organ bath (5 ml), were connected to a force-transducer (TB-612T, Nihon Koden, Japan). During the fura-2 equilibration period, the strips were stimulated with 118 mM K+ PSS every 15 min and the resting tension was increased in a stepwise manner. After equilibration, the resting tension was adjusted to 2.94 mN. The responsiveness of each strip to 118 mM K+ PSS was recorded before starting the experimental protocol. The developed tension was expressed as a percentage, while assigning the value at rest in the normal PSS and that at the steady state of pre-contraction just before the applications of the relaxants to be 0% and 100%, respectively, unless otherwise specified. Fura-2-loaded arterial strips induced a similar tension development to that observed in the unloaded strips. As a result, loading the arterial strips with fura-2, per se, did not affect the contractility (Hirano et al., 1990).
Treatment of the arterial and valvular strips with brefeldin A
In most measurements, the arterial and valvular strips were treated with 20 μg ml−1 brefeldin A in the normal PSS at 25°C after fura-2 loading. This concentration was selected according to Bauersachs et al. (1997). The fura-2-loaded and brefeldin A-treated strips were thereafter mounted in the organ bath and equilibrated in the normal PSS as described above, and then, the experimental protocol was started. During the experimental protocol, the strips were no longer exposed to brefeldin A. When the effect of 20 min treatment with brefeldin A on the endothelium-dependent relaxation in the arterial strips was determined, the fura-2-loaded strips were mounted onto a force transducer and equilibrated in the normal PSS, and then the experimental protocol was started. The strips were treated with 20 μg ml−1 brefeldin A 20 min before and during the pre-contraction by 100 nM U46619, and the effect of brefeldin A on the subsequent relaxation induced by bradykinin and substance P was determined.
Evaluation of the type of K+ channels involved in the EDHF-mediated relaxations
The arterial strips were pre-contracted with 100 nM U46619 in the presence of 10 μM indomethacin and 30 μM L-NOARG, and then the endothelium-dependent relaxations were initiated by successive application of 100 nM bradykinin and 10 nM substance P. Indomethacin and L-NOARG were applied 10 min before initiating the pre-contraction. When the effects of the K+ channel blockers on these relaxation were examined, the blockers were applied 10 min before the initiation of the pre-contraction, namely at the same time when indomethacin and L-NOARG were applied.
Drugs and solutions
The composition of normal PSS was as follows (mM): NaCl 123, KCl 4.7, NaHCO2 15.5, KH2 PO2 1.2, MgCl2 1.2, CaCl2 1.25 and D-glucose 11.5. High K+ PSS was prepared by replacing NaCl with equimolar KCl. PSS was bubbled with a mixture of 95% O2 and 5% CO2, and the resulting pH was 7.4. Fura-2/AM was purchased from Dojindo Laboratories (Kumamoto, Japan). Indomethacin was purchased from Wako (Osaka, Japan) and L-NOARG was from the Aldrich Chemical Company, Inc. (Milwaukee, WI, U.S.A.). Adenosine 5′-triphosphate (ATP) was purchased from Boehringer Mannheim GmBH (Germany). Brefeldin A, 4-aminopyridine (4-AP), ionomycin, probenecid, glibenclamide, iberiotoxin, apamin and tetrabutylammonium (TBA) were purchased from Sigma (St. Louis, MO, U.S.A.). Bradykinin and substance P were purchased from the Peptide Institute Inc. (Osaka, Japan). U46619 (9,11-dideoxy-11a,9a-epoxymethano-prostaglandin F2α), a thromboxane A2 analogue, was purchased from Funakoshi (Tokyo, Japan).
Data analysis
The values are expressed as the mean±s.e.mean. The analysis of variance (ANOVA) including a post hoc analysis (Dunnett's method) was used to determine statistical significance. One strip obtained from one animal was used for each experiment, therefore the number of experiments (n value) indicates the number of animals. P values of less than 0.05 were considered to demonstrate a statistically significant difference. All data were collected using a computerized data acquisition system (MacLab; Analog Digital Instruments, Australia, Macintosh; Apple Computer, U.S.A.).
Results
Effects of brefeldin A on the contractility of the arterial strips
In the porcine coronary artery with endothelium, both 118 mM K+-depolarization and 100 nM U46619 induced sustained increases in [Ca2+]i and tension, which were sustained for more than 30 min (data not shown). The developed tension induced by 118 mM K+-depolarization and 100 nM U46619 were 13.30±1.10 mN and 10.66±1.32 mN (n=17), respectively. The tension induced by 118 mM K+-depolarization and 100 nM U46619 in the presence of 10 μM indomethacin and 30 μM L-NOARG were 15.59±0.92 mN and 13.00±0.88 mN (n=32), respectively. A significant enhancement of the developed tension was observed in the presence of indomethacin and L-NOARG. When the levels of tension at rest and those obtained during the sustained contraction induced by 118 mM K+-depolarization (with no treatment) were assigned to be 0% and 100%, respectively, the tension induced by 118 mM K+-depolarization in the presence of 10 μM indomethacin and 30 μM L-NOARG was 136.2±3.8% (n=32). The extents of tension developed by 100 nM U46619 in the absence and presence of indomethacin and L-NOARG were 68.4±7.6% (n=17) and 106.6±7.6% (n=32), respectively. Therefore, the contractions induced by 118 mM K+-depolarization and 100 nM U46619 were both significantly augmented by treatment with indomethacin and L-NOARG. These findings were consistent with reports suggesting the basal release of NO and/or PGI2 from the endothelium of vascular strips (Kuroiwa et al., 1995).
In the strips treated with brefeldin A, the extent of tension induced by 118 mM K+-depolarization and 100 nM U46619 in the absence of indomethacin and L-NOARG was 15.07±0.41 mN and 12.33±1.02 mN (n=9), while those obtained in the presence of 10 μM indomethacin and 30 μM L-NOARG were 16.34±1.15 mN and 13.74±1.02 mN (n=15), respectively. There was no statistically significant difference in the extent or in the time course of the developed tension (data not shown) between brefeldin A-treated and untreated strips. In the strips treated with brefeldin A, indomethacin and L-NOARG also augmented the tension development induced by 118 mM K+-depolarization (from 100% to 129.4±3.9%) and 100 nM U46619 (from 77.2±8.6% to 110.5±12.5%), while assigning the level of tension at rest and that obtained during the sustained contraction induced by 118 mM K+-depolarization in the absence of indomethacin and L-NOARG to be 0% and 100%, respectively. These findings suggest that brefeldin A treatment per se had a negligible effect on the contractility of smooth muscle and on the basal release of NO and PGI2 from endothelial cells.
Effects of brefeldin A on the endothelium-dependent relaxations during the U46619-induced contractions
In the present study, bradykinin and substance P were used as agonists to induce endothelium-dependent relaxation in porcine left circumflex coronary arterial strips as the following reasons: both bradykinin and substance P showed no direct effect on [Ca2+]i and tension of smooth muscle (data not shown), and both bradykinin and substance P induced similar patterns of reduction in [Ca2+]i and tension of smooth muscle in strips with endothelium. When bradykinin (Figure 1a) or substance P (data not shown) were applied during the sustained contraction induced by 100 nM U46619, [Ca2+]i and tension decreased to reach a maximum reduction, and then gradually returned to sustained levels which were slightly lower than those observed prior to the application of bradykinin or substance P. The maximum reduction of [Ca2+]i and tension induced by bradykinin and substance P were concentration-dependent (Figure 2; controls).
Figure 1.
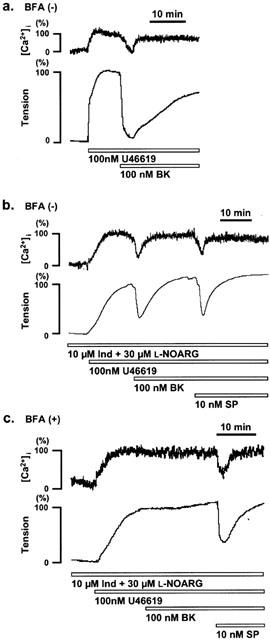
Effect of brefeldin A on bradykinin- and substance P-induced relaxation during U46619-induced contraction in the porcine coronary artery. (a) Representative recordings of the changes in [Ca2+]i and tension induced by bradykinin (BK) during the contraction induced by 100 nM U46619 in strips without brefeldin A (BFA) pretreatment. (b and c) changes in [Ca2+]i and tension induced by BK and substance P (SP) in the presence of 10 μM indomethacin (Ind) and 30 μM NG-nitro-L-arginine (L-NOARG), in strips without (b) and with (c) 6 h pretreatment of 20 μg ml−1 BFA. The levels of [Ca2+]i and tension obtained at rest and during the sustained phase of the contraction induced by 100 nM U46619 just before the application of the BK and SP were assigned to be 0% and 100%, respectively. The raw data traces are from the different preparations of different individuals.
Figure 2.
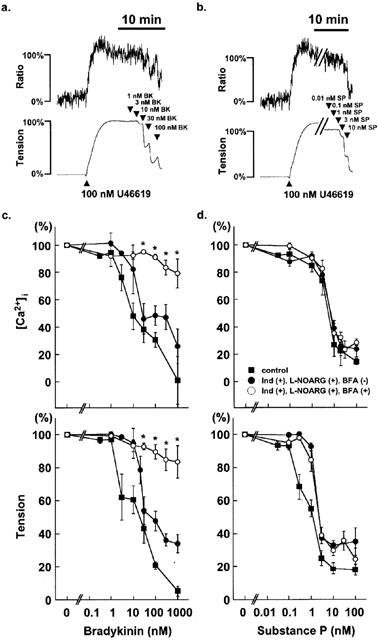
Effect of brefeldin A on the concentration-dependent relaxation induced by bradykinin and substance P. (a,b) Representative traces of the changes in [Ca2+]i (upper traces) and tension (lower traces) induced by cumulative applications of bradykinin (a) and substance P (b) during the 100 nM U46619-induced contraction in the absence of indomethacin and L-NOARG in the control, brefeldin A-untreated strips of the porcine coronary artery. Three – five concentrations of bradykinin and substance P were applied in one strip. (c,d) The concentration-response curves for the decrease in [Ca2+]i and tension induced by bradykinin (c) and substance P (d) during the 100 nM U46619-induced contractions were obtained in the absence (control) and the presence of 10 μM indomethacin (Ind) and 30 μM L-NOARG in the strips with no treatment by brefeldin A (BFA), and in the presence of 10 μM Ind and 30 μM L-NOARG in the strips treated with 20 μg ml−1 BFA for 6 h. The levels of [Ca2+]i and tension obtained at rest and during the sustained phase of the contraction induced by 100 nM U46619 just before the application of the bradykinin and substance P were assigned to be 0% and 100%, respectively. The raw data traces are from the different preparations of different individuals. The data are the mean±s.e.mean (n=3 – 21). * P<0.005, compared to the data obtained with Ind and L-NOARG in the strips with no treatment by BFA.
To determine the presence of a EDHF-mediated relaxation, the relaxation was induced in the presence of 10 μM indomethacin and 30 μM L-NOARG, which completely inhibit the production of PGI2 and NO, respectively. In the presence of 10 μM indomethacin and 30 μM L-NOARG, an application of bradykinin (Figure 1b) or substance P also induced rapid decreases of [Ca2+]i and tension. However, this reduction was transient, and [Ca2+]i and tension rapidly returned to control levels just prior to the application of bradykinin or substance P. The time required for [Ca2+]i and tension to return to the control levels was significantly shorter (P<0.05) than that observed in the absence of indomethacin and L-NOARG. In both cases, [Ca2+]i recovered faster than tension (Figure 1). In addition, in the presence of both 10 μM indomethacin and 30 μM L-NOARG, the maximum reduction of [Ca2+]i and tension induced by bradykinin and substance P was concentration-dependent (Figure 2). The decreases in tension were significantly attenuated by the treatment with both 10 μM indomethacin and 30 μM L-NOARG (Figure 2). In the presence of both 10 μM indomethacin and 30 μM L-NOARG, when [Ca2+]i and tension returned to the pre-application levels during the application of bradykinin, an additional application of substance P could still induce rapid and transient decreases of [Ca2+]i and tension again (Figure 1b). The extent of these changes were similar to those without bradykinin pretreatment. The permutation of the order of applications of bradykinin and substance P had no effect on the extent of changes in [Ca2+]i and tension (data not shown), indicating that there is no interference in the transient relaxing effect between bradykinin and substance P.
As shown in Figure 1c, the treatment of the strips with 20 μg ml−1 brefeldin A for 6 h completely abolished the relaxation induced by bradykinin in the presence of 10 μM indomethacin and 30 μM L-NOARG. However, the subsequent application of substance P induced a relaxation which was not significantly different from that observed in the strips untreated with brefeldin A (Figure 2d). On the other hand, in the absence of indomethacin and L-NOARG, the 6 h treatment with brefeldin A had no significant effect on the relaxation induced by bradykinin. Shorter treatment periods (20 min to 4 h) to brefeldin A had no effect on the bradykinin-induced relaxation either in the presence or absence of indomethacin and L-NOARG (data not shown). We therefore performed the 6 h treatment with brefeldin A in the following experiments.
Figure 2 summarizes the effects of brefeldin A on the concentration-dependent decreases in [Ca2+]i and tension induced by bradykinin and substance P. Without brefeldin A treatment in the absence of indomethacin and L-NOARG, bradykinin induced decreases in [Ca2+]i and tension in the concentration range between 1 nM and 1 μM (Figure 2c; control). The concentrations of bradykinin required to induce the half maximal decrease (IC50) in [Ca2+]i and tension were 15.5±9.4 nM and 15.0±10.0 nM (n=3 – 16), respectively, thus suggesting that the decrease in tension at the maximum was associated with the decrease in [Ca2+]i. In the presence of indomethacin and L-NOARG, the IC50 values for inhibition of [Ca2+]i elevation and tension development were 14.1±4.5 nM and 23.2±1.8 nM, respectively. The treatment with 20 μg ml−1 brefeldin A substantially abolished the bradykinin-induced decreases in [Ca2+]i and tension in the range of concentrations between 1 nM and 1 mM (Figure 2c).
Substance P induced relaxation in the concentration range between 0.1 nM and 10 nM without treatment with brefeldin A in the absence of indomethacin and L-NOARG (Figure 2d; control). The IC50 in [Ca2+]i and tension were 4.7±0.8 nM and 0.7±0.2 nM, respectively (n=4 – 16). The treatment with indomethacin and L-NOARG significantly attenuated the substance P-induced decreases in tension (IC50; 1.5±0.1 nM, n=8 – 18) but had little effect on the decrease in [Ca2+]i (IC50; 5.4±1.0 nM, n=8 – 18). Treatment with brefeldin A had no significant effect on the decrease of [Ca2+]i (IC50; 4.8±0.7 nM, n=3 – 19) and tension (IC50; 1.6±0.2 nM) induced by substance P in the presence of indomethacin and L-NOARG (Figure 2d).
Effects of brefeldin A on the bradykinin-induced relaxation during the contraction induced by high K+-depolarization
To examine the effects of brefeldin A on the endothelium-dependent relaxations due to factors other than EDHF, the relaxations induced by bradykinin and substance P were evaluated during the contractions induced by 118 mM K+-depolarization. It has been shown that high external K+ inhibits the membrane hyperpolarization induced by EDHF (Feletou & Vanhoutte, 1999). Therefore, during the high K+-induced contraction, the effects of EDHF on smooth muscle can be eliminated, and the endothelium-dependent relaxation is mainly due to either NO or PGI2 (Kuroiwa et al., 1995).
During the 118 mM K+-induced contraction, bradykinin induced a rapid, and then, sustained decrease in tension, while there was no significant decrease in [Ca2+]i (Figure 3a). The treatment with 10 μM indomethacin alone had no effect on this bradykinin-induced relaxation during the K+-depolarization (Figure 3b,f), while the combination of 10 μM indomethacin and 30 μM L-NOARG completely inhibited the relaxation (Figure 3c,f). The administration of 30 μM L-NOARG considered to be sufficient to block NO-dependent relaxation. As a result, the bradykinin-induced relaxation during the K+-depolarization was considered to be dependent mainly on NO production. In the strips treated with 20 μg ml−1 brefeldin A, bradykinin induced a similar relaxation to that seen in the strips untreated with brefeldin A (Figure 3d). The extent of decreases in [Ca2+]i and tension induced by 100 nM bradykinin were 88.7±4.5% and 63.4±10.7%, respectively (n=10; Figure 3f). This relaxation was also completely abolished by treatment with indomethacin and L-NOARG in the brefeldin A-treated strips (Figure 3e,f).
Figure 3.
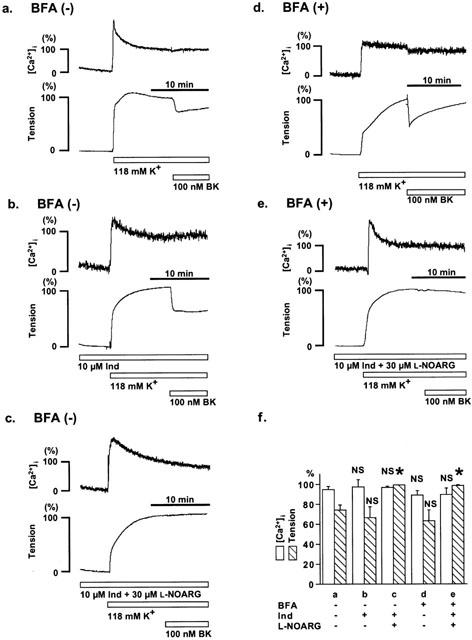
Effect of brefeldin A on the bradykinin-induced relaxation during 118 mM K+-depolarization. (a – e) Representative recordings of the bradykinin-induced decreases in [Ca2+]i and tension during the contraction induced by 118 mM K+-depolarization in the strips without (a – c) and with (d, e) 6 h pretreatment of 20 μg ml−1 brefeldin A (BFA), either in the absence (a,d) or presence of 10 μM indomethacin (Ind) and/or 30 μM L-NOARG (b,c,e). (f) Summary of five independent measurements for each protocol (a – e). The maximum decreases in [Ca2+]i and tension induced by bradykinin were shown. The levels of [Ca2+]i and tension obtained at rest and during the sustained phase of the contraction induced by 118 mM K+ just before the application of bradykinin were assigned to be 0% and 100%, respectively. The raw data traces are from the different preparations of different individuals. The data are the mean±s.e.mean. * Significantly different (P<0.05); NS, not significant, compared with the data obtained by protocol a.
Effects of brefeldin A on the [Ca2+]i transients in the endothelial cells in situ
To determine the intracellular mechanism mediating the inhibition of bradykinin-induced EDHF-dependent relaxation by brefeldin A, we examined the effects of brefeldin A on the [Ca2+]i transients induced by bradykinin and substance P in the endothelial cells. For this purpose, fura-2-loaded strips of porcine aortic valve were used to monitor changes in [Ca2+]i in the endothelial cells in situ. Figure 4 shows the representative recordings of the [Ca2+]i elevations induced by 10 μM ATP, 100 nM bradykinin and 10 nM substance P in the absence (Figure 4a) and presence (Figure 4b) of 10 μM indomethacin and 30 μM L-NOARG. The extent of [Ca2+]i elevation induced by bradykinin and substance P were 120.6±5.9% (n=6) and 78.2±9.0% (n=9) of that obtained with 10 μM ATP (100%). In the strips treated with brefeldin A for 6 h, the extent of [Ca2+]i elevation induced by bradykinin and substance P were 122.9±18.7% (n=8) and 86.5±8.8% (n=5), respectively (Figure 4c). These [Ca2+]i elevations observed in brefeldin A-treated strips did not differ from those seen in the non-treated strips. The level of [Ca2+]i elevation induced by 10 μM ATP was 11.0±2.4% of that obtained with 10 μM ionomycin in the strips with 6 h treatment by brefeldin A, and 12.1±2.1% in the strips without treatment. Ionomycin is a Ca2+ ionophore and used to induce a saturation of fura-2 fluorescence (Hirano et al., 1990). The level of fluorescence ratio obtained with ionomycin thus was considered to be the same regardless of the treatment with brefeldin A. Therefore, there was no significant difference in the response to ATP between brefeldin A-treated and untreated strips.
Figure 4.
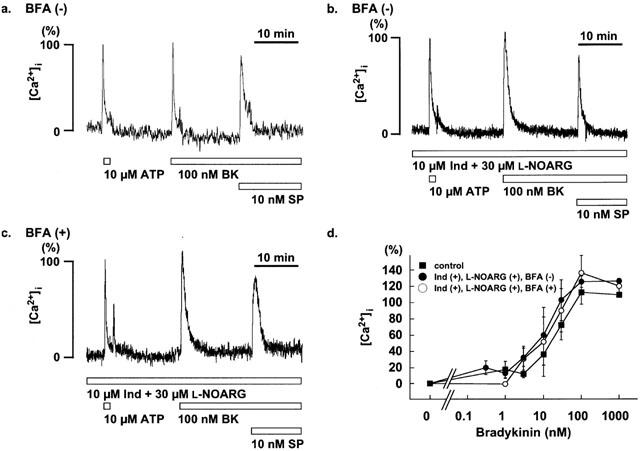
Effect of brefeldin A on [Ca2+]i of in situ endothelial cells of the porcine aortic valve. (a) Representative recordings of the changes in [Ca2+]i induced by 10 μM ATP, 100 nM bradykinin (BK) and 10 nM substance P (SP) in the strips without brefeldin A (BFA) pretreatment. (b and c) changes in [Ca2+]i induced by BK and SP in the presence of 10 μM indomethacin (Ind) and 30 μM NG-nitro-L-arginine (L-NOARG), in strips without (b) and with (c) 6 h pretreatment of 20 μg ml−1 BFA. The levels of [Ca2+]i at rest and that at the peak response induced by 10 μM ATP, were assigned to be 0% and 100%, respectively. (c) The concentration-response curves for the BK-induced [Ca2+]i elevations in the absence and presence of 10 μM Ind and 30 μM L-NOARG in the strips with no treatment by BFA, and in the presence of 10 μM Ind and 30 μM L-NOARG in the strips treated with BFA. The raw data traces are from the different preparations of different individuals. The data are the mean±s.e.mean (n=3 – 14).
Effects of brefeldin A on the bradykinin-increased [Ca2+]i elevation was further examined by evaluating concentration-response curves (Figure 4d). In the absence of indomethacin and L-NOARG, bradykinin (1 – 100 nM) induced an elevation of the [Ca2+]i, and the concentration required to induce a half maximum elevation was 20.4±3.9 nM. The treatment with indomethacin and L-NOARG had no effect on the bradykinin-induced [Ca2+]i elevations in the entire concentration range examined (Figure 4d). The concentration-response curve for the bradykinin-induced [Ca2+]i elevation obtained in the strips treated with brefeldin A did not significantly differ from those obtained in the strips without treatment (Figure 4d).
Effects of K+ channel blockers on the endothelium-dependent L-NOARG/indomethacin-resistant relaxations during the U46619-induced contractions
To determine the type of K+ channels involved in the bradykinin- and substance P-induced EDHF-mediated relaxations, we examined the effect of various K+ channel blockers on the control relaxation as shown in Figure 1b. The K+ channel blockers examined are 1 μM apamin (a blocker of the small conductance Ca2+-activated K+ channel), 1 μM iberiotoxin (a blocker of the large conductance Ca2+-activated K+ channel), 3 mM TBA (non-selective blocker of the Ca2+-activated K+ channel), 1 mM 4-AP (a blocker of the voltage-dependent K+ channel) and 1 μM glibenclamide (a blocker of the ATP-sensitive K+ channel). The K+ channel blockers were applied 10 min prior to the pre-contraction by 100 nM U46619 in the presence of 10 μM indomethacin and 30 μM L-NOARG, and then the relaxations were induced by successive application of 100 nM bradykinin and 10 nM substance P. Apamin, iberiotoxin, and glibenclamide had no significant effect on the decrease in [Ca2+]i and tension induced by bradykinin and substance P (data not shown). TBA and 4-AP had no effect on the resting tension, but the resting [Ca2+]i progressively increased in the presence of TBA. However, [Ca2+]i elevation induced by 100 nM U46619 in the presence of either TBA or 4-AP were similar to those obtained in the absence of TBA and 4-AP (79.0±9.9%, n=32 in the absence of TBA and 4-AP versus 68.8±6.8% in the presence of TBA alone, n=21; 91.6±10.9% in the presence of 4-AP alone, n=4; 73.7±5.0% in the presence of TBA and 4-AP, n=3) (Figure 5a – c). 4-AP (1 mM) alone had no effect on the relaxations induced by bradykinin and substance P (Figure 5a,d). TBA alone completely inhibited the bradykinin-induced decrease in [Ca2+]i and tension (Figure 5b). On the other hand, TBA only partially inhibited the substance P-induced decrease in [Ca2+]i, while it had no significant effect on decrease in tension (Figure 5b,d). The permutation of the order of application of bradykinin and substance P had no effect on the inhibitory effect of TBA (data not shown). The combination of TBA and 4-AP completely inhibited the substance P-induced decrease in [Ca2+]i and tension (Figure 5b).
Figure 5.
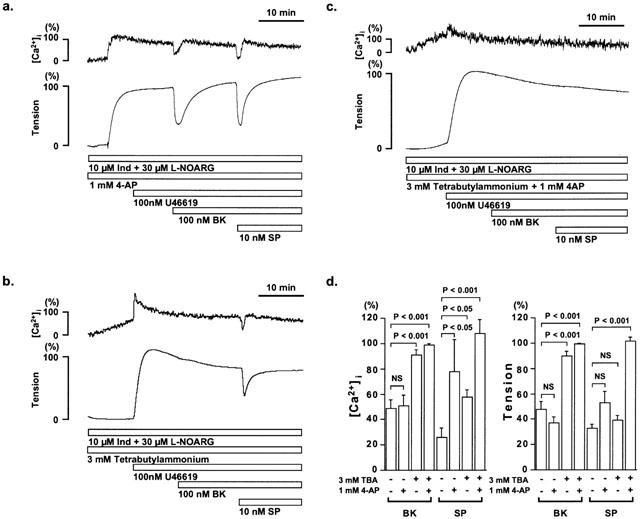
Effect of tetrabutylammonium and 4-aminopyridine on the EDHF-mediated relaxation during U46619-induced contraction in the porcine coronary artery. (a – c) Representative recordings of changes in [Ca2+]i and tension induced by 100 nM bradykinin (BK) and 10 nM substance P (SP) during the 100 nM U46619-induced contraction in the presence of 10 μM indomethacin (Ind) and 30 μM L-NOARG, with 1 mM 4-aminopyridine (4-AP) (a), 3 mM tetrabutylammonium (TBA) (b), and 4-AP plus TBA (c). The control relaxations in the absence of K+ channel blockers is as shown in Figure 1b. (d) The effect of 3 mM TBA and 1 mM 4-AP on the maximum reduction of [Ca2+]i and tension induced by BK or SP. The levels of [Ca2+]i and tension obtained at rest and during the sustained phase of the contractions induced by 100 nM U46619 just before the application of bradykinin and substance P were assigned to be 0% and 100%, respectively. The raw data traces are from the different preparations of different individuals. The data are the mean±s.e.mean. (n=3 – 21). NS, not significantly different.
We also investigated the effect of 3 mM TBA on the [Ca2+]i transients induced by bradykinin in the endothelial cells in situ. TBA had no effect on both the resting levels of [Ca2+]i in endothelial cells and the [Ca2+]i elevations induced by 100 nM bradykinin (155.0±18.7%, n=5) (data not shown).
Discussion
The most important finding in the present study is that brefeldin A inhibits the production by bradykinin of endothelium-derived relaxing factor, which is different from NO or PGI2, in the porcine coronary artery. This finding is consistent with those of a previous report (Bauersachs et al., 1997). However, in the present study, we demonstrated that brefeldin A had no effect on the NO/PGI2-independent relaxation induced by substance P. The NO-mediated relaxations induced by bradykinin and substance P were resistant to brefeldin A as reported by Bauersachs et al. (1997). Thus, it is suggested that the inhibitory effect of brefeldin A was specific to the bradykinin-induced NO/PGI2-independent relaxation, although the NO/PGI2-independent relaxations induced by bradykinin and substance P were both mediated mainly by hyperpolarization of smooth muscle, because they were inhibited by elevating extracellular K+ concentration or by K+ channel blockers. Therefore, we suggest that the bradykinin-induced EDHF-mediated relaxation in the porcine coronary artery depends on a brefeldin A-sensitive mechanism. Moreover, we suggest that the mechanism of the substance P-induced EDHF-mediated relaxation differs from that seen with bradykinin.
The sustained phase of the relaxations induced by bradykinin and substance P was accompanied by a slight decrease in [Ca2+]i, and most of the sustained relaxation was abolished by L-NOARG. On the other hand, the early transient relaxations observed in the presence of indomethacin and L-NOARG were accompanied by decreases in [Ca2+]i, and these relaxations were inhibited by elevating extracellular K+ concentration or K+ channel blockers. These observations suggest that EDHF is involved in the early transient phase of the bradykinin and substance P-induced relaxations, and that the sustained phase is mainly dependent on NO.
The sensitivity of the EDHF-mediated relaxation toward various K+ channel blockers differed with the type of vessel and agonist used to induce relaxation, thus suggesting a different type of K+ channel is involved in the different EDHF-mediated relaxation in the different vessel (Triggle et al., 1999). Nevertheless, many reports support the involvement of Ca2+-activated K+ channel in the EDHF-mediated relaxations (Edwards & Weston, 1998; Feletou & Vanhoutte, 1999; Graier et al., 1996), and a small conductance type of the Ca2+-activated K+ channel was suggested to be involved in the bradykinin-induced relaxation in the porcine coronary artery (Cowan & Cohen, 1991; Hecker et al., 1994). In the present study, the bradykinin-induced EDHF-mediated relaxation was completely abolished by TBA, while iberiotoxin, apamin, glibenclamide or 4-AP alone had no effect. As a result, we suggest that the major K+ channel involved in the bradykinin-induced EDHF-mediated relaxation was an intermediate conductance Ca2+-activated K+ channel (Latorre et al., 1989). It is unlikely that a large and small conductance Ca2+-activated K+ channels played a major role. Regarding the complete inhibition by TBA of the bradykinin-induced EDHF-mediated relaxation, there is a possibility that TBA inhibited the action of bradykinin at the receptor level in the endothelial cells (Colden-Stanfield et al., 1990). However, this was not the case because TBA had no effect on the [Ca2+]i elevations induced by 100 nM bradykinin in the endothelial cells. On the other hand, the substance P-induced EDHF-mediated relaxation showed different sensitivity toward K+ channel blockers, and the voltage-dependent K+ channel in addition to the Ca2+-activated K+ channel was suggested to be involved. The most important implication from these observations is that the type of K+ channel activated by bradykinin-induced EDHF differs from that activated by substance P-induced EDHF, even in the same artery. Our observation further suggested that the nature of the EDHF produced by bradykinin and thus the mechanism of production of the EDHF differed from that obtained with substance P. These differences between bradykinin and substance P may be related to the difference in the sensitivity of the EDHF-mediated relaxation toward brefeldin A.
The intracellular membrane transport plays a critical role in targeting membrane-bound proteins such as secretary proteins and microsomal proteins to appropriate locations where they perform their physiological functions (Klionsky & Ohsumi, 1999). Brefeldin A inhibits microtubule-dependent vesicle transport from endoplasmic reticulum to Golgi apparatus and intercisternal Golgi transport (Misumi et al., 1986; Oda et al., 1987). Brefeldin A could thus cause an impaired distribution of proteins in the secretary vesicle and microsome (Chardin & McCormick, 1999). It is reported that brefeldin A induces such reorganization of intracellular organelle within two min in the cultured cells including MDCK, rat hepatocytes and murine T cell hybridoma, and that the effect of brefeldin A is reversible upon its removal (Klausner et al., 1992). Bauersachs et al. (1997) demonstrated that 90 min treatment with 35 μmol l−1 (approximately 10 μg ml−1) brefeldin A inhibited the bradykinin-induced EDHF-mediated relaxation in the porcine coronary artery. They also showed that 3 h washout of brefeldin A resulted in the almost complete restoration of the relaxation. However, in the present study, 6 h pre-treatment with brefeldin A was required to inhibit the bradykinin-induced EDHF-mediated relaxation, and this inhibitory effect was observed a few hours after the removal of brefeldin A. These findings thus suggest that not only the impaired distribution but also the substantial decrease in the amount of any components involved in EDHF production was required for inhibition of the EDHF-mediated relaxation. The recovery of the decreased components may not be achieved within a few hours after removal of brefeldin A, even although the altered organization of the intracellular organelle can be reversed. The effect of 6 h pretreatment with brefeldin A, however, was not considered to be due to non-specific effects, because NO-mediated relaxation induced by bradykinin or substance P-induced relaxation remained intact. Furthermore, we also demonstrated that the signal transduction from the bradykinin receptor to the Ca2+ signal in endothelial cells of the aortic valve remained intact after 6 h treatment with brefeldin A. Accordingly, brefeldin A is considered to reduce the enzyme(s) responsible for production of EDHF by bradykinin.
Cytochrome P450 is located on endothelial microsomal membranes (Abraham et al., 1985). Brefeldin A was shown to cause impaired distribution and subsequent reduction of cytochrome P450 (Bauersachs et al., 1997). Several reports have suggested EDHF to be cytochrome P450 metabolites of arachnidonic acid such as EETs (Dong et al., 1997; Fisslthaler et al., 1999; Graier et al., 1996). However, controversy remains regarding the chemical identity of EDHF, and candidates other than EETs, including K+ (Edwards et al., 1998) and cannabinoides (Randall & Kendall, 1998), have been suggested. Collectively, EDHF does not seem to be a single entity, but its chemical identity may differ with the type of vessel and agonist. Our observation of the differential effect of brefeldin A on the bradykinin- and substance P-induced EDHF-mediated relaxation are consistent with the suggestion that EDHF is not a single entity.
The endothelial constitutive nitric-oxide synthase (eNOS) is cytosolic enzyme and plays a major role in NO production in endothelial cells. eNOS was shown to bind to caveolin, a membrane protein specifically located at caveolae, small invaginations of the plasma membrane (Shaul et al., 1996). Binding to caveolin inhibits the activity of eNOS. The Ca2+-calmodulin complex causes dissociation of eNOS from caveolin complex and releases eNOS activity from inhibition by caveolin (Michel et al., 1997). Our findings indicated that brefeldin A had no effect on these NO-producing systems as a whole. Bauersachs et al. (1997) demonstrated that brefeldin A had no significant effect on the total cellular activity of eNOS, but altered the distribution of eNOS activity and protein between the caveolae fraction and the cytosolic fraction.
In conclusion, brefeldin A specifically inhibited the bradykinin-induced EDHF-mediated relaxation with no effect on the intracellular Ca2+ signalling in endothelial cells. The requirement of 6 h pre-treatment with brefeldin A suggested that not only an impaired distribution but also a substantial reduction of the components involved in the production of EDHF are necessary to inhibit the EDHF-mediated relaxation. This observation was consistent with the notion that the metabolites of cytochrome P450 function as EDHF. However, since the EDHF-mediated relaxation induced by substance P showed a different sensitivity toward the inhibition by K+ channel blockers and brefeldin A, even in the same artery, these findings also suggest that EDHF is more than a single entity.
Acknowledgments
We thank Mr Brian Quinn for his critical reading of the manuscript. This study was supported in part by Grants-in-Aid for Scientific Research (No. 10557072, 11838013, 11670687) and for Scientific Research on Priority Area (No. 12213103) from the Ministry of Education, Science, Sports and Culture, Japan, by the Research Grant for Cardiovascular Diseases (11C-1) from the Ministry of Health and Welfare, Japan, and by grants from the Foundation for the Promotion of Clinical Medicine, the Suzuken Memorial Foundation and KANZAWA Medical Research Foundation.
Abbreviations
- 4-AP
4-aminopyridine
- ATP
adenosine 5′-triphosphate
- [Ca2+]i
cytosolic Ca2+ concentration
- DMEM
Dulbecco's modified Eagle's medium
- EDHF
endothelium-derived hyperpolarizing factor
- EETs
epoxyeicosatrienoic acids
- fura-2/AM
acetoxymethyl ester form of fura-2
- Ind
indomethacin
- L-NOARG
NG-nitro-L-arginine
- NO
nitric oxide
- NOS
NO synthase
- PGI2
prostaglandin I2
- PSS
physiological salt solution
- TBA
tetrabutylammonium
References
- ABRAHAM N.G., PINTO A., MULLANE K.M., LEVERE R.D., SPOKAS E. Presence of cytochrome P-450-dependent monooxygenase in intimal cells of the hog aorta. Hypertension. 1985;7:899–904. doi: 10.1161/01.hyp.7.6.899. [DOI] [PubMed] [Google Scholar]
- AOKI H., KOBAYASHI S., NISHIMURA J., KANAIDE H. Sensitivity of G-protein involved in endothelin-1-induced Ca2+ influx to pertussis toxin in porcine endothelial cells in situ. Br. J. Pharmacol. 1994;111:989–996. doi: 10.1111/j.1476-5381.1994.tb14841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAUERSACHS J., FLEMING I., SCHOLZ D., POPP R., BUSSE R. Endothelium-derived hyperpolarizing factor, but not nitric oxide, is reversibly inhibited by brefeldin A. Hypertension. 1997;30:1598–1605. doi: 10.1161/01.hyp.30.6.1598. [DOI] [PubMed] [Google Scholar]
- CHARDIN P., MCCORMICK F. Brefeldin A: the advantage of being uncompetitive. Cell. 1999;97:153–155. doi: 10.1016/s0092-8674(00)80724-2. [DOI] [PubMed] [Google Scholar]
- COLDEN-STANFIELD M., SCHILLING W.P., POSSANI L.D., KUNZE D.L. Bradykinin-induced potassium current in cultured bovine aortic endothelial cells. J. Membr. Biol. 1990;116:227–238. doi: 10.1007/BF01868462. [DOI] [PubMed] [Google Scholar]
- COWAN C.L., COHEN R.A. Two mechanisms mediate relaxation by bradykinin of pig coronary artery: NO-dependent and -independent responses. Am. J. Physiol. 1991;261:H830–H835. doi: 10.1152/ajpheart.1991.261.3.H830. [DOI] [PubMed] [Google Scholar]
- DI VIRGILIO F., STEINBERG T.H., SC S.Organic-anion transport inhibitors to facilitate measurement of cytosolic free Ca2+ with fura-2 Methods in Cell Biol. 1989San Diego: Academic Press; 453–462.ed. Tartakoff, A.M. pp [DOI] [PubMed] [Google Scholar]
- DONG H., WALDRON G.J., GALIPEAU D., COLE W.C., TRIGGLE C.R. NO/PGI2-independent vasorelaxation and the cytochrome P450 pathway in rabbit carotid artery. Br. J. Pharmacol. 1997;120:695–701. doi: 10.1038/sj.bjp.0700945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., DORA K.A., GARDENER M.J., GARLAND C.J., WESTON A.H. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- EDWARDS G., WESTON A.H. Endothelium-derived hyperpolarizing factor–a critical appraisal. Prog. Drug Res. 1998;50:107–133. doi: 10.1007/978-3-0348-8833-2_2. [DOI] [PubMed] [Google Scholar]
- FELETOU M., VANHOUTTE P.M. Endothelium-derived hyperpolarizing factor. Clin. Exper. Pharmacol. Physiol. 1996;23:1082–1090. doi: 10.1111/j.1440-1681.1996.tb01174.x. [DOI] [PubMed] [Google Scholar]
- FELETOU M., VANHOUTTE P.M. The alternative: EDHF. J. Mol. Cell. Cardiol. 1999;31:15–22. doi: 10.1006/jmcc.1998.0840. [DOI] [PubMed] [Google Scholar]
- FISSLTHALER B., POPP R., KISS L., POTENTE M., HARDER D.R., FLEMING I., BUSSE R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- GRAIER W.F., HOLZMANN S., HOEBEL B.G., KUKOVETZ W.R., KOSTNER G.M. Mechanisms of L-NG-nitroarginine/indomethacin-resistant relaxation in bovine and porcine coronary arteries. Br. J. Pharmacol. 1996;119:1177–1186. doi: 10.1111/j.1476-5381.1996.tb16020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HECKER M., BARA A.T., BAUERSACHS J., BUSSE R. Characterization of endothelium-derived hyperpolarizing factor as a cytochrome P450-derived arachidonic acid metabolite in mammals. J. Physiol. 1994;481:407–414. doi: 10.1113/jphysiol.1994.sp020449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HIRANO K., KANAIDE H., ABE S., NAKAMURA M. Effects of diltiazem on calcium concentrations in the cytosol and on force of contractions in porcine coronary arterial strips. Br. J. Pharmacol. 1990;101:273–280. doi: 10.1111/j.1476-5381.1990.tb12700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JARAI Z., WAGNER J.A., VARGA K., LAKE K.D., COMPTON D.R., MARTIN B.R., ZIMMER A.M., BONNER T.I., BUCKLEY N.E., MEZEY E., RAZDAN R.K., ZIMMER A., KUNOS G. Cannabinoid-induced mesenteric vasodilation through an endothelial site distinct from CB1 or CB2 receptors. Proc. Natl. Acad. Sci. U.S.A. 1999;96:14136–14141. doi: 10.1073/pnas.96.24.14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KANAIDE H. Measurement of [Ca2+]i in smooth muscle strips using front-surface fluorimetry. Methods in Mol. Biol. 1999;114:269–277. doi: 10.1385/1-59259-250-3:269. [DOI] [PubMed] [Google Scholar]
- KLAUSNER R.D., DONALDSON J.G., LIPPINCOTT-SCHWARTZ J. Brefeldin A: insights into the control of membrane traffic and organelle structure. J. Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLIONSKY D.J., OHSUMI Y. Vacuolar import of proteins and organelles from the cytoplasm. Annu. Rev. Cell Dev. Biol. 1999;15:1–32. doi: 10.1146/annurev.cellbio.15.1.1. [DOI] [PubMed] [Google Scholar]
- KUHBERGER E., GROSCHNER K., KUKOVETZ W.R., BRUNNER F. The role of myoendothelial cell contact in non-nitric oxide-, non-prostanoid-mediated endothelium-dependent relaxation of porcine coronary artery. Br. J. Pharmacol. 1994;113:1289–1294. doi: 10.1111/j.1476-5381.1994.tb17138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUROIWA M., AOKI H., KOBAYASHI S., NISHIMURA J., KANAIDE H. Role of GTP-protein and endothelium in contraction induced by ethanol in pig coronary artery. J. Physiol. 1993;470:521–537. doi: 10.1113/jphysiol.1993.sp019873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUROIWA M., AOKI H., KOBAYASHI S., NISHIMURA J., KANAIDE H. Mechanism of endothelium-dependent relaxation induced by substance P in the coronary artery of the pig. Br. J. Pharmacol. 1995;116:2040–2047. doi: 10.1111/j.1476-5381.1995.tb16409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LATORRE R., OBERHAUSER A., LABARCA P., ALVAREZ O. Varieties of calcium-activated potassium channels. Ann. Rev. Physiol. 1989;51:385–399. doi: 10.1146/annurev.ph.51.030189.002125. [DOI] [PubMed] [Google Scholar]
- MICHEL J.B., FERON O., SACKS D., MICHEL T. Reciprocal regulation of endothelial nitric-oxide synthase by Ca2+-calmodulin and caveolin. J. Biol. Chem. 1997;272:15583–15586. doi: 10.1074/jbc.272.25.15583. [DOI] [PubMed] [Google Scholar]
- MISUMI Y., MISUMI Y., MIKI K., TAKATSUKI A., TAMURA G., IKEHARA Y. Novel blockade by brefeldin A of intracellular transport of secretory proteins in cultured rat hepatocytes. J. Biol. Chem. 1986;261:11398–11403. [PubMed] [Google Scholar]
- ODA K., HIROSE S., TAKAMI N., MISUMI Y., TAKATSUKI A., IKEHARA Y. Brefeldin A arrests the intracellular transport of a precursor of complement C3 before its conversion site in rat hepatocytes. FEBS Lett. 1987;214:135–138. doi: 10.1016/0014-5793(87)80028-5. [DOI] [PubMed] [Google Scholar]
- RANDALL M.D., KENDALL D.A. Anandamide and endothelium-derived hyperpolarizing factor act via a common vasorelaxant mechanism in rat mesentery. Eur. J. Pharmacol. 1998;346:51–53. doi: 10.1016/s0014-2999(98)00003-x. [DOI] [PubMed] [Google Scholar]
- SHAUL P.W., SMART E.J., ROBINSON L.J., GERMAN Z., YUHANNA I.S., YING Y., ANDERSON R.G., MICHEL T. Acylation targets endothelial nitric-oxide synthase to plasmalemmal caveolae. J. Biol. Chem. 1996;271:6518–6522. doi: 10.1074/jbc.271.11.6518. [DOI] [PubMed] [Google Scholar]
- TRIGGLE C.R., DONG H., WALDRON G.J., COLE W.C. Endothelium-derived hyperpolarizing factor(s): species and tissue heterogeneity. Clin. Exp. Pharmacol. Physiol. 1999;26:176–179. doi: 10.1046/j.1440-1681.1999.03007.x. [DOI] [PubMed] [Google Scholar]
- VANHOUTTE P.M., SHIMOKAWA H. Endothelium-derived relaxing factor and coronary vasospasm. Circulation. 1989;80:1–9. doi: 10.1161/01.cir.80.1.1. [DOI] [PubMed] [Google Scholar]