

Abstract
The serotonin2C (5-HT2C) receptor couples to both phospholipase C (PLC)-inositol phosphate (IP) and phospholipase A2 (PLA2)-arachidonic acid (AA) signalling cascades. Agonists can differentially activate these effectors (i.e. agonist-directed trafficking of receptor stimulus) perhaps due to agonist-specific receptor conformations which differentially couple to/activate transducer molecules (e.g. G proteins). Since editing of RNA transcripts of the human 5-HT2C receptor leads to substitution of amino acids at positions 156, 158 and 160 of the putative second intracellular loop, a region important for G protein coupling, we examined the capacity of agonists to activate both the PLC-IP and PLA2-AA pathways in CHO cells stably expressing two major, fully RNA-edited isoforms (5-HT2C-VSV, 5-HT2C-VGV) of the h5-HT2C receptor.
5-HT increased AA release and IP accumulation in both 5-HT2C-VSV and 5-HT2C-VGV expressing cells. As expected, the potency of 5-HT for both RNA-edited isoforms for both responses was 10 fold lower relative to that of the non-edited receptor (5-HT2C-INI) when receptors were expressed at similar levels.
Consistent with our previous report, the efficacy order of two 5-HT receptor agonists (TFMPP and bufotenin) was reversed for AA release and IP accumulation at the non-edited receptor thus demonstrating agonist trafficking of receptor stimulus. However, with the RNA-edited receptor isoforms there was no difference in the relative efficacies of TFMPP or bufotenin for AA release and IP accumulation suggesting that the capacity for 5-HT2C agonists to traffic receptor stimulus is lost as a result of RNA editing.
These results suggest an important role for the second intracellular loop in transmitting agonist-specific information to signalling molecules.
Keywords: RNA-editing; G protein coupled receptors; serotonin receptors; efficacy; receptor-effector coupling; signal transduction, agonist trafficking
Introduction
The serotonin2C (5-HT2C) receptor is a member of the 5-HT2 seven transmembrane spanning (7-TMS) receptor family which characteristically activates second messenger signal transduction cascades via pertussis toxin insensitive G proteins of the Gq and possibly G12/13 families (Berg et al., 1999; Chang et al., 2000; Gohla et al., 1999). Activation of the 5-HT2C receptor leads to both phospholipase A2 (PLA2)-mediated arachidonic acid (AA) release and phospholipase C (PLC)-phosphatidylinositol (PI) hydrolysis in brain (Conn et al., 1986; Kaufman et al., 1995) and in heterologous expression systems (Berg et al., 1996; 1998). Recent studies have shown that the 5-HT2C receptor subtype is widely expressed at significant densities in numerous brain regions (Mengod et al., 1997) and there is increasing evidence that this receptor plays a significant role in many physiological functions and behaviours, such as sleep, affective state, feeding behaviour and temperature regulation as well as being likely targets for hallucinogen drugs of abuse (for review see Roth et al., 1998).
The post-transcriptional process of editing of mRNA can generate unique isoforms of proteins in a cell and/or tissue specific manner (Simpson & Emeson, 1996; Smith et al., 1997). Recently, mRNA transcripts of the rat and human (h) 5-HT2C receptor have been found to undergo adenosine-to-inosine editing events at five sites which encompass amino acids 156 – 160 within the putative second intracellular loop of the encoded human receptor (Burns et al., 1997; Niswender et al., 1999). In human brain, the non-edited receptor contains the amino acids isoleucine, asparagine, and isoleucine (i.e., INI) at positions 156, 158 and 160, respectively, while two of the principal edited isoforms expressed have valine, serine and valine (i.e., VSV) or valine, glycine, and valine (i.e., VGV) corresponding to these amino acid positions (156, 158 and 160, respectively). In general, 5-HT has lower affinity for, and consequently is less potent at eliciting PI hydrolysis from, 5-HT2C-VSV or 5-HT2C-VGV receptor isoforms in comparison with the non-edited 5-HT2C-INI receptor (Burns et al., 1997; Fitzgerald et al., 1999; Niswender et al., 1999). Further, guanine nucleotide-sensitive, high affinity binding is reduced (Fitzgerald et al., 1999; Herrick-Davis et al., 1999; Niswender et al., 1999) as is ligand-independent (constitutive) receptor activity (Herrick-Davis et al., 1999; Niswender et al., 1999) with the 5-HT2C-VSV or 5-HT2C-VGV isoforms. Since the second intracellular loop of 7-TMS receptors is known to play a role in receptor-G protein coupling (Gudermann et al., 1997), these data suggest that RNA-edited 5-HT2C receptor isoforms have reduced G protein-coupling efficiency.
Recently it has been suggested that 7-TMS receptor agonists may have the capacity to promote unique receptor conformations which can differentially couple to/activate each of multiple signalling cascades coupled to a single receptor. This hypothesis has been termed ‘agonist-directed trafficking of receptor stimulus' (ADTRS) (Berg et al., 1998; Clarke & Bond, 1998; Kenakin, 1995), because agonists can direct (traffic) the receptor stimulus differentially to individual signalling cascades. Thus, in contrast to the doctrine of traditional receptor theory (Furchgott, 1966), within the framework of ADTRS agonist intrinsic efficacy is effector pathway-dependent. Support for the ADTRS hypothesis has stemmed from studies in which reversal of agonist potency order (Robb et al., 1994; Spengler et al., 1993) and differences in agonist relative efficacy (Berg et al., 1998; Brink et al., 2000; Cordeaux et al., 2000) have been observed. For the 5-HT2C (and 5-HT2A) receptor system, using an unambiguous measure of agonist efficacy (maximal response to partial agonists), we have found that agonist relative efficacy differs depending upon whether PLC or PLA2 activity is measured and that agonist efficacy order also is response-dependent (Berg et al., 1998), providing strong support for ADTRS. Although the mechanism by which agonists differentially activate effector pathways has not been established, the currently favoured hypothesis for this action is that agonist-specific receptor conformations differentially couple to and/or activate transducer molecules, such as G proteins.
The amino acid changes in the 5-HT2C receptor as a result of editing occur in a region of the receptor known to participate in G protein coupling, however to date, the effect of RNA editing of the 5-HT2C receptor on signal transduction has been studied only for the PLC-PI effector system. The purpose of this study was to investigate the capacity of the fully RNA-edited h5-HT2C receptor isoforms (5-HT2C-VSV and 5-HT2C-VGV) to activate the PLA2-AA effector system as well as the PLC-PI system. Additionally, we examined the impact of h5-HT2C receptor RNA-editing on the capacity of agonists to differentially activate the PLC-PI versus PLA2-AA pathways (i.e., agonist-directed trafficking of receptor stimulus).
Methods
Materials
The following materials were purchased from commercial sources: [3H]-myo-inositol, [14C]-arachidonic acid were purchased from New England Nuclear (Boston, MA, U.S.A.). 5-HT HCl, (±)-2,5-dimethoxy-4-iodoamphetamine hydrobromide (DOI), lysergic acid diethylamide (LSD), bufotenin, quipazine maleate, 3-trifluoromethylphenyl-piperazine (TFMPP) were purchased from Research Biochemicals, Inc (Natick, MA, U.S.A.). Foetal bovine serum was from Gemini Bioproducts (Calabasas, CA, U.S.A.). All other tissue culture reagents were purchased from GIBCO (Grand Island, NY, U.S.A.). All other drugs and chemicals (reagent grade) were purchased from Sigma Chemical Co. (St. Louis, MO, U.S.A.).
Transfection
Stable co-transfection of CHO-K1 cells was performed in 35 mm dishes using lipofectamine (15 μl per well) and the mammalian expression vector pCMVII encoding either non-edited (5-HT2C-INI) or RNA-edited (5-HT2C-VSV, 5-HT2C-VGV) h5-HT2C receptors (2 μg DNA) with 0.2 μg of the expression vector, pZeoSV (Invitrogen), encoding zeocin resistance. For some experiments, cDNA inserts encoding 5-HT2C-VSV and 5-HT2C-VGV isoforms were cloned into the kpnI site of pZeoSV and expressed stably. Approximately 30 – 40 zeocin resistant colonies were isolated and screened for each construct. Stable clones were selected for their resistance to zeocin (250 μg ml−1), their ability to bind [3H]-mesulergine and the ability to release AA and/or accumulate inositol phosphates (IP) in response to 5-HT, as described below.
Cell culture
CHO-INI, CHO-VSV and CHO-VGV cell lines are CHO-K1 cells which stably express non-edited (5-HT2C-INI) or RNA-edited (5-HT2C-VSV, 5-HT2C-VGV) human 5-HT2C receptors, respectively. Cells were maintained in alpha-MEM supplemented with 5% FBS and 100 μg ml−1 zeocin. For all experiments, cells were seeded into 12 or 24 well tissue culture vessels for functional studies or 15 cm dishes for radioligand binding studies at a density of 4×104 cells per cm2. Following a 24 h plating period, cells were washed with Hank's balanced salt solution (HBSS) and placed into D-MEM/F-12 [1 : 1] with 5 μg ml−1 insulin, 5 μg ml−1 transferrin, 30 nM selenium, 20 nM progesterone and 100 μM putrescine (serum-free media) and cultured for an additional 24 h prior to experimentation. Because some of the agonists used in this study (e.g., LSD, TFMPP) have some affinity for the 5-HT1B receptor which is naturally expressed in CHO-K1 cells (Berg et al., 1994; Giles et al., 1996), all functional experiments were done in cells previously treated with pertussis toxin (24 h, 50 ng ml−1) during the serum-free culture period.
Radioligand binding
Saturation binding assays using [3H]-mesulergine in cell membrane preparations (50 μg protein per tube) were done using 15 concentrations of radioligand in duplicate over a 3-log unit range as previously described (Berg et al., 1994). Incubations were carried out for 1 h at 37°C followed by rapid filtration with 0.3% polyethyleneamine coated filters using a Brandel cell harvester. Non-specific binding was determined in the presence of 1 μM mianserin.
Competition binding assays were done with cell (CHO-VSV; 2300 fmol mg−1 protein) membrane preparations (50 μg protein per tube) using half-log unit concentrations of the competing ligand over a range from 1×10−10 M to 1×10−3 M in the presence of [3H]-mesulergine (1 nM). Incubations were carried out for 1 h at 37°C followed by rapid filtration with 0.3% polyethyleneamine coated filters using a Brandel cell harvester.
IP accumulation and AA release measurements
Cells were labelled with 1 μCi ml−1 [3H]-myo-inositol in serum free medium for 24 h and with 0.1 μCi ml−1 [3H]-arachidonic acid for 4 h at 37°C prior to experiments. Measurements of total [3H]-IP accumulation were made from the same multiwell (simultaneously) as [3H]-AA release measurements as described previously (Berg et al., 1998).
Assessment of agonist-directed trafficking of receptor stimulus (ADTRS)
To assess the capacity of agonists to differentially activate the PLC-IP vs the PLA2-AA signalling pathways, relative efficacies of the test agonists were calculated for each response using the ratio of the response to maximal concentrations of the test agonist (at least 100×Ki to produce full receptor occupancy) to that of the reference agonist 5-HT (i.e. intrinsic activity), as we have done before (Berg et al., 1998). Since all of the test ligands used in this study were partial agonists, the intrinsic activity of the ligands provides an unambiguous measure of relative efficacy (Kenakin, 1997). Traditional receptor theory necessitates that agonist relative efficacies must be independent of the response measured (Furchgott, 1966; Kenakin, 1997) and therefore the relative efficacies of the test ligands should not be different between responses. Consequently, a difference in the relative efficacy of an agonist determined from measurement of AA release or IP accumulation under identical conditions (see above) is interpreted as support for ADTRS. Although data demonstrating reversal of potency order of agonists for different responses (Robb et al., 1994; Spengler et al., 1993) has been interpreted as support for ADTRS (Kenakin, 1995), the use of potency measurements to assess agonist efficacy is potentially confounded, since potency of full (but not partial) agonists is determined by affinity as well as efficacy (see Kenakin, 1995).
Data analysis
For saturation binding experiments, data were fit with nonlinear regression to the model:
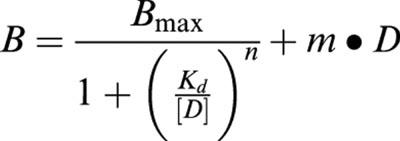 |
where B is the measured amount of radioligand bound (fmol mg−1 protein) in the presence of various concentrations of radioligand [D], Bmax is the maximal amount bound, Kd is the concentration producing half-maximal binding, n is the slope factor, and m is the slope of the linear regression line for ‘non-specific' binding.
IC50 values were derived from non-linear regression analysis of competition binding data using the program Prism 3.0 for Macintosh (Graphpad). Data were fit with non-linear regression analysis to one-site and two-site models and the best fit determined with an F-test by Prism software. The competition curves for all of the test ligands used were best fit with a one-site model. Ki values were calculated from IC50 values using the transformation of Cheng – Prusoff (Cheng & Prusoff, 1973).
Concentration response data were fit with nonlinear regression to the model:
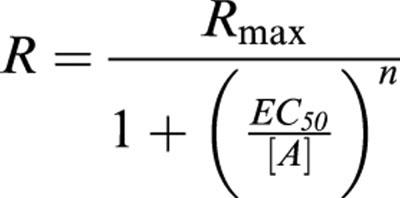 |
where R is the measured response at a given agonist concentration (A), Rmax=maximal response, EC50=the concentration of agonist producing half-maximal response, and n=slope index.
The Student t-test (paired) was used for statistical comparisons. Asterisks (*) denote P values <0.05.
Results
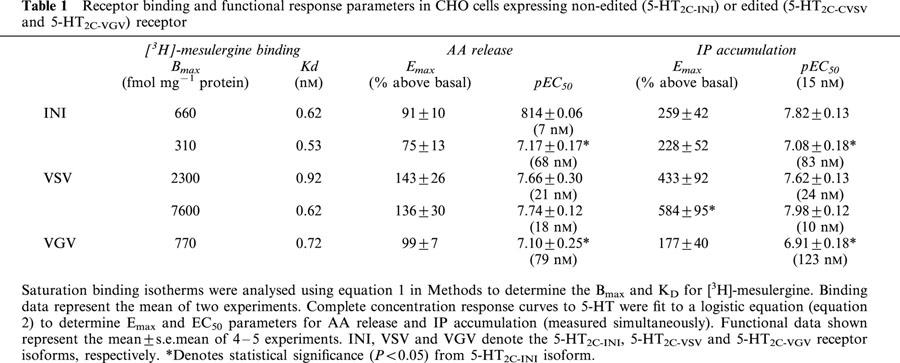
5-HT-mediated AA release and IP accumulation were measured simultaneously from the same cells expressing stably either the non-edited (5-HT2C-INI) or RNA-edited (5-HT2C-VSV or 5-HT2C-VGV) receptors. As shown in Figure 1 and summarized in Table 1, 5-HT produced a concentration-dependent increase in both AA release and IP accumulation from each receptor isoform. For each receptor isoform the potency (EC50) of 5-HT for IP accumulation was not different from that for AA release, however the potency for 5-HT at the edited isoforms was approximately 10 fold less than that for the non-edited receptor.
Figure 1.
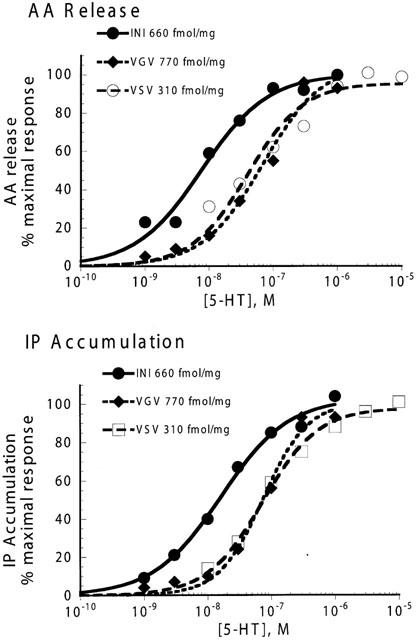
Comparison of concentration response curves to 5-HT between non-edited (5-HT2C-INI) and RNA-edited (5-HT2C-VSV and 5-HT2C-VGV) 5-HT2C receptor isoforms. CHO cells expressing stably the 5-HT2C-INI, 5-HT2C-VSV or 5-HT2C-VGV receptor isoforms were incubated with various concentrations of 5-HT for 10 min followed by measurement of IP accumulation or AA release simultaneously from the same multiwell. Data shown are normalized to the per cent of Emax from each individual experiment and represent the mean of 4 – 5 experiments measured in triplicate. Individual concentration curves were fit to the logistic equation 2 described in Methods to determine Emax and EC50 values (provided in Table 1).
Table 1.
Receptor binding and functional response parameters in CHO cells expressing non-edited (5-HT2C-INI) or edited (5-HT2C-CVSV and 5-HT2C-VGV) receptor
The relative efficacy of a series of partial agonists (at least 100×Ki; see Table 2), with respect to 5-HT, was measured for the 5-HT2C-VSV receptor stably expressed at moderate levels (310 fmol mg−1 protein). In contrast to results obtained with the non-edited 5-HT2C receptor (see Berg et al., 1998, and Figure 3B), agonist relative efficacy at the 5-HT2C-VSV receptor was not different for IP accumulation and AA release (Table 2). Because the intrinsic activity of LSD was very low, the ability of LSD to elicit AA release and IP accumulation was measured in two additional clones where the 5-HT2C-VSV receptor isoform was overexpressed (2300 fmol mg−1 and 7600 fmol mg−1). As expected due to the increased receptor expression, the response to LSD was enhanced in these clones and therefore more accurately measured. Figure 2 shows that the relative efficacy of LSD on AA release was not different from that for IP accumulation in either clone.
Table 2.
Relative efficacies of various 5-HT2C receptor agonists
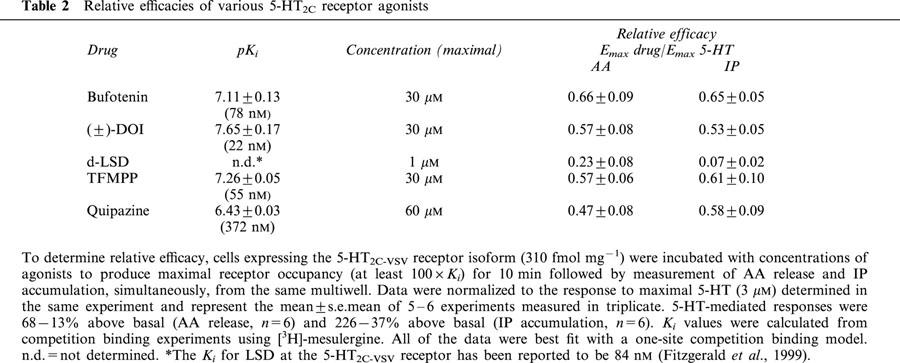
Figure 3.
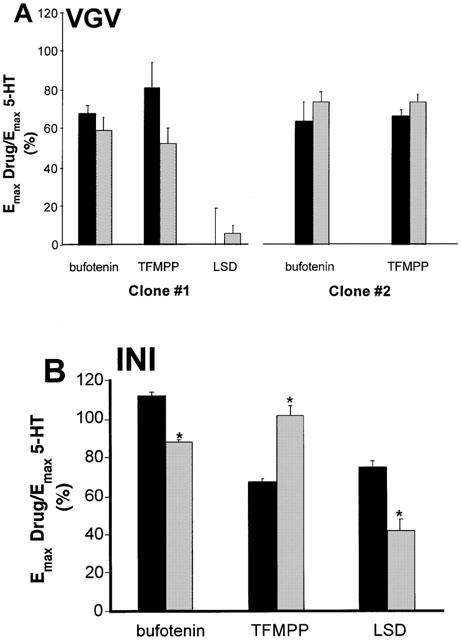
Comparison of relative efficacies of bufotenin, TFMPP and LSD between 5-HT2C-INI and 5-HT2C-VGV receptor isoforms. Cells were incubated for 10 min with maximal concentrations of LSD (300 nM), bufotenin (10 μM for 5-HT2C-INI and 30 μM for 5-HT2C-VGV), TFMPP (10 μM for 5-HT2C-INI and 30 μM for 5-HT2C-VGV) or 5-HT (1 μM for 5-HT2C-INI and 3 μM for 5-HT2C-VGV). AA release and IP accumulation were measured simultaneously from the same multiwell. Data were normalized to the 5-HT response determined in the same experiment and represent the mean±s.e.mean of 3 – 6 experiments. 5-HT-mediated responses for the 5-HT2C-INI isoform were 85±13% and 285±47%, for the 5-HT2C-VGV clone #1, 86±15% and 169±10% and for 5-HT2C-VGV clone #2, 47±6% and 189±4%, AA release and IP accumulation, respectively. *Denotes statistical significance between agonist-mediated AA release and IP accumulation.
Figure 2.
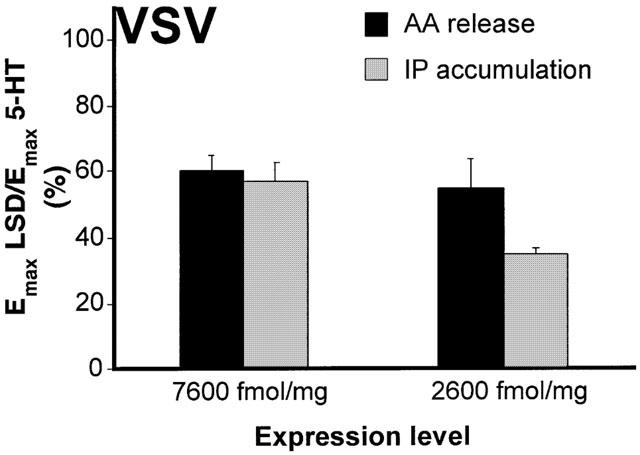
Relative efficacy of LSD for AA release and IP accumulation in cells expressing the 5-HT2C-VSV isoform at high densities. Cells were incubated with 300 nM LSD (maximal concentration) for 10 min followed by simultaneous measurement of AA release or IP accumulation. Bars show the intrinsic activity of LSD, referenced to the maximal 5-HT response determined in the same experiment, and represent the mean±standard error of three experiments. 5-HT-mediated AA release was 70±7% and 85±7% and 5-HT-mediated IP accumulation was 293±33% and 429±40%, for cells expressing 2300 and 7600 fmol mg−1, respectively.
ADTRS also did not occur with the 5-HT2C-VGV receptor isoforms (Figure 3). As we have shown previously (Berg et al., 1998), with the non-edited receptor (5-HT2C-INI), the relative efficacy of bufotenin and of LSD was greater for AA release than for IP accumulation, whereas the reverse was true for TFMPP (i.e. agonist efficacy order was reversed; Figure 3B). However, in cells stably expressing the 5-HT2C-VGV isoform, using two different clones, there was no difference in the relative efficacies for bufotenin or TFMPP. Interestingly, LSD failed to elicit either AA release or IP accumulation in CHO cells expressing the 5-HT2C-VGV isoform.
Discussion
RNA-edited 5-HT2C receptor isoforms, 5-HT2C-VSV and 5-HT2C-VGV, couple to both PLA2 and PLC
Amino acid changes in the second intracellular loop of 7-TMS receptors might be expected to alter coupling to or activation of G proteins (Gudermann et al., 1997) or other transducer molecules which may interact with the receptor in this region (Hall et al., 1999). The 5-HT2C-VGV and 5-HT2C-VSV isoforms of the 5-HT2C receptor have different amino acids in the second intracellular loop from the 5-HT2C-INI (non-edited) receptor and these edited receptor isoforms have reduced capacity to signal to the PLC-PI effector pathway (Burns et al., 1997; Niswender et al., 1999; Fitzgerald et al., 1999). In addition to the PLC-PI effector pathway, the 5-HT2C receptor also couples to the PLA2-AA signalling cascade, however the effect of RNA editing on the ability of the 5-HT2C receptor to activate PLA2 signalling has not been studied. We found that 5-HT increased both AA release as well as IP accumulation (measured simultaneously from the same cells) from each edited receptor isoform. For each receptor isoform (edited and non-edited) the potency of 5-HT did not differ between effector pathways as might be expected since both responses stem from activation of the same receptor. As reported previously (Burns et al., 1997; Niswender et al., 1999; Fitzgerald et al., 1999), the potency for 5-HT to elicit IP accumulation at the edited receptors (5-HT2C-VSV or 5-HT2C-VGV) was less than that for the non-edited receptor (5-HT2C-INI) when expressed at similar densities. Similarly, the potency for 5-HT to stimulate AA release was also less for the edited receptors. This result is consistent with the finding that affinity of 5-HT for 5-HT2C-VSV and 5-HT2C-VGV isoforms is reduced in comparison to that of non-edited 5-HT2C-INI receptors (Niswender et al., 1999; Fitzgerald et al., 1999; Herrick-Davis et al., 1999). The higher potency of 5-HT in the cell lines expressing the 5-HT2C-VSV isoform at high levels is consistent with the presence of receptor reserve in these cells.
Agonist-directed trafficking of receptor stimulus (ADTRS) does not occur at the RNA-edited h5-HT2C receptor isoforms
Although traditional receptor theory allows for receptors to couple to multiple effector pathways, it does not accommodate the capacity for agonists to differentially activate effectors. Within the context of traditional receptor theory, the intrinsic efficacy of agonists (and consequently agonist relative efficacy) must be independent of the response measured (Furchgott, 1966). Since potency of full agonists is, in part, influenced by efficacy, recent reports that the potency order of agonists differs depending upon the response measured (Robb et al., 1994; Spengler et al., 1993) suggested that agonist relative efficacy may, in fact, be dependent on response and prompted Kenakin to propose a new hypothesis he termed ‘agonist-directed trafficking of receptor stimulus' (Kenakin, 1995). The main premise of this hypothesis is that agonists are able to promote the formation of agonist-specific receptor conformations and that these conformations have different capacities to activate effector pathways. Within this framework, agonist intrinsic efficacy would not be independent of the response measured. Although the ADTRS hypothesis, as formulated by Kenakin, does not presuppose a mechanism, the simplest way agonists could differentially activate effectors coupled to the same receptor is if agonist-specific conformations have different capacities to couple to or activate transducer molecules, such as G proteins.
Using an unambiguous measure of agonist relative efficacy (intrinsic activity of partial agonists), we recently found that for the unedited 5-HT2C receptor (5-HT2C-INI) agonist relative efficacy differed depending on whether AA release or IP accumulation was measured (Berg et al., 1998). Furthermore, for some agonists, efficacy order was reversed. Relative to 5-HT, DOI, bufotenin and LSD preferentially activated the PLA2-AA pathway, while quipazine and TFMPP favoured IP accumulation. In the present study using a different clonal cell line transfected with a different expression vector from those used in our original report, we replicated our original finding that not only was agonist relative efficacy different for different responses coupled to the non-edited 5-HT2C receptor, but efficacy order was reversed for bufotenin and TFMPP. This difference in agonist efficacy order is strong evidence in support of the ADTRS hypothesis.
Since the changes in amino acids produced as a result of RNA editing of the 5-HT2C receptor mRNA are in a region of the receptor which has been shown to be involved in signal transduction and G protein coupling for several 7-TMS receptors (Gudermann et al., 1997), we examined the ability of agonists to differentially activate PLC-IP versus PLA2-AA in cells expressing the 5-HT2C-VSV or 5-HT2C-VGV isoforms. Consistent with previous reports showing that LSD does not increase PLC activity in NIH-3T3 cells expressing the 5-HT2C-VGV isoform (Backstrom et al., 1999; Fitzgerald et al., 1999), we also found that LSD did not elicit either AA release or IP accumulation in CHO cells with the 5-HT2C-VGV receptor. In contrast to our results with the non-edited 5-HT2C receptor, we found that agonist relative efficacy was not different between responses for the edited receptors. These results suggest that agonists cannot differentially traffic receptor stimulus to each of the two effector pathways coupled to the receptor when amino acids are changed at positions 156, 158 and 160 in the second intracellular loop of the 5-HT2C receptor.
In conclusion, using an unambiguous measure of relative efficacy, we have shown that the relative efficacy for agonists to activate PLC-PI hydrolysis is not different from their relative efficacy to activate PLA2-AA release upon activation of the 5-HT2C-VSV and 5-HT2C-VGV RNA-edited isoforms of the 5-HT2C receptor. This is in contrast to the different relative efficacies and different efficacy order for agonists acting at the non-edited (5-HT2C-INI) receptor. The lack of effector pathway-dependence of agonist relative efficacy indicates that the potential for agonists to traffic receptor stimuli differentially to effector mechanisms is missing from the edited 5-HT2C receptor isoforms and suggests that the second intracellular loop may play an important role in transmitting agonist-specific information to signalling molecules. The currently favoured hypothesis for the mechanism of ADTRS is that agonists promote the formation of ligand-specific receptor conformations which have different capacities to couple to/activate signalling molecules (e.g., G proteins). The loss of ADTRS with RNA-edited receptor isoforms suggests either that the edited isoforms are not capable of adopting ligand-specific conformations or that the capacity of ligand-specific conformations to differentially interact with signalling molecules is impaired. The loss of spontaneous receptor activity as a result of RNA editing (Niswender et al., 1999; Herrick-Davis et al., 1999) may suggest that these isoforms are more restricted in the conformations they adopt and thus, perhaps, the ability of ligands to promote different receptor conformations is also impaired. However, since it has been shown that the coupling efficiency of edited isoforms to G proteins and to the PLC pathway is reduced (Niswender et al., 1999; Fitzgerald et al., 1999), it seems likely that changes in the ability of ligand-specific receptor conformations to differentially interact with signalling molecules is the most likely explanation for the lack of agonist-directed trafficking. Support for this hypothesis must await identification of the transducing molecule(s) that couples the 5-HT2C receptor to PLA2.
Acknowledgments
This work was supported by United States Public Health Service Grants DA 09094 (K.A. Berg), GM 58652 (W.P. Clarke); MH 34005 (E. Sanders-Bush), NS 36891 (R.B. Emeson) and grants from the Pharmaceutical Research and Manufacturers of America Foundation (C.M. Niswender) and the Texas Advanced Research Program (3659-0044; W.P. Clarke, K.A. Berg). The authors would like to thank Drs David McLoughlin and Brian Stout for helpful comments and Blythe King and Bonnie Garcia for expert technical assistance.
Abbreviations
- AA
arachidonic acid
- ADTRS
agonist-directed trafficking of receptor stimulus
- CHO
Chinese hamster ovary
- DOI
(±)-1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane
- G protein
guanine nucleotide binding protein
- HBSS
Hank's balanced salt solution
- 5-HT
5-hydroxytryptamine, serotonin
- IP
inositol phosphates
- LSD
lysergic acid diethylamide
- PI
phosphatidylinositol
- PLA2
phospholipase A2
- PLC
phospholipase C
- TFMPP
3-trifluoromethylphenyl-piperazine
- 7-TMS
seven transmembrane spanning
References
- BACKSTROM J.R., CHANG M.S., CHU H., NISWENDER C.M., SANDERS-BUSH E. Agonist-directed signaling of serotonin 5-HT2C receptors: differences between serotonin and lysergic acid diethylamide (LSD) Neuropsychopharmacology. 1999;21:77S–81S. doi: 10.1016/S0893-133X(99)00005-6. [DOI] [PubMed] [Google Scholar]
- BERG K.A., CLARKE W.P., SAILSTAD C., SALTZMAN A., MAAYANI S. Signal transduction differences between 5-hydroxytryptamine type 2A and type 2C receptor systems. Mol. Pharmacol. 1994;46:477–484. [PubMed] [Google Scholar]
- BERG K.A., MAAYANI S., CLARKE W.P. 5-hydroxytryptamine 2C receptor activation inhibits 5-hydroxytryptamine 1B-like receptor function via arachidonic acid metabolism. Mol. Pharmacol. 1996;50:1017–1023. [PubMed] [Google Scholar]
- BERG K.A., MAAYANI S., GOLDFARB J., SCARAMELLINI C., LEFF P., CLARKE W.P. Effector pathway-dependent relative efficacy at serotonin type 2A and 2C receptors: evidence for agonist-directed trafficking of receptor stimulus. Mol. Pharmacol. 1998;54:94–104. [PubMed] [Google Scholar]
- BERG K.A., STOUT B.D., CROPPER J.D., MAAYANI S., CLARKE W.P. Novel actions of inverse agonists on 5-HT2C receptor systems. Mol. Pharmacol. 1999;55:863–872. [PubMed] [Google Scholar]
- BRINK C.B., WADE S.M., NEUBIG R.R. Agonist-directed trafficking of porcine alpha(2A)-adrenergic receptor signaling in Chinese hamster ovary cells: 1-isoproterenol selectively activates G(s) J. Pharmacol. Exp. Ther. 2000;294:539–547. [PubMed] [Google Scholar]
- BURNS C.M., CHU H., RUETER S.M., HUTCHINSON L.K., CANTON H., SANDERS-BUSH E., EMESON R.B. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature. 1997;387:303–308. doi: 10.1038/387303a0. [DOI] [PubMed] [Google Scholar]
- CHANG M., ZHANG L., TAM J.P., SANDERS-BUSH E. Dissecting G protein-coupled receptor signaling pathways with membrane-permeable blocking peptides. Endogenous 5-HT(2C) receptors in choroid plexus epithelial cells. J. Biol. Chem. 2000;275:7021–7029. doi: 10.1074/jbc.275.10.7021. [DOI] [PubMed] [Google Scholar]
- CHENG Y., PRUSOFF W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- CLARKE W.P., BOND R.A. The elusive nature of intrinsic efficacy. Trends. Pharmacol. Sci. 1998;19:270–276. doi: 10.1016/s0165-6147(97)01138-3. [DOI] [PubMed] [Google Scholar]
- CONN P.J., SANDERS-BUSH E., HOFFMAN B.J., HARTIG P.R. A unique serotonin receptor in choroid plexus is linked to phosphatidylinositol turnover. Proc. Natl. Acad. Sci. U.S.A. 1986;83:4086–4088. doi: 10.1073/pnas.83.11.4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CORDEAUX Y., BRIDDON S.J., MEGSON A.E., MCDONNELL J., DICKENSON J.M., HILL S.J. Influence of receptor number on functional responses elicited by agonists acting at the human adenosine A(1) receptor: evidence for signaling pathway-dependent changes in agonist potency and relative intrinsic activity. Mol. Pharmacol. 2000;58:1075–1084. doi: 10.1124/mol.58.5.1075. [DOI] [PubMed] [Google Scholar]
- FITZGERALD L.W., IYER G., CONKLIN D.S., KRAUSE C.M., MARSHALL A., PATTERSON J.P., TRAN D.P., JONAK G.J., HARTIG P.R. Messenger RNA editing of the human serotonin 5-HT2C receptor. Neuropsychopharmacology. 1999;21:82S–90S. doi: 10.1016/S0893-133X(99)00004-4. [DOI] [PubMed] [Google Scholar]
- FURCHGOTT R.F. The use of ß-haloalkylamines in the differentiation of receptors and in the determination of dissociation constants of receptor-agonist complexes Advances in Drug Research 1966London: Academic Press; 21–55.ed. Harper, N.J. & Simmonds, A.B. pp [Google Scholar]
- GILES H., LANSDELL S.J., BOLOFO M.L., WILSON H.L., MARTIN G.R. Characterization of a 5-HT1B receptor on CHO cells: functional responses in the absence of radioligand binding. Br. J. Pharmacol. 1996;117:1119–1126. doi: 10.1111/j.1476-5381.1996.tb16705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOHLA A., OFFERMANNS S., WILKIE T.M., SCHULTZ G. Differential involvement of Galpha12 and Galpha13 in receptor -mediated stress fiber formation. J. Biol. Chem. 1999;274:17901–17907. doi: 10.1074/jbc.274.25.17901. [DOI] [PubMed] [Google Scholar]
- GUDERMANN T., SCHONEBERG T., SCHULTZ G. Functional and structural complexity of signal transduction via G - protein-coupled receptors. Annu. Rev. Neurosci. 1997;20:399–427. doi: 10.1146/annurev.neuro.20.1.399. [DOI] [PubMed] [Google Scholar]
- HALL R.A., PREMONT R.T., LEFKOWITZ R.J. Heptahelical receptor signaling: beyond the G protein paradigm. J. Cell. Biol. 1999;145:927–932. doi: 10.1083/jcb.145.5.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HERRICK-DAVIS K., GRINDE E., NISWENDER C.M. Serotonin 5-HT2C receptor RNA editing alters receptor basal activity: implications for serotonergic signal transduction. J. Neurochem. 1999;73:1711–1717. doi: 10.1046/j.1471-4159.1999.731711.x. [DOI] [PubMed] [Google Scholar]
- KAUFMAN M.J., HARTIG P.R., HOFFMAN B.J. Serotonin 5-HT2C receptor stimulates cyclic GMP formation in choroid plexus. J. Neurochem. 1995;64:199–205. doi: 10.1046/j.1471-4159.1995.64010199.x. [DOI] [PubMed] [Google Scholar]
- KENAKIN T. Agonist-receptor efficacy II: agonist trafficking of receptor signals. Trends. Pharmacol. Sci. 1995;16:232–238. doi: 10.1016/s0165-6147(00)89032-x. [DOI] [PubMed] [Google Scholar]
- KENAKIN T. Pharmacologic analysis of drug-receptor interaction 1997New York: Raven Press; 3rd edition [Google Scholar]
- MENGOD G., PALACIOS J.M., WIEDERHOLD K.H., HOYER D. 5-hydroxytryptamine receptor histochemistry: Comparison of receptor mRNA distribution and radioligand autoradiography in the brain Serotonergic. Neurons and 5-HT Receptors in the CNS 1997Berlin: Springer-Verlag; 213–237.ed. Baumgarten, H.G. & Gothert, M. pp [Google Scholar]
- NISWENDER C.M., COPELAND S.C., HERRICK-DAVIS K., EMESON R.B., SANDERS-BUSH E. RNA editing of the human serotonin 5-hydroxytryptamine 2C receptor silences constitutive activity. J. Biol Chem. 1999;274:9472–9478. doi: 10.1074/jbc.274.14.9472. [DOI] [PubMed] [Google Scholar]
- ROBB S., CHEEK T.R., HANNAN F.L., HALL L.M., MIDGLEY J.M., EVANS P.D. Agonist-specific coupling of a cloned Drosophila octopamine/tyramine receptor to multiple second messenger systems. EMBO J. 1994;13:1325–1330. doi: 10.1002/j.1460-2075.1994.tb06385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROTH B.L., WILLINS D.L., KRISTIANSEN K., KROEZE W.K. 5-Hydroxytryptamine2-family receptors (5-hydroxytryptamine2A, 5-hydroxytryptamine2B, 5-hydroxytryptamine2C): where structure meets function. Pharmacol. Ther. 1998;79:231–257. doi: 10.1016/s0163-7258(98)00019-9. [DOI] [PubMed] [Google Scholar]
- SIMPSON L., EMESON R.B. RNA editing. Annu. Rev. Neurosci. 1996;19:27–52. doi: 10.1146/annurev.ne.19.030196.000331. [DOI] [PubMed] [Google Scholar]
- SMITH H.C., GOTT J.M., HANSON M.R. A guide to RNA editing. RNA. 1997;3:1105–1123. [PMC free article] [PubMed] [Google Scholar]
- SPENGLER D., WAEBER C., PANTALONI C., HOLSBOER F., BOCKAERT J., SEEBURG P.H., JOURNOT L. Differential signal transduction by five splice variants of the PACAP receptor. Nature. 1993;365:170–175. doi: 10.1038/365170a0. [DOI] [PubMed] [Google Scholar]