

Abstract
Several different molecular species of phosphatidic acid (PA) bind to a G-protein coupled receptor (GPCR) to induce activation of the p42/p44 mitogen-activated protein kinase (p42/p44 MAPK) pathway in HEK 293 cells. PA is active at low nanomolar concentrations and the response is sensitive to pertussis toxin (which uncouples GPCRs from Gi/o). The de-acylated product of PA, lysophosphatidic acid (LPA), which binds to members of the endothelial differentiation gene (EDG) family of receptors also stimulated p42/p44 MAPK in a pertussis toxin sensitive manner, but with an ∼100 – 1000 fold lower potency compared with the different molecular species of PA. RT – PCR using gene-specific primers showed that HEK 293 cells express EDG2 and PSP24, the latter being a lipid binding GPCR out with the EDG cluster. We conclude that PA is a novel high potency GPCR agonist.
Keywords: Phosphatidate, lysophosphatidate, kinases, G-protein, signalling
Introduction
Over the past few years there has been a major advance in our understanding of how certain lipids can act as intercellular mediators by binding to new classes of GPCRs to regulate cell function. Two lipid agonists, sphingosine 1-phosphate (S1P) and lysophosphatidic acid (LPA) have been shown to bind to the endothelial differentiation gene (EDG) family of GPCRs to regulate cell growth, differentiation, migration and apoptosis (for review see Pyne & Pyne, 2000). In contrast, phosphatidic acid (PA) has traditionally been implicated in regulating intracellular signalling events. For instance, PA is involved in the membrane recruitment of Raf-1, which contains a specific PA binding region (amino acids 390 – 426) (Rizzo et al., 2000).
The group of EDG receptors that bind LPA with high-affinity include EDG2, EDG4 and EDG7 (35% identity to the S1P binding EDG receptors, which include EDG1, 3, 5, 6 and 8). EDG2 (or Vzg-1) was the first family member to have its ligand identified as LPA. EDG2 over-expression in a neuronal-like cell (TSM) exaggerated LPA-stimulated cytoskeletal changes (Rho-mediated), Gi-mediated stimulation of p42/p44 MAPK activation and inhibition of adenylate cyclase (Hecht et al., 1996). EDG2 and EDG4 transfected HTC4 hepatoma cells exhibited LPA-stimulated calcium transients (An et al., 1998). LPA can also bind to GPCRs that are not within the EDG cluster, such as the PSP24. Initial characterization studies have shown that Xenopus oocyte PSP24 bound LPA, plasmalogen-glycerophosphate and cyclic PA (Guo et al., 1996; Fischer et al., 1998). However, two mammalian PSP24 homologues (75% similarity with xPSP24) did not bind LPA (Kawasawa et al., 2000). Significantly, in the context of the current study, PA has not been tested against these receptors. Moreover, there are studies, which suggest that exogenous PA may signal via a GPCR (Sliva et al., 2000), although micromolar concentrations are required.
In this study, we have shown that PA acts via a GPCR to regulate the p42/p44 MAPK pathway in HEK 293 cells. This response was ∼100 – 1000 fold more sensitive to various PA species compared with LPA. We conclude that PA might act on a receptor distinct from that binding LPA.
Alternatively, it is possible that the additional acyl side chain in PA binds to an exo-site on the LPA receptor or in the neighbouring lipid milieu to increase the overall potency for PA at this receptor.
Methods
Cell culture
HEK 293 cells were maintained in Minimal Essential Medium (MEM) containing 20% (v v−1) foetal calf serum (FCS) and placed in MEM for 24 h before experimentation.
p42/p44 MAPK assays
The phosphorylated forms of p42/p44 MAPK were detected by Western blotting cell lysates with anti-phospho-p42/p44 MAPK antibody. Immunoblotting was performed as described by us previously (Rakhit et al., 2000).
RT – PCR
First strand synthesis was carried out using 5 μg extracted total RNA and Superscript II reverse transcriptase. The PCR reaction for EDG transcripts was carried out using the following protocol: 94°C for 3 min and 30 cycles of 94°C for 1 min, 48°C for 1 min and 72°C for 2 min. This was followed by a final extension of 10 min at 72°C. The following reactions were performed using specific primers constructed to EDG2, 4 and 7. The EDG2 forward primer was 5′-TGG CTG CCA TCT CTA CTT CC-3′; reverse primer, 5′-AAC CAA TCC AGG AGT CCA GC-3′ (predicted size of product is 0.9 kB). The EDG4 forward primer was 5′-TGG TCA TCA TGG GCC AGT GC-3′; reverse primer, 5′-TAG TGG ACA GAC TCG CGG GT-3′ (predicted size of product is 0.955 kB). The EDG7 forward primer was 5′-TGA ATG AGT GTC ACT ATG ACA AGC-3′; reverse primer, 5′-TTT TAT TGC AGA CTG CAC CTT GGC-3′ (predicted size of product is 1 kB). The PCR reaction for amplification of the full open reading frame (∼1.5 kB) of PSP24 transcript was carried out using the following protocol: 95°C for 5 min and 35 cycles of 95°C for 1 min, 54°C for 30 s and 68°C for 3 min. The forward PCR primer was XbaI hPSP24 for: 5′-GCT CTA GAG CAC CAT GGT CTT CTC GGC AGT G-3′ and the reverse primer was HindIII hPSP24 rev: 5′-CCC AAG CTT GGG ATT CAC ACC ACC GTC CGA TG-3′. These primers were designed against human PSP24 from brain (AB030566).
Materials
All biochemicals, including pertussis toxin, suramin, PAs and LPA were from Sigma Chemical Co. (Dorset, U.K.). Cell culture supplies were from Life Technologies (Paisley, U.K.). Anti-phospho-p42/p44 MAPK antibodies from New England Biolabs (Hitchin, U.K.). Anti-p42 MAPK antibodies were Transduction Laboratories (Kentucky, U.S.A.). Reporter HRP-anti rabbit/mouse antibodies were from the Scottish Antibody Protection Unit (Carluke, U.K.).
Results
PA Stimulation of p42/p44 MAPK in HEK 293 cells
We have found that low concentrations (3 nM) of exogenous dioctanoyl PA stimulated p42/p44 MAPK activation in HEK 293 cells (Figure 1a). Moreover, the pre-treatment of these cells with pertussis toxin (0.1 μg ml−1 for 18 h substantially reduced (>90%) the dioctanoyl PA-dependent activation of p42/p44 MAPK (Figure 1a). Pertussis toxin did not affect the stimulation of p42/p44 MAPK by PMA (Figure 1a), which short circuits GPCRs by activating Raf via a PKC-dependent mechanism. The extent to which p42 MAPK was phosphorylated in response to PA exceeded that of p44 MAPK, which on some Western blots was barely detectable. Molecular species analysis revealed that the nature of the acyl chains (stearoyl/arachidonoyl, dipalmitoyl, dioctanoyl or dioleoyl) affected the potency of the response. For instance, dioctanoyl PA (C8:0) stimulated p42/p44 MAPK at 0.3 nM and 3 nM, with no activation observed at 30 nM at t=10 min cell stimulation (Figure 1b). Dipalmitoyl PA (C16:0) was without effect at 0.3 nM, but stimulated p42/p44 MAPK activation at 3 nM and 30 nM (Figure 1b). Dioleoyl PA (C18:1) and stearoyl-arachidonoyl (C18:0, 20:4) were not effective at 0.3 nM or 3 nM, but did stimulate p42/p44 MAPK at 30 nM (Figure 1b). Prolonged incubation with these PA species induces activation of p42/p44 MAPK at lower concentrations (data not shown). Rank order potencies are therefore: dioctanoyl PA>dipalmitoyl PA>stearoyl/arachidonoyl=dioleoyl PA. Furthermore, the pre-treatment of cells with suramin abolished the stimulation of p42/p44 MAPK by dioctanoyl PA and dipalmitoyl PA, reduced the effect of stearoyl/arachidonoyl PA, but had little effect against dioleoyl PA (Figure 1c). Rank order effects of suramin were therefore: dioctanoyl PA=dipalmitoyl PA>stearoyl/arachidonoyl>dioleoyl PA. The differential effect of suramin on the stimulatory effect of different PA species remains to be established. We recently showed that growth factor receptors are tethered to GPCRs (e.g. EDG1/PDGFβ receptor) and that this enables stronger stimulation of p42/p44 MAPK in HEK 293 cells (Alderton et al., 2001a). Thus, the ability of different molecular species of PA to induce activation of p42/p44 MAPK may be related to their ability to promote PA receptor-growth factor receptor signal complexes. Moreover, suramin can also block growth factor receptor signalling. Therefore, the differential effect of suramin on the stimulatory effect of different PA species may also be related to the ability of these lipid agonists to promote growth factor-PA receptor complexes. Further studies are required to validate this model. Suramin did not affect the stimulation of p42/p44 MAPK by PMA (Figure 1c).
Figure 1.
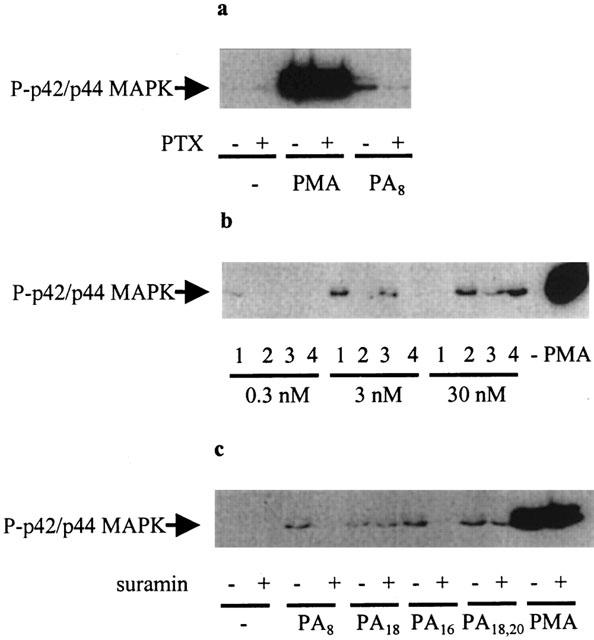
PA regulation of the p42/p44 MAPK pathway. HEK 293 cells were pre-treated with and without pertussis toxin (0.1 μg ml−1, 18 h) or suramin (50 μM, 10 min), prior to stimulation with PA (0.3 – 30 nM, 10 min) or PMA (1 μM, 10 min). Western blots probed with anti-phospho p42/p44 MAPK and anti-p42 MAPK antibodies showing (a) the effect of pertussis toxin on the stimulation of p42/p44 MAPK by dioctanoyl PA (3 nM) and PMA; (b) the concentration-dependent effect of dioctanoyl PA (1), dioleoyl PA (2), dipalmitoyl PA (3) and stearoyl/arachidonoyl PA (4); (c) the effect of suramin on the stimulation of p42/p44 MAPK by dioctanoyl PA (3 nM), dipalmitoyl PA (30 nM), stearoyl-arachidonoyl PA (30 nM), dioleoyl PA (30 nM) and PMA. Total p42 MAPK immunostaining showed equal loading of protein in each sample (data not shown). These are representative results of three independent experiments.
LPA stimulation of p42/p44 MAPK in HEK 293 cells
In contrast to PA, μM concentrations (1 – 50 μM) of LPA were required to stimulate p42/p44 MAPK activation in HEK 293 cells (Figure 2a). At a high concentration of LPA (100 μM), the extent to which p42/p44 MAPK can be activated by LPA was reduced, similar to that observed for dioctanoyl PA. The pre-treatment of cells with pertussis toxin abolished the LPA-dependent activation of p42/p44 MAPK (Figure 2b). We have previously shown that the pre-treatment of HEK 293 cells with suramin also reduced the stimulation of p42/p44 MAPK by LPA (Alderton et al., 2001b).
Figure 2.
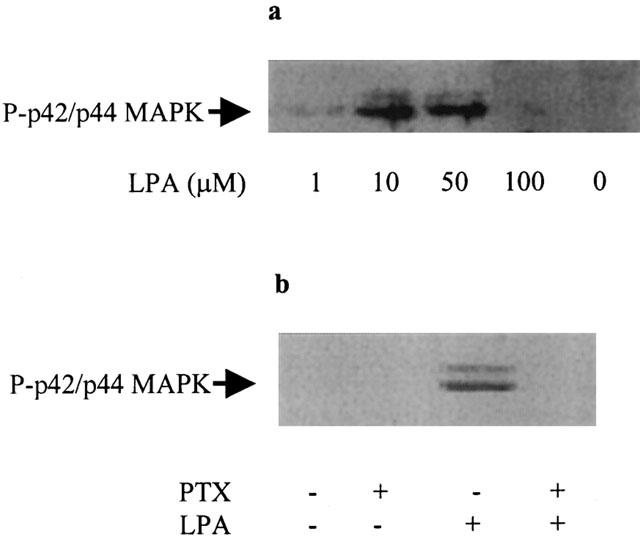
LPA regulation of the p42/p44 MAPK pathway. HEK 293 cells were pre-treated with and without pertussis toxin (0.1 μg ml−1, 18 h) prior to stimulation with LPA (1 – 100 μM, 10 min). Western blots probed with anti-phospho p42/p44 MAPK antibodies showing (a) the concentration-dependent effect of LPA; (b) the effect of pertussis toxin on the LPA- (5 μM) dependent stimulation of p42/p44 MAPK. These are representative results of three independent experiments.
RT – PCR
Using RT – PCR with gene specific primers, we have detected the presence of transcripts for the LPA binding receptor, EDG2 but not EDG4 or EDG7 in HEK 293 cells (Figure 3a). We have also detected the expression of PSP24, a novel GPCR that is outwith the EDG receptor family (Figure 3b). The identity of the PCR products was confirmed by nucleotide sequencing.
Figure 3.
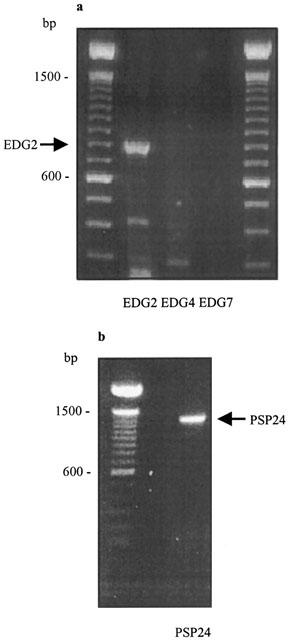
RT – PCR of EDG2, EDG4, EDG7 and PSP24. (a) RT – PCR of EDG2 but not EDG4 or EDG7 transcripts. (b) RT – PCR of PSP24 transcripts.
Discussion
A major finding of this study was that PA stimulates the activation of the p42/p44 MAPK pathway in HEK 293 cells via a pertussis toxin sensitive mechanism.
Since pertussis toxin is an inhibitor of Gi/o signalling, we propose that PA can bind to a GPCR to induce the activation of the p42/p44 MAPK pathway. These findings are supported by results showing that suramin (which blocks GPCR and growth factor receptor signal signal transduction) also reduced the effect of PA on p42/p44 MAPK, the extent of which was dependent on the molecular species of PA.
Comparison with LPA, a GPCR agonist that binds to EDG2, which is expressed in HEK 293 cells, shows that the different molecular species of PA are ∼100 – 1000 fold more effective than LPA at stimulating the p42/p44 MAPK pathway. If the binding affinities for LPA and PA at a common receptor were identical, then PA would have to induce 100 – 1000 fold amplification of the p42/p44 MAPK pathway compared with LPA.
These differences in the degree of amplification between PA and LPA cannot be explained by full and partial agonism respectively. Furthermore, both agonists appear to activate p42/p44 MAPK via a common Gi/o-dependent mechanism. Thus, changes in sensitivity are unlikely to be due to activation of distinct signalling pathways regulating p42/p44 MAPK at different receptor occupancies.
There are two possibilities that might account for the data. First, PA might bind to a high-affinity receptor distinct from the LPA receptor. In this regard, we have shown that PSP24 is expressed in HEK 293 cells. Second, both PA and LPA may bind to the same receptor, but that the additional acyl group in PA binds to a second site, through a hydrophobic interaction. The ecto-site binding would effectively clamp PA to the receptor and increase overall potency. This site may be on the receptor or in the lipid milieu surrounding the receptor. Our proposal has precedent with the long acting β-adrenoceptor agonists (e.g. salmeterol). Therefore, the findings of this study are important for two reasons. First, the data demonstrate that PA is a GPCR agonist that can activate a major mitogenic signalling pathway in mammalian cells. In addition, the 100 – 1000 fold higher potency of PA in stimulating p42/p44 MAPK compared with LPA, suggests that de-acylation of PA to LPA by soluble PLA2 would represent an effective mechanism for terminating the action of PA at a common receptor.
Second, the possibility that the acyl side chain might increase potency by clamping PA to an exo-site on or near the LPA receptor could be very instructive in terms of rationale drug design. Thus, it may be possible to develop PA derivatives with diacyl side chains that are very high affinity LPA receptor antagonists that could be usefully exploited to prevent angiogenesis (which is stimulated by LPA) and therefore tumour growth.
Acknowledgments
This study was supported by grants from The Biotechnology and Biological Sciences Research Council and Wellcome Trust and Dr S. Pyne is a Wellcome Trust Senior Fellow.
Abbreviations
- EDG
endothelial differentiation gene
- GPCR
G-protein coupled receptor
- HEK 293 cells
Human embryonic kidney 293 cells
- LPA
lysophosphatidic acid
- p42/p44 MAPK
p42/p44 mitogen-activated protein kinase
- PA
phosphatidic acid
- PMA
phorbol 12-myristate 13-acetate
- S1P
sphingosine 1-phosphate
References
- ALDERTON F., RAKHIT S., KONG K-C., PALMER T., SAMBI B., PYNE S., PYNE N.J. Tethering of the platelet-derived growth factor β receptor to G-protein-coupled receptors: A novel platform for integrative signalling by these receptor classes in mammalian cells. J. Biol. Chem. 2001a;276:28578–28585. doi: 10.1074/jbc.M102771200. [DOI] [PubMed] [Google Scholar]
- ALDERTON F., DARROCH P., SAMBI B., MCKIE A., AHMED I.S., PYNE N.J., PYNE S. G-protein coupled receptor stimulation of the p42/p44 mitogen activated protein kinase pathway is attenuated by lipid phosphate phosphatases 1, 1a and 2 in HEK 293 cells. J. Biol. Chem. 2001b;276:13452–13460. doi: 10.1074/jbc.M006582200. [DOI] [PubMed] [Google Scholar]
- AN S., BLEU T., HALLMARK O.G., GOETZL E.J. Characterization of a novel subtype of human G-protein coupled receptor for lysophosphatidic acid. J. Biol. Chem. 1998;273:7906–7910. doi: 10.1074/jbc.273.14.7906. [DOI] [PubMed] [Google Scholar]
- FISCHER D.J., LILIOM K., GUO Z., NUSSER N., VIRAG T., MURAKAMI-MUROFUSHI K., KOBAYASHI S., ERICKSON J.R., SUN G., MILLER D.D., TIGYI G. Naturally occurring analogs of lysophosphatic acid elicit different cellular responses through selective activation of multiple receptor subtypes. Mol. Pharmacol. 1998;54:979–988. doi: 10.1124/mol.54.6.979. [DOI] [PubMed] [Google Scholar]
- GUO Z., LILIOM K., FISCHER D.J., BATHURST I.C., TOMEI L.D., KIEFER M.C., TIGYI G. Molecular cloning of a high affinity receptor for the growth factor-like lipid mediator lysophosphatidic acid from Xenopus oocytes. Proc. Natl. Acad. Sci. (U.S.A.). 1996;93:14367–14372. doi: 10.1073/pnas.93.25.14367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HECHT J.H., WEINER J.A., POST S.R., CHUN J. Ventricular gene zone-1 (vzg-1) encodes a lysophosphatidic acid receptor expressed in neurogenic regions of the developing cerebral cortex. J. Cell. Biol. 1996;135:1071–1083. doi: 10.1083/jcb.135.4.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAWASAWA Y., KUME K., IZUMI T., SHIMIZU T. Mammalian PSP24s (α and β isoforms) are not responsive to lysophosphatidic acid in mammalian expression systems. Biochem. Biophys. Res. Commun. 2000;276:957–964. doi: 10.1006/bbrc.2000.3570. [DOI] [PubMed] [Google Scholar]
- PYNE S., PYNE N.J. Sphingosine 1-phosphate signalling in mammalian cells. Biochem. J. 2000;349:385–402. doi: 10.1042/0264-6021:3490385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAKHIT S., PYNE S., PYNE N.J. The platelet-derived growth factor stimulation of p42/p44 mitogen-activated protein kinase in airway smooth muscle involves a G-protein-mediated tyrosine phosphorylation of Gab1. Mol. Pharmacol. 2000;58:413–420. doi: 10.1124/mol.58.2.413. [DOI] [PubMed] [Google Scholar]
- RIZZO M.A., SHOME K., WATKINS S.C., ROMERO G. The recruitment of Raf-1 to membranes is mediated by direct interaction with phosphatidic acid and is independent of association with Ras. J. Biol. Chem. 2000;275:23911–23918. doi: 10.1074/jbc.M001553200. [DOI] [PubMed] [Google Scholar]
- SLIVA D., MASON R., XIAO H., ENGLISH D. Enhancement of the migration of metastatic human breast cancer cells by phosphatidic acid. Biochem. Biophys. Res. Comm. 2000;268:471–479. doi: 10.1006/bbrc.2000.2111. [DOI] [PubMed] [Google Scholar]