

Abstract
Inhibitors of the angiotensin converting enzyme (ACE) have been shown to exert their cardioprotective actions through a kinin-dependent mechanism. ACE is not the only kinin degrading enzyme in the rat heart.
Since aminopeptidase P (APP) has been shown to participate in myocardial kinin metabolism to the same extent as ACE, the aims of the present study were to investigate whether (a) inhibition of APP leads to a reduction of myocardial infarct size in a rat model of acute ischaemia and reperfusion, (b) reduction of infarct size is mediated by bradykinin, and (c) a combination of APP and ACE inhibition leads to a more pronounced effect than APP inhibition alone.
Pentobarbital-anaesthetized rats were subjected to 30 min left coronary artery occlusion followed by 3 h reperfusion. The APP inhibitor apstatin, the ACE-inhibitor ramiprilat, or their combination were administered 5 min before ischaemia. Rats receiving HOE140, a specific B2 receptor antagonist, were pretreated 5 min prior to enzyme inhibitors. Myocardial infarct size (IS) was determined by tetrazolium staining and expressed as percentage of the area at risk (AAR).
IS/AAR% was significantly reduced in rats that received apstatin (18±2%), ramiprilat (18±3%), or apstatin plus ramiprilat (20±4%) as compared with those receiving saline (40±2%), HOE (43±3%) or apstatin plus HOE140 (49±4%).
Apstatin reduces IS in an in vivo model of acute myocardial ischaemia and reperfusion to the same extent than ramiprilat. Cardioprotection achieved by this selective inhibitor of APP is mediated by bradykinin. Combined inhibition of APP and ACE did not result in a more pronounced reduction of IS than APP-inhibition alone.
Keywords: Apstatin, aminopeptidase P, APP, bradykinin, infarct size, ischaemia, ramiprilat, angiotensin converting enzyme, ACE
Introduction
Bradykinin is a small vasoactive peptide which is involved in a variety of biological processes. Kinins participate in the regulation of blood pressure, renal and cardiac functions which has been studied intensively by using specific kinin receptor antagonists (Bhoola et al., 1992). Because of the very short half-life of kinins their specific efficiency depends on local formation by tissue kallikrein-kinin systems and is restricted by the spectrum and potency of local kinin degrading enzymes. A number of peptidases have been identified to possess kininase activity the inhibition of which increases the availability and effectiveness of kinins. There is increasing evidence that part of the cardiovascular actions of angiotensin converting enzyme (ACE) inhibitors, including reduction of infarct size, improvement of performance and energetic state of ischaemic myocardium, and prevention of ventricular hypertrophy and remodelling are due to the potentiation of the effects of kinins (reviewed by Dendorfer et al., 1999). ACE is not the only kinin-degrading enzyme and recent studies focus on the modulation of other peptidases. In rats, aminopeptidase P (APP) has been shown to participate in myocardial kinin metabolism to the same extent as ACE whereas neutral endopeptidase 24.11 (NEP) only plays a minor role (Dendorfer et al., 1997). Inhibition of APP may therefore be a sufficient means to potentiate cardiovascular effects of kinins. Indeed, the potentiation of kinin-induced coronary vasodilation during APP-inhibition in the isolated perfused rat heart was equivalent to that provoked by ACE inhibition (Dendorfer et al., 2000). Likewise, infusion of the selective APP-inhibitor apstatin in anaesthesized rats leads to a marked potentiation of the bradykinin-induced decrease in mean arterial blood pressure (Kitamura et al., 1999). In order to investigate further whether inhibition of APP has similar cardioprotective effects as inhibition of ACE, we now studied the influence of apstatin on the reduction of myocardial infarct size in an in situ rat model of acute ischaemia and reperfusion. Furthermore, the aims of the present study were to investigate whether cardioprotection by apstatin is mediated by bradykinin and whether a combination of APP and ACE inhibition leads to a more pronounced effect than inhibition of either one enzyme alone.
Methods
Surgical preparations
The present study has been carried out in accordance with the guide for the care and use of laboratory animals as adopted by the ‘Ministerium für Natur und Umwelt des Landes Schleswig-Holstein, Germany'. Male Wistar rats (270 – 310 g body weight, Charles River, Sulzfeld, Germany) were anaesthetized with pentobarbitone (100 mg kg−1, i.p.), tracheotomized, and ventilated with room air (tidal volume: 10 ml kg−1, 50 strokes per minute) enriched with oxygen to maintain arterial oxygen tension in the normal range. The left jugular vein was cannulated in order to compensate for fluid loss (NaCl 0.9%, 4 ml kg−1 h−1), to maintain anaesthesia with pentobarbitone (150 μg kg−1 min−1) or to inject drugs. Another catheter was placed in the left carotid artery to measure mean arterial blood pressure (MAP). Core temperature was continuously monitored and maintained at 37.0 – 37.7°C. A lateral thoracotomy was performed, and a 6-0 suture was looped under the left descending coronary artery for later induction of coronary artery occlusion (CAO).
Experimental protocols
After surgical preparation, rats were allowed 10 min for stabilization before baseline haemodynamics were recorded. Rats were randomized to one of six protocols. All protocols included 30 min of CAO followed by 180 min of reperfusion. This was preceded by application of drugs, 5 min prior to CAO. The different groups received either saline, the ACE inhibitor ramiprilat (50 μg kg−1), the aminopeptidase P inhibitor apstatin (1 mg kg−1), apstatin plus the B2-receptor antagonist HOE140 (500 μg kg−1), HOE140 alone, or apstatin plus ramiprilat. When applied together with apstatin, HOE140 was administered intravenously 5 min prior to the enzyme inhibitor. One hundred i.u. heparin were given 1 min before CAO. Doses used for apstatin (Kitamura et al., 1999), ramiprilat (Hartman et al., 1993) and HOE140 (Gohlke et al., 1998) have been shown to be effective previously.
Measurement of area at risk and infarct size
After 30 min of CAO and 180 min of reperfusion, hearts were removed and the aorta was quickly cannulated. The coronary artery ligature was retied, and hearts were perfused with black Chinese ink at a constant pressure (80 mmHg) to stain the perfused myocardium black, whereas the area at risk (AAR) remained unstained. Atria and the right ventricle were removed and the left ventricle (LV) including the septum was cut into slices (1 mm thickness) from apex to base. Slices were then incubated for 30 min at room temperature in 2,3,5-triphenyltetrazolium chloride (1% in 0.1 M phosphate buffer, pH 7.4), which stained viable tissue red and so delineated the pale area of infarct size (IS). Areas of LV, AAR, and IS were quantified using computer assisted planimetry.
Substances
Apstatin, ramiprilat, and HOE140 were generously supplied by Aventis Pharma (Frankfurt, Germany). 2,3,5- triphenyltetrazolium chloride was purchased from Sigma (Deisenhofen, Germany), heparin as Liquemin® N 25 000 from Roche (Grenzach-Wyhlen, Germany) and salts from Merck (Darmstadt, Germany). Pentobarbitone was obtained from the pharmacy of the Medical University of Lübeck.
Calculations and statistics
All quantitative data are given as means±s.e.mean of five independent experiments. Parameters of infarct sizes were compared among the treatment groups using a one-way ANOVA with Bonferroni's correction. Haemodynamics were tested for differences between the groups by using a two-way ANOVA for repeated measurements. Temporal changes in haemodynamic parameters within one group were tested with a one-way ANOVA for repeated measurements followed by paired t-tests with Bonferroni's correction when differences were significant. Differences were considered as being statistically significant at an error level of P<0.05.
Results
Haemodynamics
Data for MAP and heart rate (HR) are listed in Table 1. There were no differences in MAP or HR among all groups at the different time points of the experiment. Inhibition of APP by apstatin, inhibition of ACE by ramiprilat or B2 receptor blockade by HOE140 did not alter MAP or HR, while the combined enzyme inhibition with apstatin and ramiprilat significantly decreased MAP without affecting HR if compared with the baseline haemodynamics. Occlusion of the left coronary artery significantly decreased MAP in all groups except the one treated with apstatin and ramiprilat. In all groups MAP decreased slightly throughout the experiment which might be indicative for developing heart failure.
Table 1.
Systemic haemodynamics in rats
Area at risk
The ratio between AAR and LV did not significantly differ between all groups (mean: 67%), indicating a constant placement of the coronary ligature (Figure 1).
Figure 1.
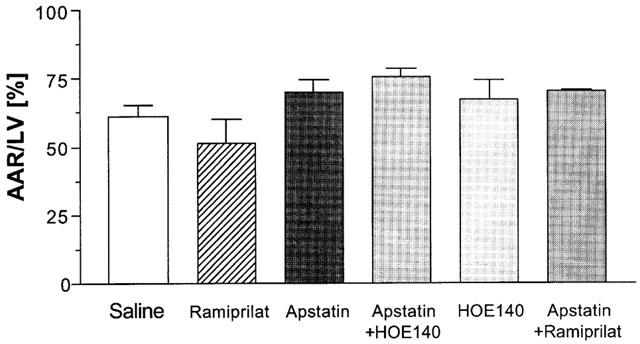
Pretreatment with either apstatin, an inhibitor of aminopeptidase P, ramiprilat, an inhibitor of angiotensin converting enzyme, or their combination was followed by 30 min of coronary artery occlusion and 3 h of reperfusion. HOE140, an antagonist of B2-receptors, was used to investigate the influence of kinins. The ratio between area at risk (AAR) and left ventricle (LV) was used as an indicator for a constant placement of the coronary ligature. There were no significant differences between the groups. Data are given as means±s.e.mean (n=5).
Infarct size
Figure 2 demonstrates the ratio between IS and AAR (IS/AAR%). As described previously (Hartman et al., 1993), IS/AAR% was significantly reduced in rats that received ramiprilat (18±3%) as compared with those receiving saline (40±2%). Inhibition of APP by apstatin also led to a significant reduction in IS/AAR% (18±2%) which did not differ to that obtained by ramiprilat. Blockade of B2 receptors with HOE140 completely abolished the cardioprotective effect of APP inhibition by apstatin (49±4%), while HOE140 alone did not influence IS/AAR% (43±3%). Inhibition of ACE in addition to APP reduced IS/AAR% to the same extent (20±4%) as inhibition of either APP or ACE alone.
Figure 2.
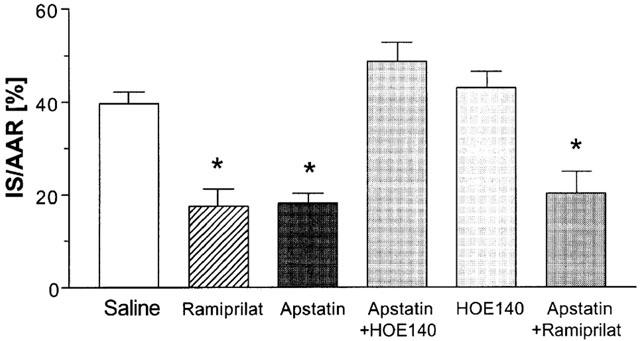
Ischaemic injury is given as the ratio between infarct size (IS) and area at risk (AAR). Rats were either treated with apstatin, an inhibitor of aminopeptidase P, ramiprilat, an inhibitor of angiotensin converting enzyme, or their combination. The left coronary artery was occluded for 30 min followed by 3 h of reperfusion. HOE140, an antagonist of B2 receptors, was used to investigate the influence of kinins. Data are given as means±s.e.mean (n=5). *P<0.05 vs saline.
Discussion
With the present study we could show that cardioprotection due to potentiation of kinin actions can be accomplished by selective inhibition of kininases other than ACE in a whole animal model.
There is increasing evidence that kinins possess cardioprotective actions in the ischaemic heart. In isolated rat hearts with ischaemia-reperfusion injuries bradykinin (BK) reduces the duration and incidence of ventricular fibrillations and reduces release of cytosolic enzymes. In anaesthetized animals intracoronary infusion of BK is followed by comparable beneficial changes and decreases infarct size (Linz et al., 1997). ACE inhibitors besides inhibiting the formation of angiotensin II out of angiotensin I increase the efficacy of endogenous kinins to the extent that beneficial kinin dependent cardiac effects arise. Treatment with ACE inhibitors such as ramiprilat increases cardiac kinins and reduces postischaemic reperfusion injuries in isolated rat hearts as well as infarct size and remodelling in postinfarcted animals. Infarct size reduction by ACE inhibitors and BK in anaesthetized animals is reversed by HOE140 (Heusch et al., 1997) and beneficial effects of ACE inhibitors on cardiac function were not observed in kininogen deficient rats (Liu et al., 2000). There are a number of enzymes that possess kininase activity (Bhoola et al., 1992). Recently, we and others could show that ACE is not the only important kininase in the rat heart. Inhibition of both APP or ACE leads to a similar preservation of exogenous BK in rat coronary circulation and therefore APP contributes to a similar extent to myocardial kinin degradation as ACE (Dendorfer et al., 1997; Ersahin & Simmons, 1997). Whether the accumulation of kinins through kininase inhibition correlates with a potentiation of their effects has been studied in the isolated rat heart. Potentiation of BK-induced vasodilation through inhibition of APP was equal to that obtained by ACE inhibition (Dendorfer et al., 2000). Apstatin has been shown to be a selective inhibitor of APP without affecting ACE (Prechel et al., 1995). Inhibition of APP by apstatin has been shown to reduce arrhythmia and the release of cytosolic enzymes in an in vitro rat model of ischaemia (Ersahin et al., 1999). In the present study we examined the cardioprotective effects of APP-inhibition in an in situ model of acute myocardial infarction with reperfusion and compared it to the effects of ACE inhibition. Furthermore, we tried to delineate whether cardioprotection by APP inhibition is mediated by BK. For ACE inhibition we used ramiprilat, a well studied substance, which has been shown to reduce infarct size in different animal models when given as a single kininase inhibitor (Schriefer et al., 1996; Weidenbach et al., 2000; Yang et al., 1997). The dosage of apstatin used has been shown to potentate kinin-induced vasodilation in rats for at least 240 min (Kitamura et al., 1999) so that APP-inhibition in our model was maintained throughout the whole period of ischaemia and reperfusion. Application of apstatin led to a 45% reduction of myocardial infarct size which was equal to that obtained by ramiprilat and is in the range of those obtained with ACE inhibitors in other studies (Hartman et al., 1993; Weidenbach et al., 2000). Reduction of infarct size by apstatin was achieved in the absence of any haemodynamic changes. Therefore we assume that a local rather than a systemic kallikrein-kinin-system is influenced in our model. Cardioprotection by APP inhibition was fully prevented by blockade of B2 receptors with HOE140 indicating that apstatin leads to an accumulation of cardiac kinins which develop their cardioprotective action during ischaemia and reperfusion.
HOE140 did not influence infarct size when given alone, indicating that endogenous kinins without their potentiation through kininase inhibitors do not have cardioprotective effects in our model. This is in accordance with results previously obtained in pigs (Jalowy et al., 1998). Another aim of this study was to investigate whether a further accumulation of myocardial kinins through combined inhibition of ACE and APP would lead to a more pronounced reduction of infarct size than inhibition of either on enzyme alone. Additive potentiation of BK by ACE and APP inhibition has been demonstrated in isolated rat hearts, measuring coronary perfusion pressure (Dendorfer et al., 2000). There was no significant difference between reduction of myocardial infarct size by apstatin alone or its combination with ramiprilat. Similar results have also been obtained in vitro where a combination of apstatin with ramiprilat was not significantly better than either one alone in reducing the release of creatine kinase after ischaemia in the isolated rat heart (Ersahin et al., 1999). Therefore we conclude that cardioprotection in ischaemia and reperfusion achieved by inhibition of each of the major kininases in rat myocardium is most effective and potentiation of kinins through additional kininase inhibitors does not further reduce the remaining injury. The equivalent protection by inhibition of kinin degradation through either ACE or APP also implies that reduced degradation is the major cause for the enhanced efficiency of endogenous BK. As such, there is no evidence that degradation independent mechanisms of kinin potentiation, which have been reported for ACE inhibitors specifically (Minshall et al., 1997), would significantly contribute to the observed effects.
However, there are some studies indicating that reduction in infarct size by ACE inhibitors is a delay rather than a prevention of necrosis (reviewed by Przyklenk & Kloner, 1993). So although apstatin led to an impressive reduction in infarct size after 3 h of reperfusion in this study further experiments are required to assess the beneficial effects of long term treatment with apstatin after acute myocardial infarction.
Conclusion
Apstatin reduces infarct size in an in vivo model of acute myocardial ischaemia and reperfusion. Cardioprotection achieved by this selective inhibitor of APP is mediated by bradykinin. A further accumulation of kinins by combined inhibition of APP and ACE does not result in a more pronounced reduction of infarct size than APP-inhibition alone.
Abbreviations
- AAR
area at risk
- ACE
angiotensin converting enzyme
- APP
aminopeptidase P
- BK
bradykinin
- CAO
coronary artery occlusion
- HR
heart rate
- IS
infarct size
- LV
left ventricle
- MAP
mean arterial blood pressure
- NEP
neutral endopeptidase 24.11
References
- BHOOLA K.D., FIGUEROA C.D., WORTHY K. Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol. Rev. 1992;44:1–80. [PubMed] [Google Scholar]
- DENDORFER A., WOLFRUM S., DOMINIAK P. Pharmacology and cardiovascular implications of the kinin-kallikrein system. Jpn. J. Pharmacol. 1999;79:403–426. doi: 10.1254/jjp.79.403. [DOI] [PubMed] [Google Scholar]
- DENDORFER A., WOLFRUM S., SCHÄFER U., STEWART J.M., INAMURA N., DOMINIAK P. Potentiation of the vascular response to kinins by inhibition of myocardial kininases. Hypertension. 2000;35:32–37. doi: 10.1161/01.hyp.35.1.32. [DOI] [PubMed] [Google Scholar]
- DENDORFER A., WOLFRUM S., WELLHÖNER P., KORSMAN K., DOMINIAK P. Intravascular and interstitial degradation of bradykinin in isolated perfused rat heart. Br. J. Pharmacol. 1997;122:1179–1187. doi: 10.1038/sj.bjp.0701501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ERSAHIN C., EULER D.E., SIMMONS W.H. Cardioprotective effects of the aminopeptidase P inhibitor apstatin: studies on ischemia/reperfusion injury in the isolated rat heart. J. Cardiovasc. Pharmacol. 1999;34:604–611. doi: 10.1097/00005344-199910000-00019. [DOI] [PubMed] [Google Scholar]
- ERSAHIN C., SIMMONS W.H. Inhibition of both aminopeptidase P and angiotensin-converting enzyme prevents bradykinin degradation in the rat coronary circulation. J. Cardiovasc. Pharmacol. 1997;30:96–101. doi: 10.1097/00005344-199707000-00014. [DOI] [PubMed] [Google Scholar]
- GOHLKE P., PEES C., UNGER T. AT2 receptor stimulation increases aortic cyclic GMP in SHRSP by a kinin-dependent mechanism. Hypertension. 1998;31:349–355. doi: 10.1161/01.hyp.31.1.349. [DOI] [PubMed] [Google Scholar]
- HARTMAN J.C., WALL T.M., HULLINGER T.G., SHEBUSKI R.J. Reduction of myocardial infarct size in rabbits by ramiprilat: reversal by the bradykinin antagonist HOE 140. J. Cardiovasc. Pharmacol. 1993;21:996–1003. doi: 10.1097/00005344-199306000-00022. [DOI] [PubMed] [Google Scholar]
- HEUSCH G., ROSE J., EHRING T. Cardioprotection by ACE inhibitors in myocardial ischaemia/reperfusion. The importance of bradykinin. Drugs. 1997;54 Suppl 5:31–41. doi: 10.2165/00003495-199700545-00006. [DOI] [PubMed] [Google Scholar]
- JALOWY A., SCHULZ R., DÖRGE H., BEHRENDS M., HEUSCH G. Infarct size reduction by AT1-receptor blockade through a signal cascade of AT2-receptor activation, bradykinin and prostaglandins in pigs. J. Am. Coll. Cardiol. 1998;32:1787–1796. doi: 10.1016/s0735-1097(98)00441-0. [DOI] [PubMed] [Google Scholar]
- KITAMURA S., CARBINI L.A., SIMMONS W.H., SCICLI A.G. Effects of aminopeptidase P inhibition on kinin-mediated vasodepressor responses. Am. J. Physiol. 1999;276:H1664–H1671. doi: 10.1152/ajpheart.1999.276.5.H1664. [DOI] [PubMed] [Google Scholar]
- LINZ W., WIEMER G., SCHÖLKENS B.A. Beneficial effects of bradykinin on myocardial energy metabolism and infarct size. Am. J. Cardiol. 1997;80:118A–123A. doi: 10.1016/s0002-9149(97)00466-9. [DOI] [PubMed] [Google Scholar]
- LIU Y.H., YANG X.P., MEHTA D., BULAGANNAWAR M., SCICLI G.M., CARRETERO O.A. Role of kinins in chronic heart failure and in the therapeutic effect of ACE inhibitors in kininogen-deficient rats. Am. J. Physiol: Heart Circ. Physiol. 2000;278:H507–H514. doi: 10.1152/ajpheart.2000.278.2.H507. [DOI] [PubMed] [Google Scholar]
- MINSHALL R.D., ERDÖS E.G., VOGEL S.M. Angiotensin I-converting enzyme inhibitors potentiate bradykinin's inotropic effects independently of blocking its inactivation. Am. J. Cardiol. 1997;80:132A–136A. doi: 10.1016/s0002-9149(97)00468-2. [DOI] [PubMed] [Google Scholar]
- PRECHEL M.M., ORAWSKI A.T., MAGGIORA L.L., SIMMONS W.H. Effect of a new aminopeptidase P inhibitor, apstatin, on bradykinin degradation in the rat lung. J. Pharmacol. Exp. Ther. 1995;275:1136–1142. [PubMed] [Google Scholar]
- PRZYKLENK K., KLONER R.A. “Cardioprotection” by ACE-inhibitors in acute myocardial ischemia and infarction. Basic Res. Cardiol. 1993;88 Suppl 1:139–154. doi: 10.1007/978-3-642-72497-8_10. [DOI] [PubMed] [Google Scholar]
- SCHRIEFER J.A., BROUDY E.P., HASSEN A.H. Endopeptidase inhibitors decrease myocardial ischemia/reperfusion injury in an in vivo rabbit model. J. Pharmacol. Exp. Ther. 1996;278:1034–1039. [PubMed] [Google Scholar]
- WEIDENBACH R., SCHULZ R., GRES P., BEHRENDS M., POST H., HEUSCH G. Enhanced reduction of myocardial infarct size by combined ACE inhibition and AT(1)-receptor antagonism. Br. J. Pharmacol. 2000;131:138–144. doi: 10.1038/sj.bjp.0703544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG X.P., LIU Y.H., PETERSON E., CARRETERO O.A. Effect of neutral endopeptidase 24.11 inhibition on myocardial ischemia/reperfusion injury: the role of kinins. J. Cardiovasc. Pharmacol. 1997;29:250–256. doi: 10.1097/00005344-199702000-00014. [DOI] [PubMed] [Google Scholar]