

Abstract
The effect of activation of protein kinase C (PKC) or adenylyl cyclase on release of glutamate has been investigated in a perfused slice preparation from the rat caudal trigeminal nucleus. Stimulation of PKC by phorbol 12-myristate 13-acetate (PMA) produced a concentration-dependent increase in K+-evoked release of [2H]-glutamate (maximum increase 45%, EC50 11.8 nM), but in the presence of gabapentin (30 μM) the facilitation of release was blocked. The adenylyl cyclase activator forskolin (FSK) also induced a concentration-dependent increase in K+-evoked release of [3H]-glutamate (maximum increase 36%, EC50 2.4 μM), and again this facilitatory effect was blocked by gabapentin (30 μM). We suggest that these results may be of relevance to the antihyperalgesic properties of gabapentin, in conditions where concomitant release of substance P and CGRP produces activation of PKC and adenylyl cyclase respectively.
Keywords: gabapentin, glutamate release, trigeminal nucleus, protein kinase C, adenylyl cyclase
Introduction
Gabapentin (Neurontin®), which was originally designed as an anti-epileptic drug and was approved for this use in 1993, has more recently been shown to be effective in reducing the pain of neuropathy or neuralgia in man (Backonja et al., 1998; Rowbotham et al., 1998). These findings have been supported by the results from preclinical models (e.g. Hwang & Yaksh, 1997; Field et al., 1999) and have created much interest in the pharmacology of gabapentin. Although the binding site for gabapentin in the brain has been identified as the α2δ-subunit of voltage-gated calcium channels (Gee et al., 1996), it is not yet clear how this finding relates to the antihyperalgesic, and other properties of the drug.
We have recently reported the use of a brainstem slice, containing the caudal sensory subnucleus of the spinal trigeminal nucleus (Sp5C), as a model for testing the effects of gabapentin on release of sensory transmitters (Maneuf et al., 2000). We showed that while gabapentin did not reduce the normal level of K+-evoked release of [3H]-glutamate, it was effective in inhibiting a facilitatory effect of substance (SP) or calcitonin gene related peptide (CGRP) on glutamate release. The present work investigated the consequence of direct activation of the second-messenger pathways linked to NK1 and CGRP receptors, on glutamate release in the Sp5C slice and the action of gabapentin on this effect. Phorbol 12-myristate 13-acetate (PMA) was used to activate protein kinase C (PKC), and forskolin (FSK) to activate adenylyl cyclase.
Methods
Consecutive coronal sections (400 μm) running caudally from 14 mm posterior to bregma (Paxinos & Watson, 1986) were cut from the brainstem of male Hooded Lister rats (250 g) using a McIllwain tissue chopper. Slices 1 mm in diameter containing Sp5C were pooled and incubated in aerated (95% O2/5% CO2) artificial cerebrospinal fluid (aCSF, composition mM: NaCl, 118; KCl, 4.8; CaCl2, 1.3; MgSO4, 1.2; NaHCO3, 25; KH2 PO4, 1.2; ascorbic acid, 0.6; glucose, 11; captopril, bestatin and phosphoramidon, all 0.01) containing 0.1 μM [3H]-glutamate for 30 min at room temperature (pH 7.4).
Slices were transferred to a Brandel SF-20 superfusion system (2 per chamber) and washed for 40 min with aCSF at 1 ml min−1) prior to collection of aliquots every 5 min (flow rate now 0.5 ml min−1) for the next 60 min. Thirty minutes after starting collection, a single 5-min pulse of aCSF containing a 24 mM excess of K+ (as KCl) was applied to evoke the release of [3H]-glutamate. Drugs or vehicle were added 5 min before, and during the high K+ pulse in the presence of 0.4% BSA. At the end of the experiment slices were solubilized in 0.5 ml dimethylsulphoxide (DMSO) in 4.5 ml of scintillation fluid (Ultima Gold MV, Packard Bioscience), and the radioactivity in both the solubilized slices and perfusates (0.5 ml volume) counted overnight in a liquid scintillation counter.
[3H]-glutamate release was expressed as a fractional rate, with the radioactivity released during a 5-min time bin divided by the total radioactivity in the slice at the beginning of that period. The amplitude of the peak of glutamate release in the presence of test treatment was expressed as a percentage of that in parallel controls. Each mean data point is the result of 4 – 6 separate experiments, with four replicates per experiment. Statistical analysis was performed using either a non-linear regression analysis (curve fit in Graphpad, Prism 2.01) or analysis of variance (ANOVA) followed by a Tukey's multiple-comparison test. PMA and FSK (Sigma, Poole, U.K.) were dissolved at 10 mM in DMSO and diluted as required. Gabapentin was dissolved in water and diluted to concentration in aCSF and was obtained from Pfizer Global R&D, Cambridge, U.K.
Results
The effect of PMA on the K+-evoked release of [3H]-glutamate was determined from 4 – 6 experiments for each concentration of PMA. In this series of experiments the basal fractional rate of release was 3.54±0.22%, which was increased to 6.65±0.42% after the high K+-stimulus. The addition of PMA for 10 min, starting 5 min before the increase in K+ caused a concentration-dependent increase in the evoked release of [3H]-glutamate, without affecting the basal rate. The maximum increase in the K+-evoked release was 44.5% above the baseline level of evoked release (control, Figure 1) with a value of EC50 for this effect of 11.8 nM (95% c.l.: 5.3, 25.7 nM), derived by curve fitting.
Figure 1.
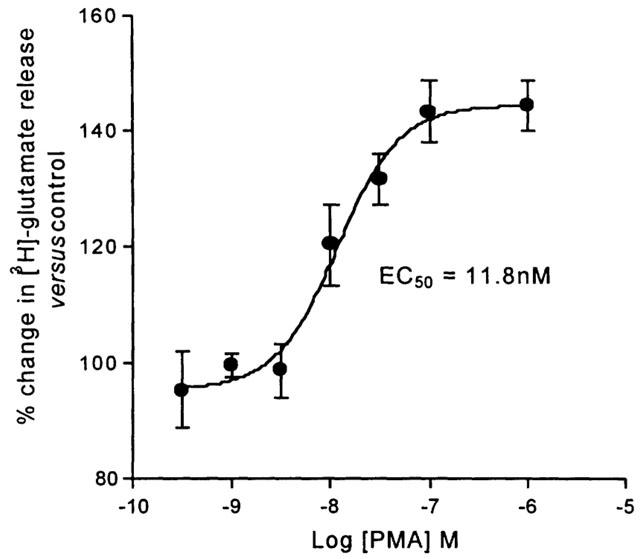
The facilitatory effect of the PKC activator phorbol 12-myristate 13-acetate (PMA) on K+-evoked [3H]-glutamate release from the Sp5C slice. Release is expressed as a percentage of the control (without PMA, 100%). Each data point is the mean from 4 – 6 different experiments±one s.e.mean.
When gabapentin was co-applied with a sub-maximal concentration of PMA (30 nM), the facilitatory effect of the phorbol ester was inhibited. At 30 μM, gabapentin blocked the increase in the evoked release of [3H]-glutamate (103±3.45% of control versus 131.6±4.45% for PMA alone, P<0.01). At 10 μM the effect of gabapentin was less, but a significant inhibition was still achieved (112.8±5.9% of control, P<0.05, Figure 2).
Figure 2.
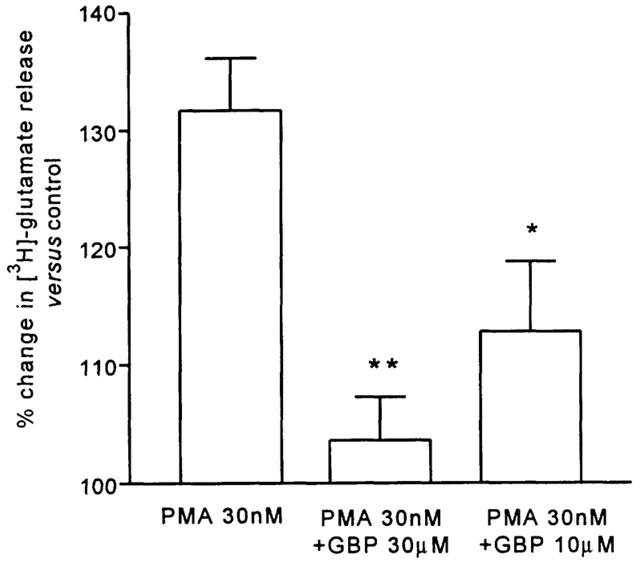
The effect of gabapentin (GBP) at 10 or 30 μM on the increase in the evoked release of glutamate from the Sp5C slice by phorbol 12-myristate 13-acetate (PMA) 30 nM. Histograms show mean values from 4 – 6 observations±s.e.mean (**P<0.01 and *P<0.05, Tukey's post hoc test).
The presence of forskolin (FSK) in the aCSF also caused a concentration-dependent increase in the level of K+-evoked release of [3H]-glutamate above the control level (6.00±0.60%, compared to the basal fractional rate of release of 2.83±0.27%). From curve fitting the values for EC50 and maximum for the effect of FSK were 2.44 μM (95% c.l.: 0.47, 12.54 μM) and 136.1% (Figure 3).
Figure 3.
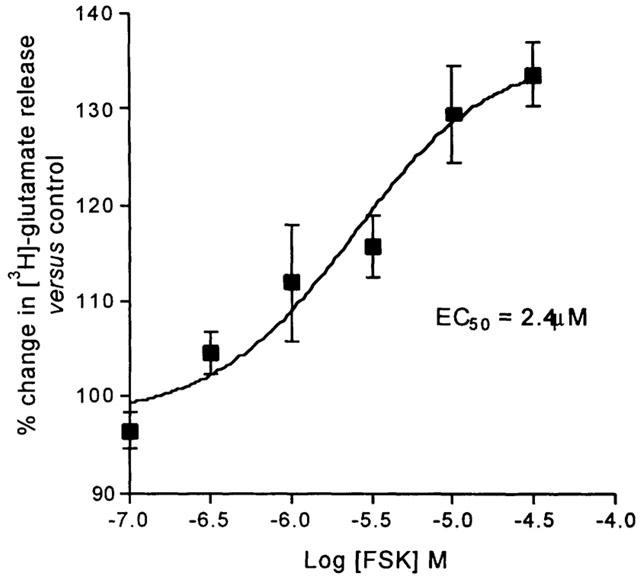
The facilitatory effect of the adenylyl cyclase activator forskolin (FSK) on K+-evoked [3H]-glutamate release from the Sp5C slice. Release is expressed as a percentage of the control (without FSK). Each data point is the mean from 4 – 6 different experiments±one s.e.mean.
Thirty μM gabapentin co-applied with 10TM FSK blocked the facilitation of K+-evoked release of [3H]-glutamate (93.63±5% versus 133.4±7.57% in the presence of FSK alone, P<0.001); gabapentin at 10 μM also produced a significant reduction of the FSK-facilitated release (109.1±7.5%, P<0.05, Figure 4).
Figure 4.
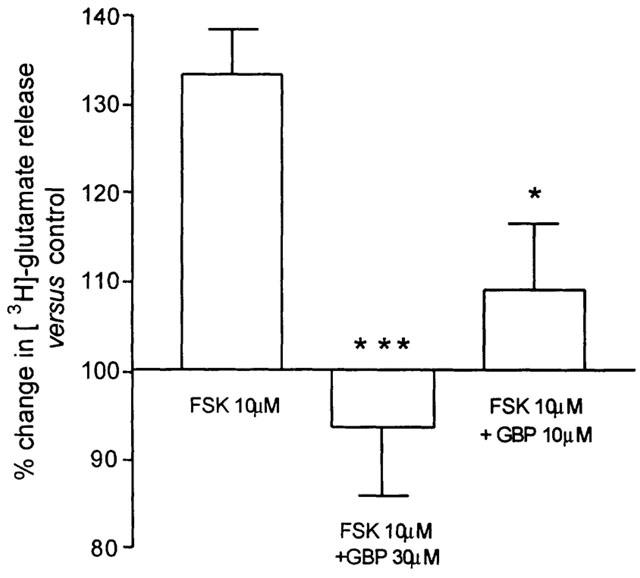
The effect of gabapentin (GBP) at 10 or 30 μM on the increase in the evoked release of glutamate from the Sp5C slide by forskolin (FSK) 10 μM. Histograms show mean values from 4 – 6 observations±one s.e.mean (***P<0.001 and *P<0.05, Tukey's post hoc test).
Discussion
The evoked release of glutamate from the Sp5C slice is increased in the presence of SP or CGRP and gabapentin is able to block this facilitatory action, although the drug has no effect on the normal level of K+-evoked [3H]-glutamate release (Maneuf & Yaksh, 2000). We have taken these findings to correlate with the ability of gabapentin to reduce nocifensive responses in models of hyperalgesia and allodynia, where it has no effect against ‘acute pain' (Hwang & Yaksh, 1997; Field et al., 1997; 1999). The present experiments extended our first study, and approached the mechanism of action of gabapentin, by investigating the effect on direct activation of the second-messenger systems associated with NK1 and CGRP receptors.
We have now found that stimulation of PKC by PMA, or of adenylyl cyclase using FSK produces an increase in the level of K+-evoked release of [3H]-glutamate to the same extent as was seen with the peptide agonists. We did not try the effect of PMA and FSK together, to test for a larger facilitation of release, because we had previously seen that the effects of SP and CGRP in combination were not additive (Maneuf & McKnight, unpublished). In either case the facilitated component of release is blocked by gabapentin, suggesting that the original receptor-mediated increases were not transduced by G-protein subunits directly, but may be the consequences of the actions of second-messenger activated kinases. Increased release of transmitters by phorbol esters has been reported for many neuronal systems, and in the context of the present experiments the reported enhancement of glutamate release from spinal cord synaptosomes (Shinomura et al., 1999) is of relevance. In this latter study FSK had no effect, although the release of glutamate is reported to be increased by activation of adenylyl cyclase or protein kinase A (PKA) in other sites, including hippocampus and cortex (see e.g. Bouron & Reuter, 1999). Gabapentin has been shown to inhibit K+-evoked release of glutamate in slice preparations from both hippocampus and cortex (Dooley et al., 2000).
The mechanism of the facilitatory effects of PMA and FSK on release of glutamate cannot be readily arrived at from such crude experiments as ours, but some speculation may be appropriate. The very fact that the effect is sensitive to block by gabapentin, invites the conclusion that an increase in influx through voltage-dependent Ca2+-channels is involved. This conclusion has any viability so long as we accept that the site of action of gabapentin is the α2-δ-subunit, and that the interaction is of functional relevance to the conductance properties of the α1 subunit. Although we have not gone so far in the present study, we have shown that the facilitatory effect of SP in Sp5C is also blocked by S-(+)-isobutylgaba (pregabalin), but not by the R-(-) antipode (Maneuf et al., 2000). This pattern of stereospecificity is the same as for displacement of specific binding of gabapentin in brain (Taylor et al., 1993), which is purportedly to the α2δ-subunit.
That the release of glutamate must be increased, here by the activation of PKC or adenylyl cyclase (and so PKA), before the action of gabapentin is seen presumably points to a requirement for phosphorylation at some key intracellular site. The only potential phosphorylation site in α2δ is extracellular (Gurnett et al., 1996), leaving cytoplasmic β-subunits, which have multiple consensus sites for phosphorylation, or the cytoplasmic loops of the α1 subunits as the potential substrates (Isom et al., 1994). How the binding of gabapentin to the extracellular domain of α2δ (Brown & Gee, 1998) could be translated to the functional effect, must remain the subject of conjecture.
It remains to be established whether other possible mechanisms could underlie the effects of gabapentin, and in particular the recent report of a subtype-selective agonist action of gabapentin at the GABAB receptor (Ng et al., 2001) cannot be ignored. Future studies on the pharmacology of the effects of gabapentin alone will shed light here. For the GABAB receptor as the site of action, we can say that the effect of baclofen in the Sp5C is quite different in that the ‘unfacilitated' release of glutamate is powerfully inhibited by this agent (Maneuf & McKnight, unpublished).
In conclusion, we have found that activation of PKC or adenylyl cyclase increases the level of K+-evoked release of glutamate in the Sp5C slice, and that gabapentin restores the release to the control level. It is clear that these findings are consistent with our earlier observations that SP and CGRP also produce a gabapentin-sensitive increase in release in Sp5C (Maneuf et al., 2000). We proposes that the increase in glutamate release, and the conferring of its sensitivity to gabapentin, relies on phosphorylation of a protein subunit that regulates Ca2+-channel function. There is ample evidence to implicate PKC and PKA in the hyperalgesia and/or allodynia in tests where gabapentin is active (e.g. Ahlgren & Levine, 1994; Aley & Levine, 1999; Cunha et al., 1999; Hua & Yaksh, 1999; Ohsawa & Kamei, 1999). It is tempting, therefore, to speculate that such a putative mechanism explains the antihyperalgesic action of the drug.
Abbreviations
- CGRP
calcitonin gene-related peptide
- FSK
forskolin
- GBP
gabapentin
- PKC
protein kinase C
- PMA
phorbol 12-myristate 13-acetate
- SP
substance P
- Sp5C
caudal sensory subnucleus of the spinal trigeminal nucleus
References
- AHLGREN S.C., LEVINE J.D. Protein kinase C inhibitors decrease hyperalgesia and C-fiber hyperexcitability in the streptozotocin-diabetic rat. J. Neurophysiol. 1994;72:684–692. doi: 10.1152/jn.1994.72.2.684. [DOI] [PubMed] [Google Scholar]
- ALEY K.O., LEVINE J.D. Role of protein kinase A in the maintenance of inflammatory pain. J. Neurosci. 1999;19:2181–2186. doi: 10.1523/JNEUROSCI.19-06-02181.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BACKONJA M., BEYDOUN A., EDWARDS K.R., SCHWARTZ S.L., FONSECA V., HES M., LAMOREAUX L., GAROFALO E. Gabapentin for the symptomatic relief of painful neuropathy in patients with diabetis mellitus: a randomized controlled trial. J. Am. Med. Assoc. 1998;280:1831–1836. doi: 10.1001/jama.280.21.1831. [DOI] [PubMed] [Google Scholar]
- BROWN J.P., GEE N.S. Cloning and deletion mutagenesis of the α2Λ calcium channel subunit from porcine cerebral cortex. Expression of a soluble form of the protein that retains [3H]gabapentin binding activity. J. Biol. Chem. 1998;273:25458–25465. doi: 10.1074/jbc.273.39.25458. [DOI] [PubMed] [Google Scholar]
- BOURON A., REUTER H. The D1 dopamine receptor agonist SKF-38393 stimulates the release of glutamate in the hippocampus. Neuroscience. 1999;94:1063–1070. doi: 10.1016/s0306-4522(99)00352-8. [DOI] [PubMed] [Google Scholar]
- CUNHA F.Q., TEIXEIRA M.M., FERREIRA S.H. Pharmacological modulation of secondary mediator systems-cyclic AMP and cyclic GMP-on inflammatory hyperalgesia. Br. J. Pharmacol. 1999;127:671–678. doi: 10.1038/sj.bjp.0702601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOOLEY D.J., MIESKE C.A., BOROSKY S.A. Inhibition of K+-evoked glutamate release from rat neocortical and hippocampal slices by gabapentin. Neurosci. Lett. 2000;280:107–110. doi: 10.1016/s0304-3940(00)00769-2. [DOI] [PubMed] [Google Scholar]
- FIELD M.J., MCCLEARY S., HUGHES J., SINGH L. Gabapentin and pregablin, but not morphine and amitriptyline, block both static and dynamic components of mechanical allodynia induced by streptozocin in the rat. Pain. 1999;80:391–398. doi: 10.1016/s0304-3959(98)00239-5. [DOI] [PubMed] [Google Scholar]
- FIELD M.J., OLES R.J., LEWIS A.S., MCCLEARY S., HUGHES J., SINGH L. Gabapentin (neurontin) and S-(+)-3-isobutylgaba represent a novel class of selective antihyperalgesic agents. Br. J. Pharmacol. 1997;121:1513–1522. doi: 10.1038/sj.bjp.0701320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GEE N.S., BROWN J.P., DISSANAYAKE V.U., OFFORD J., THURLOW R., WOODRUFF G.N. The novel anticonvulsant drug, gabapentin (Neurontin), binds to the α2Λ subunit of a calcium channel. J. Biol. Chem. 1996;271:5768–5776. doi: 10.1074/jbc.271.10.5768. [DOI] [PubMed] [Google Scholar]
- GURNETT C.A., DE WAARD M., CAMPBELL K.P. Dual function of the voltage-dependent Ca2+ channel α2Λ subunit in current stimulation and subunit interaction. Neuron. 1996;16:431–440. doi: 10.1016/s0896-6273(00)80061-6. [DOI] [PubMed] [Google Scholar]
- HUA X.Y., YAKSH T.L. Inhibition of spinal protein kinase C reduces nerve injury-induced tactile allodynia in neuropathic rats. Neurosci. Lett. 1999;276:99–102. doi: 10.1016/s0304-3940(99)00818-6. [DOI] [PubMed] [Google Scholar]
- HWANG J.H., YAKSH T.L. Effect of subarachnoid gabapentin on tactile-evoked allodynia in a surgically induced neuropathic pain model in the rat. Reg. Anesth. 1997;22:249–256. doi: 10.1016/s1098-7339(06)80010-6. [DOI] [PubMed] [Google Scholar]
- ISOM L.L., DE JONGH K.S., CATTERALL W.A. Auxiliary subunits of votage-gated ion channels. Neuron. 1994;12:1183–1194. doi: 10.1016/0896-6273(94)90436-7. [DOI] [PubMed] [Google Scholar]
- MANEUF Y.P., HUGHES J., MCKNIGHT A.T. Gabapentin inhibits substance P- and calcitonin gene-related peptide-facilitated K+-evoked release of [3H]-glutamate from rat caudal trigeminal nudeus slices. Soc. Neurosc. Abstr. 2000;26:722.9. [Google Scholar]
- OHSAWA M., KAMEI J. Possible involvement of protein kinase C in thermal allodynia and hyperalgesia in diabetic mice. Eur. J. Pharmacol. 1999;372:221–228. doi: 10.1016/s0014-2999(99)00228-9. [DOI] [PubMed] [Google Scholar]
- NG G.Y.K., BERTRAND S., SULLIVAN R., ETHIER N., WANG J., YERGEY J., BELLEY M., TRIMBLE L., BATEMAN K., ALDER L., SMITH A., MCKEMAN R., METTERS K., O'NEILL G.P., LACAILLE J.-C., HERBERT T.E. γ-aminobutyric acid type B receptors with specific heterodimer composition and postsynaptic actions in hippocampal neurons are targets of anticonvulsant gabapentin action. Mol. Pharmacol. 2001;59:144–152. [PubMed] [Google Scholar]
- PAXINOS G., WATSON C. The Rat Brain in Stereotaxic Coordinates 1986Sydney: Academic Press; Second edition [Google Scholar]
- ROWBOTHAM M., HARDEN N., STACEY B., BERNSTEIN P., MAGNUS-MILLER L. Gabapentin for the treatment of postherpetic neuralgia: a randomized controlled trial. J. Am. Med. Assoc. 1998;280:1837–1842. doi: 10.1001/jama.280.21.1837. [DOI] [PubMed] [Google Scholar]
- SHINOMURA T., NAKAO S., ADACHI T., SHINGU K. Clonidine inhibits and phorbol acetate activates glutamate release from rat spinal cord synaptosomes. Anaesth. Analg. 1999;88:1401–1405. doi: 10.1097/00000539-199906000-00037. [DOI] [PubMed] [Google Scholar]
- TAYLOR C.P., VARTANIAN M.G., YUEN P.W., BIGGE C., SUMAN-CHAUHAN N., HILL D.R. Potent and stereospecific anticonvulsant activity of 3-isobutyl GABA relates to in vitro binding at a novel site labelled by tritiated gabapentin. Epilepsy Res. 1993;14:11–15. doi: 10.1016/0920-1211(93)90070-n. [DOI] [PubMed] [Google Scholar]