

Abstract
Arteriolar myogenic tone shows a marked dependency on extracellular Ca2+. The contribution played by mechanisms such as intracellular Ca2+ release and capacitative entry, however, are less certain. The present studies aimed to demonstrate functional evidence for involvement of such mechanisms in myogenic tone and reactivity.
Single cremaster arterioles were denuded of endothelium, pressurized under no-flow conditions and loaded with fura 2-AM for measurement of changes in intracellular Ca2+ [Ca2+]i. The cell permeable, putative, IP3 receptor antagonist 2APB (2 aminoethoxydiphenyl borate) was used to determine the possible role of IP3 receptor-mediated mechanisms in arteriolar myogenic tone and reactivity.
Arterioles dilated in response to increasing concentrations of 2APB (1 – 300 μM) without a concomitant change in global [Ca2+]i. Also 2APB (50 μM) completely inhibited the myogenic constriction in response to a step change in luminal pressure (50 – 120 mmHg) with no apparent effect on pressure-mediated increases in [Ca2+]i. 2APB markedly attenuated the constrictor response and [Ca2+]i increase stimulated by phenylephrine but not KCl.
Capacitative Ca2+ influx in arterioles was demonstrated either by re-addition of extracellular [Ca2+] following pre-treatment with 1 or 10 μM nifedipine in Ca2+ free buffer or exposure of vessels to thapsigargin (1 μM) to induce store depletion. In both cases 2APB inhibited the increase in [Ca2+]i. Capacitative Ca2+ entry showed an inverse relationship with intraluminal pressure over the range 10 – 120 mmHg.
Consistent with an effect on a Ca2+ entry pathway, 2APB had no effect on intracellular (caffeine releasable) Ca2+ stores while decreasing the rate of Mn2+ quench of fura 2 fluorescence.
The results provide functional evidence for capacitative Ca2+ entry in intact arteriolar smooth muscle. The effectiveness of 2APB in inhibiting both non-voltage gated Ca2+ entry and responsiveness to an acute pressure step is consistent with the involvement of an axis involving IP3-mediated and or capacitative Ca2+ entry mechanisms in myogenic reactivity. Given the lack of effect of 2APB on pressure-induced changes in global [Ca2+]i it is suggested that such mechanisms participate on a localized level to couple the myogenic stimulus to contraction.
Keywords: Myogenic reactivity, arteriolar smooth muscle, capacitative Ca2+ entry, IP3 receptor, cell signalling
Introduction
Arterioles typically exhibit a state of partial constriction, or myogenic tone, that is related to the level of intraluminal pressure. This contractile response is known to be dependent on extracellular Ca2+ entry, Ca2+-calmodulin mediated activation of myosin light chain kinase and phosphorylation of the regulatory light chains of myosin (for review see Davis & Hill, 1999). While the entry of Ca2+ into the arteriolar smooth muscle cells is well known to involve L-type voltage gated channels (for example, Knot et al., 1998; Wesselman et al., 1996; Potocnik et al., 2000) the contributions from intracellular Ca2+ sources and non-voltage dependent entry mechanisms are uncertain (Hill et al., 2001).
Data describing the importance of inositol trisphosphate (IP3) in acute myogenic contraction or sustained myogenic tone is limited. Narayanan et al. (1994) demonstrated in renal arcuate arteries that both IP3 and diacylglycerol increase as intraluminal pressure is raised. Technical limitations due to sample size have limited the temporal resolution in such studies so that definitive conclusions regarding the involvement of these molecules in a signalling role during different phases of the myogenic response have not been made. However, supporting a role for second messengers such as IP3 and diacylglycerol, the putative PLC inhibitor, U-73122, has been reported to inhibit myogenic responsiveness in cerebral (Osol et al., 1993) and renal (Inscho et al., 1998) arterioles.
Studies of the close relationship between myogenic tone and changes in intracellular Ca2+ have largely involved measurements of global cytosolic Ca2+ (Meininger et al., 1991; Zou et al., 1995) although recent studies have begun to consider spatial aspects of Ca2+ signalling in pressurized vessels (Jaggar et al., 1998; Miriel et al., 1999). Evidence from a variety of cell types suggests that Ca2+ changes within cellular compartments may activate distinct intracellular responses (Fagan et al., 1998). In arteriolar smooth muscle it is likely that Ca2+ regulates activities other than contraction such as activity of ion channels and sarcoplasmic reticulum function (van breemen et al., 1995; Laporte & Laher, 1997; Jaggar et al., 1998). Thus given the varying roles that Ca2+ can potentially play in myogenically active vessels it is important to have an understanding of the various Ca2+ supply mechanisms and compartmentalization that may exist.
The present studies aimed to examine the contribution of capacitative Ca2+ entry to arteriolar myogenic tone and reactivity and, further, to determine whether such Ca2+ entry occurs as a result of IP3 receptor activation and subsequent Ca2+ store depletion. While capacitative Ca2+ entry has been demonstrated for example in cells isolated from resistance arterioles (Fellner & Arendshorst, 1999; 2000) and cell lines derived from conduit vessels (Skutella & Ruegg, 1997) data is lacking as to its role in myogenically active vessels. To accomplish these aims studies were performed using protocols designed to isolate non-voltage gated mechanisms of Ca2+ entry and the recently described, small molecular weight, membrane permeable, modulator of the IP3 receptor, 2 amino ethoxy diphenylborate (2APB). This molecule has been shown to inhibit agonist-induced (IP3-mediated) Ca2+ release and capacitative Ca2+ entry in a number of cell types including myometrial and large artery smooth muscle (Maruyama et al., 1997; Ascher-Landsberg et al., 1999; Ma et al., 2000; Wu et al., 2000). Studies were performed in isolated and cannulated myogenically active arterioles in the presence and absence of the fluorescent indicator Fura 2 to measure changes in intracellular Ca2+.
Methods
Animals
The studies used male Sprague-Dawley rats weighing 242±4g (n=56). Prior to experiments, rats were housed in an animal facility equipped with a 12:12 h light-dark cycle. Rats were allowed free access to a standard rat chow and drinking water. All procedures were approved by the Animal Care and Use Committees at RMIT University.
Isolated arteriole preparation
Rats were anaesthetized (100 mg kg−1, Pentothal, Abbott Australasia) and the right or left cremaster muscle exteriorized, excised from the animal and placed in a cooled (4°C) chamber containing dissection buffer (in mM): 3-N-morpholino propanesulphonic acid (MOPS) 3; NaCl 145; KCl 5; CaCl2 2.5; MgSO4 1; NaH2PO4 1; EDTA 0.02; pyruvate 2; glucose 5 and 1% albumin (Duling et al., 1981). Segments of the main intramuscular arteriole (IA) were dissected from the muscle as previously described (Meininger et al., 1991). Individual vessel segments were then cannulated with glass micropipettes, secured using 10-0 monofilament suture and mounted in a 5 ml volume tissue chamber. The vessel preparation was positioned on the stage of an inverted microscope (Olympus) and the arterioles continuously superfused (2 – 4 ml min−1) with a physiologic salt solution (PSS) containing (in mM): NaCl 111, NaHCO3 25.7, KCl 4.9, CaCl2 2.5, MgSO4 1.2, KH2PO4 1.2, glucose 11.5 and 2-N-hydroxyethylpiperazine-N-2-ethanesulphonic acid (HEPES) 10. Arteriole segments were gradually pressurized to 70 mmHg and warmed to 34°C during a 60 min equilibration period. During this period vessels were checked for pressure leaks and allowed to develop spontaneous basal tone. After demonstrating viability the endothelial layer was removed from all vessels by the passage of an air bubble through the lumen followed by fresh buffer to remove cellular debris. Removal of the endothelium was verified by a lack of vasodilatation in response to acetylcholine (10−5 M) while dilator responses to adenosine (3×10−4 M) were maintained.
In experiments requiring measurement of changes in [Ca2+]i, vessels were incubated (60 min, room temperature) with 1 μM fura 2-acetoxy methylester (fura 2-AM; Molecular Probes, Eugene, OR, U.S.A.) in buffer containing 0.5% DMSO and 0.01% pluronic. The fura 2 solution was applied to the albuminal surface of the vessel to restrict dye loading to the vascular smooth muscle layer (Meininger et al., 1991; Zou et al., 1995). The dye loading procedure was followed by a 30 min wash period with PSS. Fura 2-AM loaded arterioles were exposed to epi-illumination (75 W Xenon source) with excitation wavelengths of 340, 360 and 380 nm using a spinning filter wheel operating at 20 Hz. Fluorescence emission at 510 nm was measured using a photomultiplier tube (Hamematsu, Bridgewater, NJ, U.S.A.) in combination with a signal conditioner (Texas A&M University, TX, U.S.A.). Simultaneous transillumination with wavelengths greater than 610 nm provided a non-fluorescent image which enabled video caliper measurement of internal arteriolar diameter while fluorescence intensities were measured. The high wavelength image was directed, by a beam splitter, to a CCD camera. This procedure did not interfere with measurements of Ca2+-related fluorescence. Fluorescent image intensities were expressed as the 340 : 380 nm ratio to allow quantitative estimates of changes in arteriolar wall [Ca2+]i and the intensity following excitation at 360 nm was recorded during Mn2+ quench studies as an index of Ca2+ entry. Details of these procedures have been presented in previous publications (Zou et al., 1995; Hill et al., 2000; Potocnik et al., 2000). Diameter of the arterioles was measured on the video image using electronic calipers and the data stored using a MacLab A-D system.
Experimental protocols
Effect of 2APB on arteriolar tone and reactivity
Initial studies were conducted to characterize the effect of 2APB on steady-state myogenic tone. Arterioles (n=5) under isobaric conditions (70 mmHg) were superfused with increasing concentrations of 2APB over the range 1 – 300 μM; with each concentration being applied for 10 min. In separate studies fura 2 loaded vessels (n=5) were examined to determine the effects of 2APB on [Ca2+]i under the same isobaric conditions. Experiments were performed in the absence and presence of the Ca2+ indicator to control for possible Ca2+ buffering effects of the fura 2.
To determine the effect of 2APB on acute vasoconstrictor responses vessels were exposed to phenylephrine (10−9 – 3×10−4 M; n=10), KCl (15 – 60 mM; n=3), or an acute pressure step (50 – 120 mmHg; n=6) in the absence or presence of 2APB (50 μM). Concentrations of phenylephrine and KCl were applied until a maximal response occurred while pressure steps were maintained for 5 min. KCl solutions were prepared by isosmotic exchange of Na+ for K+. Vessels were superfused with 2APB for 20 min prior to, and during, the application of the vasoconstrictor stimuli.
Effect of 2APB on non-voltage gated Ca2+ entry
To specifically examine Ca2+ entry into arteriolar smooth muscle via non-voltage gated mechanisms and to identify capacitative Ca2+ entry, arterioles were exposed to 0 mM Ca2+, 0.5 mM EGTA buffer containing nifedipine (1 or 10 μM) for 10 min. Ca2+ (2.5 mM) containing buffer was then returned to the vessels in the continued presence of nifedipine. Vessel diameter and [Ca2+]i were monitored during both components of the protocol. As a measure of involvement of IP3 receptor-mediated mechanisms, Ca2+ entry was examined in the absence and presence of 2APB (50 μM) using the above protocol.
As the magnitude of Ca2+ entry into smooth muscle of cannulated arterioles is known to be related to the level of intraluminal pressure (Zou et al., 1995) a separate series of studies were performed to examine the effect of pressure on non-voltage gated Ca2+ entry. Using a similar protocol to the above, studies were conducted at 10, 70 and 120 mmHg.
Ca2+ entry following thapsigargin treatment
As a positive control, additional studies were performed using the SR Ca2+ ATPase inhibitor, thapsigargin (1 μM), which has been shown in a number of cell types to cause Ca2+ store depletion, consequently providing a stimulus for capacitative Ca2+ entry (Putney & McKay, 1999). In an initial series of studies vessels were exposed to thapsigargin while being superfused with Ca2+ containing buffer. After steady-state changes in [Ca2+]i and diameter were achieved 2APB was added to the superfusate to inhibit IP3 receptor-mediated mechanisms of Ca2+ entry. In a second series of studies arterioles were superfused with Ca2+ free buffer containing nifedipine (1 μM) after which thapsigargin (1 μM) was added to the bath. Ca2+ (2.5 mM) was then returned to the superfusate to demonstrate capacitative Ca2+ entry followed by addition of 2APB (50 μM).
All experiments using thapsigargin were performed at an intraluminal pressure of 70 mmHg.
Effect of 2APB on rate of Ca2+ entry as assessed by Mn2+ quench
To further demonstrate that 2APB specifically inhibits a Ca2+ entry component, the ability of Mn2+ to quench fura 2 fluorescence was compared in the absence and presence of 2APB (50 μM). Mn2+ is known to enter cells via pathways that conduct Ca2+ such that the rate of quench of the fura 2 fluorescence at the isosbestic point (360 nm) for the indicator may be used as a measure of expected Ca2+ entry under a given condition (Sage et al., 1989; Wang & Van Breemen, 1997). Prior to addition of a superfusate containing 1 mM Mn2+ and 0 mM Ca2+ arterioles were treated with nifedipine (10 μM in Ca2+ free buffer containing 0.5 mM EGTA) to block any contribution from voltage gated Ca2+ channels. Separate vessel groups were used for assessing Mn2+ quench in either the absence or presence of 2APB. Mn2+ quench experiments were performed at an intraluminal pressure of 70 mmHg.
Involvement of the actin cytoskeleton in arteriolar vascular smooth muscle capacitative Ca2+ entry
To examine whether capacitative Ca2+ entry into arteriolar smooth muscle was dependent on a close proximity of the SR and the plasma membrane, non-voltage gated Ca2+ entry was determined before and after treatment with the cytoskeletal disrupting agent cytochalasin D. Segments of cannulated arteriole were incubated for 60 min (34°C) with cytochalasin D (1 μM). This concentration of cytochalasin was chosen based on our own studies and those of others showing it to be effective in inhibiting capacitative Ca2+ entry in cultured endothelial cells (Holda & Blatter, 1997; Bishara et al., unpublished observations) and that confocal microscopy verified that cytochalasin (1 μM) disrupts the cytoskeleton of cultured vascular smooth muscle cells (data not shown). Further, this concentration has been used in intact smooth muscle preparations to alter agonist-induced signalling pathways and inhibit agonist-induced contractile responses (Mehta et al., 2000). Capacitative Ca2+ entry was studied following return of extracellular Ca2+ containing solutions to vessels treated with nifedipine in 0 mM Ca2+ buffer solutions (see above).
Statistical analyses
Diameter and [Ca2+]i results are presented using two approaches. Results from both concentration response studies for 2APB and phenylephrine, and the 50 – 120 mmHg pressure step, are presented with values of diameter and 340 : 380 nm fluorescence ratio normalized to values obtained in 0 mM Ca2+, 0.5 mM EGTA buffer (i.e. [Ca2+]i min and passive diameter). The results of the capacitative Ca2+ influx studies are presented as per cent change from baseline (i.e. immediately prior to 2.5 mM Ca2+ and or drug treatment).
Group data is shown as mean±s.e.mean. Statistical differences between treatments has been determined by analysis of variance (ANOVA) with appropriate post hoc tests. Statistical significance was accepted at the P<0.05 level.
Chemicals and reagents
2APB (Aldrich, New South Wales, Australia) was prepared as a 0.1 M stock solution in 100% ethanol; subsequent dilutions were made in physiological salt solution. Thapsigargin (Sigma, New South Wales, Australia) was stored as a 10 mM stock solution in DMSO with further dilutions being made in physiological salt solution. Nifedipine (Sigma) was dissolved in DMSO at a concentration of 10−2 M and diluted in physiological salt solution. Solutions containing nifedipine were protected from light. Cytochalasin D (Sigma) was dissolved in DMSO and stored as aliquots at −20°C; subsequent dilutions were made in PSS. At the dilutions used there were no effects of the vehicles alone.
Results
Effect of 2APB on arteriolar tone and reactivity
De-endothelialized arterioles showed a concentration-dependent vasodilation to 2APB with 100 μM dilating vessels from a baseline of 37±4% (% of maximal diameter) to 54±4% (P<0.05; Figure 1a). A similar vasodilator effect was observed in arterioles loaded with Fura 2 (Figure 1b). Despite causing significant vasodilatation 2APB did not cause a detectable change in global [Ca2+]i (Figure 1b).
Figure 1.
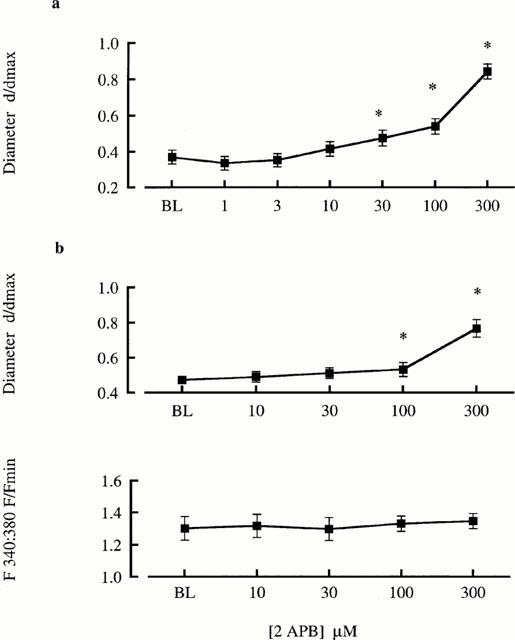
2APB concentration-response relationships for cannulated arterioles (70 mmHg) in the absence (a) and presence (b) of fura 2 loading (1 μM). Diameters are presented normalized to maximum diameter under passive conditions and changes in [Ca2+]i are presented as the change in 340 : 380 nm fluorescence ratio normalized to minimum values under passive conditions. Results are shown as mean±s.e.mean; n=5; *P<0.05.
The effect of 2APB (50 μM) on arteriolar responsiveness to phenylephrine is shown in Figures 2 and 3. 2APB caused a significant rightward shift in the concentration-diameter relationship for arterioles irrespective of fura 2 loading (Figure 2). Calculated EC50 values for the combined vessel sets were 0.12±0.4 μM under control conditions and 2.68±1.0 μM in the presence of 2APB (n=10; P<0.02). Ca2+ responses during phenylephrine exposure have been plotted in terms of the maximal and steady-state responses for each agonist concentration (Figure 3). This approach was taken as previous studies have shown that cannulated cremaster arterioles respond to adrenergic agonists with a biphasic Ca2+ response (Zou et al., 2000). 2APB caused marked inhibition of the initial peak Ca2+ response (Figure 3a) while also attenuating the much smaller increase in [Ca2+]i seen under steady-state conditions (Figure 3b).
Figure 2.
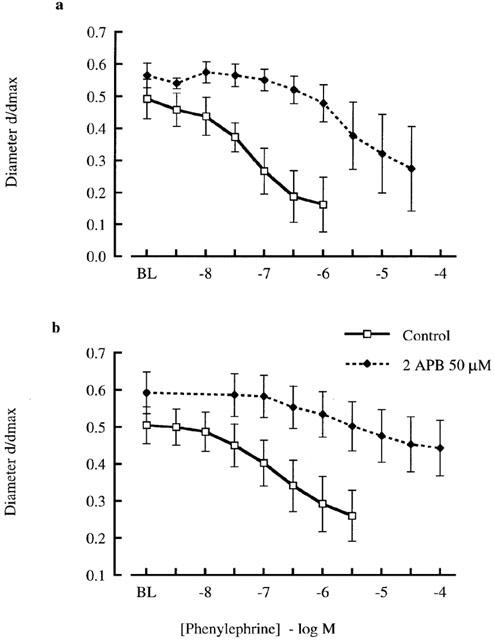
Effect of 2APB (50 μM) on phenylephrine concentration-response relationships for cannulated arterioles (70 mmHg) in the absence (a) and presence (b) of fura 2 loading (1 μM). Diameters are presented normalized to maximum diameter under passive conditions. Results are shown as mean±s.e.mean; n=5; *P<0.05.
Figure 3.
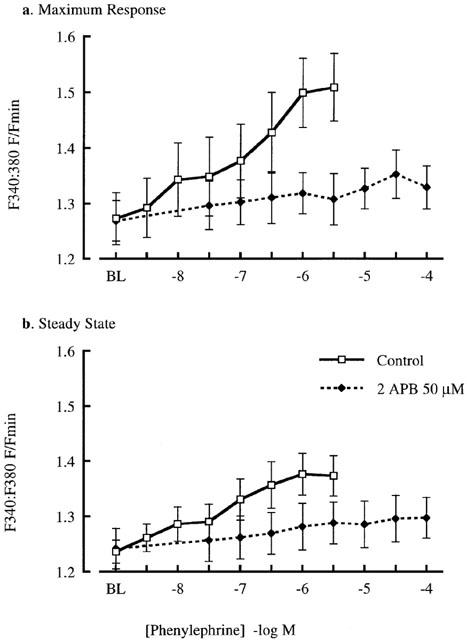
Effect of 2APB (50 μM) on phenylephrine-induced changes in [Ca2+]i. Data is presented for both the peak change in [Ca2+]i (a) and that occurring after a steady-state response had been reached (b). Changes in [Ca2+]i are presented as the change in 340 : 380 nm fluorescence ratio relative to values obtained under passive conditions. Results are shown as mean±s.e.mean; n=5; *P<0.05.
In contrast to inhibition of phenylephrine responsiveness 2APB (50 μM) had no apparent effect on the contractile response to KCl (Figure 4). For example 60 mM KCl constricted vessels to 20±3% (per cent of maximal diameter) under control conditions compared to 16±4% in the presence of 2APB (n=3; not significantly different).
Figure 4.
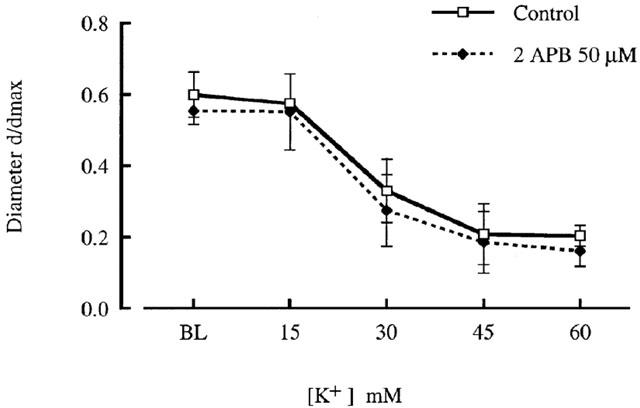
Effect of 2APB (50 μM) on KCl concentration-response relationships for cannulated arterioles (70 mmHg). Diameters are presented normalized to maximum diameter under passive conditions. Results are shown as mean±s.e.mean; n=3.
To determine the effect of 2APB on acute myogenic vasoconstriction the response to a 50 – 120 mmHg pressure step was examined in the absence and presence of the inhibitor. Under control conditions the pressure step resulted in an immediate passive distension followed by active vasoconstriction to diameters smaller than those observed prior to the pressure change (Figure 5). 2APB (50 μM) while not altering the passive distension phase completely inhibited the myogenic vasoconstriction. The inhibition of the vasoconstrictor response occurred despite similar changes in [Ca2+]i (Figure 5).
Figure 5.
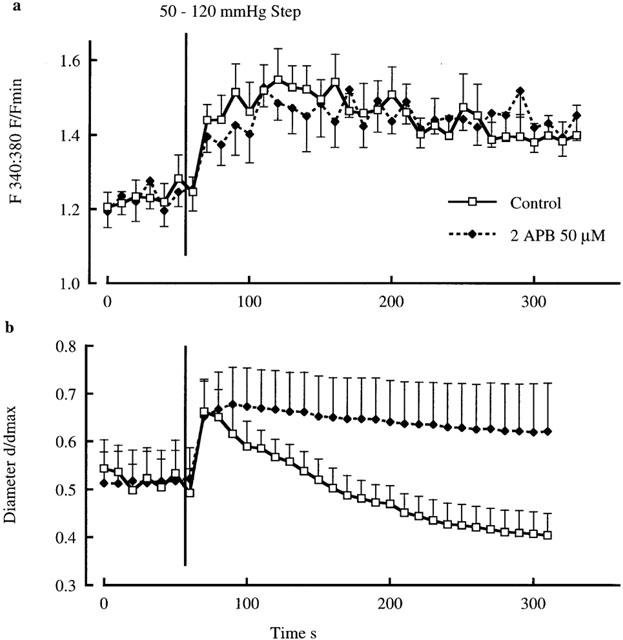
Effect of 2APB (50 μM) on diameter and [Ca2+]i responses to an acute 50 – 120 mmHg pressure step. Pressure-induced changes in [Ca2+]i are shown in (a) and changes in diameter in (b). Results are shown as mean±s.e.mean; n=6.
To determine whether 2APB depleted intracellular Ca2+ stores vessels were exposed to 20 mM caffeine in the absence and presence of 2APB (30 and 100 μM). Similar diameter and Ca2+ responses to caffeine were obtained in the presence of 2APB and under control conditions (n=3) suggesting that 2APB does not itself cause a depletion of SR Ca2+. Example responses are shown in Figure 6.
Figure 6.
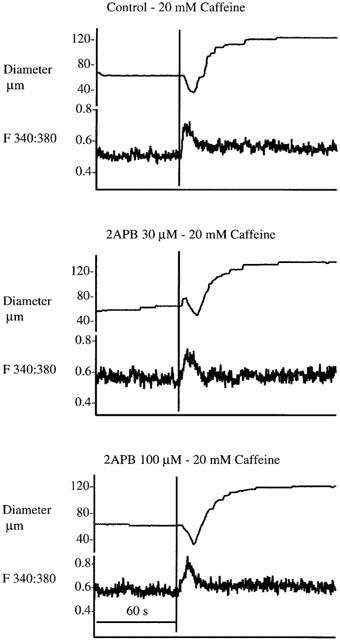
Data illustrating a lack of effect of 2APB (30 and 100 μM). Example responses typical of three separate experiments are shown. Tracings illustrate arteriole diameter responses and associated changes in [Ca2+]i following exposure to caffeine (20 mM) in the absence and presence of 2APB.
Effect of 2APB on non-voltage gated Ca2+ entry
Initial studies examined Ca2+ entry after exposing arterioles (at 70 mmHg) to 0 mM Ca2+, 0.5 mM EGTA buffer containing 1 μM nifedipine. This dose of nifedipine was chosen as it totally inhibits the contractile response to 60 mM KCl. The mean response of five arterioles to re-addition of extracellular Ca2+ (2.5 mM) in the maintained presence of nifedipine is shown in Figure 7a. Vessels initially dilated in response to 0 mM Ca2+, 0.5 mM EGTA buffer containing 1 μM nifedipine and demonstrated a decrease in intracellular Ca2+. Re-addition of extracellular Ca2+ resulted in an influx of Ca2+ with the 340:380 nm fluorescence ratio increasing from 1.00±0.01 to 1.20±0.06 (P<0.015) over a period of approximately 20 s (Figure 7). This influx of Ca2+ was totally blocked by pre-exposure to 2APB (50 μM); with the 340 : 380 nm fluorescence ratio changing from 1.02±0.02 to 1.04±0.04 (n.s., P=0.33) on re-addition of Ca2+ (Figure 7b).
Figure 7.
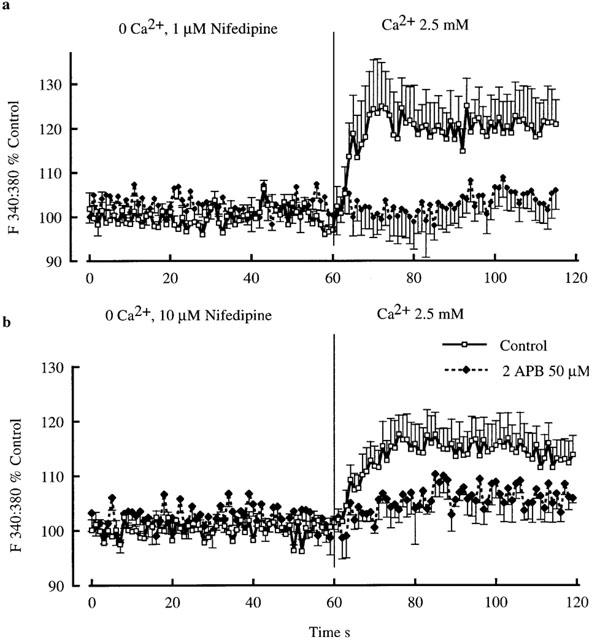
Demonstration of capacitative Ca2+ entry in cannulated and pressurized (70 mmHg) arterioles. Vessels were initially superfused with 0 mM Ca2+ buffer and 0.5 mM EGTA, containing either 1 μM (upper panel) or 10 μM (lower panel) nifedipine. Ca2+ influx was then demonstrated by addition of 2.5 mM Ca2+ to the superfusate. Results are shown as mean±s.e.mean; n=5.
Additional experiments were performed using 10 μM nifedipine in case of incomplete blockade of voltage-gated channels at the low concentration. Consistent with the above experiments a Ca2+ influx component persisted and was attenuated by 2APB (Figure 7b).
To determine whether the non-voltage gated Ca2+ entry component was affected by the level of intraluminal pressure, influx was compared at 10, 70 and 120 mmHg. As shown in Figure 8a, Ca2+ entry was significantly (P<0.02) higher at a pressure of 10 (124.3±2.4%) as compared to either 70 (117.3±5.1%) or 120 (108.9±1.2%) mmHg. Exposure to 2APB significantly attenuated Ca2+ entry (Figure 8b) at each pressure (10 mmHg, 108.9±1.0%; 70 mmHg, 101.2±3.0%; and 120 mmHg, 100.1±2.7%).
Figure 8.
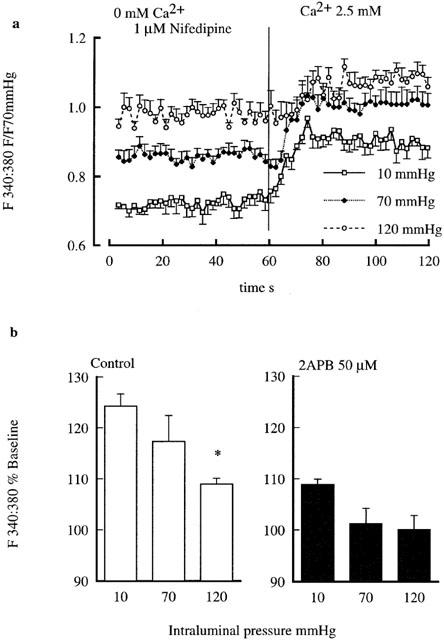
Effect of intraluminal pressure on capacitative Ca2+ entry. (a) Shows Ca2+ entry responses at intraluminal pressures of 10, 70 and 120 mmHg. Capacitative Ca2+ entry was demonstrated by re-addition of Ca2+ (2.5 mM) after prior exposure to nifedipine (1 μM) in Ca2+ free buffer. Data are presented as the 340 : 380 nm fluorescence ratio normalized to values obtained at 70 mmHg in 2.5 mM Ca2+ buffer. (b, left side) illustrates the extent of Ca2+ entry (as shown in a) normalized as per cent of the baseline response while the right hand side shows that 2APB inhibits Ca2+ entry at each of the intraluminal pressures examined. Results are shown as mean±s.e.mean; n=6 control; n=3, 2APB; *P<0.05.
Ca2+ entry following thapsigargin treatment
In the presence of Ca2+ (2.5 mM) containing PSS, thapsigargin (1 μM) caused a biphasic change in diameter with an initial phase of dilation followed by constriction to 60±2.0% (n=5) of basal diameter (Figure 9a). Intracellular Ca2+ increased over 3 – 5 min to reach a steady-state level of 154±9% of the pre-thapsigargin level (P<0.02; Figure 9a). 2APB (50 μM), added after a steady-state response was obtained caused a rapid dilation to 120±2.2% and a decrease in [Ca2+]i (113±5%). In three additional experiments thapsigargin (1 μM) was added to the vessels after superfusion with 0 Ca2+, 0.5 mM EGTA buffer containing 1 μM nifedipine. Re-addition of Ca2+ in the presence of thapsigargin resulted in a similar increase in Ca2+ influx (16±3%) compared to that occurring in the absence of the Ca2+ ATPase inhibitor (15±4%) consistent with an effect on capacitative Ca2+ entry. Ca2+ entry in the presence of nifedipine and thapsigargin was blocked by 2APB (50 μM; Figure 9b).
Figure 9.
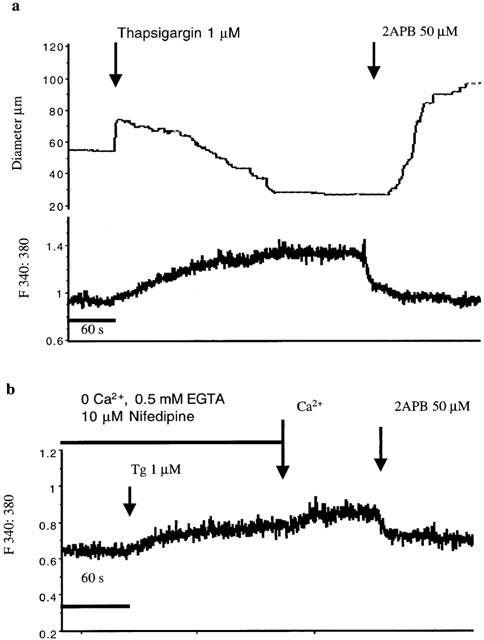
Effect of 2APB (50 μM) on capacitative Ca2+ entry in arterioles following thapsigargin (1 μM) exposure. Example tracings are shown for experiments performed in the presence of Ca2+ containing superfusate solution (upper panel) and using the Ca2+ re-addition protocol (b). Results in (a) are representative of five experiments and (b) of three experiments.
Effect of 2APB on rate of Ca2+ entry as assessed by Mn2+ quench
To further demonstrate that 2APB was exerting an effect on Ca2+ entry through non-voltage gated mechanisms the rate of Mn2+ quenching of the fura 2 signal was examined in arterioles treated with nifedipine (10 μM). As shown in Figure 10, 2APB (50 μM) decreases the rate of quench of the fluorescence signal consistent with inhibition of cation entry. Calculated slope values for the 30 s period following addition of Mn2+ were 0.0051±0.0003 in the absence of 2APB and 0.0035±0.0003 in the presence of 2APB (P<0.005).
Figure 10.
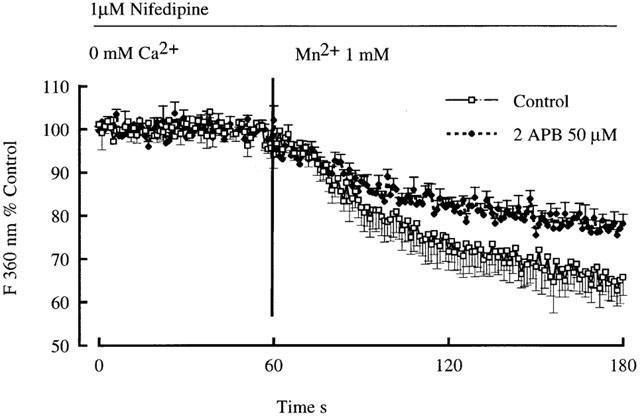
Demonstration of non-voltage gated Ca2+ entry in cannulated arterioles (70 mmHg) using the Mn2+ quench technique. Mn2+ (1 mM) was added following treatment of arterioles with Ca2+ free buffer containing 1 μM nifedipine. Results are shown as mean±s.e.mean; n=6.
Involvement of the actin cytoskeleton in arteriolar vascular smooth muscle capacitative Ca2+ entry
Capacitative Ca2+ entry was studied before and after exposure of arterioles to the cytoskeletal disrupting agent cytochalasin D (1 μM). This concentration of cytochalasin was previously shown to be effective in inhibiting capacitative Ca2+ entry in endothelial cells. Cytochalasin D treatment did not significantly decrease the extent of capacitative Ca2+ influx (Figure 11). Further, 2APB significantly inhibited Ca2+ entry in both control and cytochalasin treated vessels (Figure 11).
Figure 11.
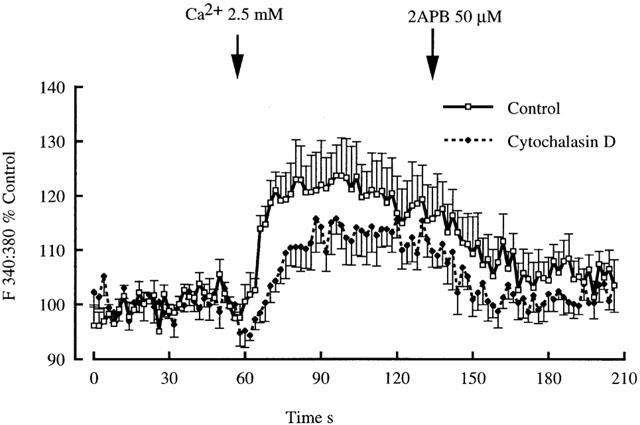
Effect of cytochalasin D (1 μM) treatment on non-voltage gated Ca2+ entry in cannulated arterioles (70 mmHg). Arterioles were pre-treated with cytochalasin for 60 min before examining capacitative Ca2+ entry as in earlier protocols. 2APB was added to demonstrate that Ca2+ entry was similarly inhibited in both groups of arterioles. Results are shown as mean±s.e.mean; n=5.
Discussion
The principal findings of these studies relate to (1) the demonstration of capacitative Ca2+ entry mechanisms in intact arterioles and (2) the involvement of IP3 receptor-mediated mechanisms in arteriolar tone and reactivity. In relation to the second of these observations it was demonstrated that the putative IP3 receptor modulator 2APB markedly inhibits the vasoconstrictor response of single cannulated arterioles to phenylephrine and attenuates the associated changes in intracellular Ca2+. This did not appear to be a non-specific inhibition of contraction as the mechanical response to KCl (15 – 60 mM), an agent which is believed to exert its effects purely via voltage-gated Ca2+ entry, was maintained in the presence of 2APB. In addition, 2APB was found to exert a modest dilator effect on arterioles maintained under isobaric (70 mmHg) conditions without altering Ca2+i while totally inhibiting the mechanical response to an acute intraluminal pressure step (50 – 120 mmHg), again without affecting [Ca2+]i. On the basis that 2APB inhibits both myogenic tone and acute myogenic constriction without altering global [Ca2+]i, it is suggested that the IP3 receptor, or related mechanisms are involved in local intracellular Ca2+-contraction coupling. Supporting the possibility that the IP3 receptor is involved in processes other than SR Ca2+ release per se the present studies also demonstrated that 2APB blocked a component of Ca2+ entry that did not involve nifedipine-sensitive mechanisms. Comparison with other cell systems would suggest that this is indicative of store-depletion or capacitative Ca2+ entry.
Studies of Maruyama et al. (1997) have characterized 2APB as an IP3 receptor antagonist or modulator. These investigators showed 2APB to inhibit IP3-induced Ca2+ release from rat cerebellar microsomes without affecting IP3 binding to its receptor. 2APB was also shown to inhibit Ca2+ entry into platelets and neutrophils while not affecting ryanodine-sensitive Ca2+ stores. In studies using cultured bovine aortic endothelial cells we have confirmed that 2APB inhibits agonist-induced (ATP and bradykinin) Ca2+ release and subsequent capacitative Ca2+ entry. Further, it does not affect agonist-induced IP3 production or alter the binding of IP3 to a binding peptide (Bishara et al., unpublished observations).
Recently, it has been suggested that 2APB may block capacitative entry at sites other than the endoplasmic reticulum IP3 receptor. In particular, a direct effect on plasma membrane Ca2+ entry channels has been proposed (Broad et al., 2001; Dobrydneva et al., 2001). Whether these studies are of direct relevance to arteriolar smooth muscle is uncertain, as given the number of TRP channels identified to date (Putney & McKay, 1999; Friel, 1996) there is the possibility of tissue-specific differences. In regard to the present results, inhibition at either site does not negate the underlying hypothesis that functional arteriolar smooth muscle exhibits capacitative Ca2+ entry. In relation to other possible sites where 2APB could impact on Ca2+ handling it is important to note that it had no apparent effect on Ca2+ entry through voltage-gated Ca2+ channels and it did not alter the extent of Ca2+ release by caffeine.
From the results of the present studies it would appear that 2APB is more effective in inhibiting the acute contractile responses to phenylephrine or an acute pressure step than steady-state isobaric arteriolar tone. Thus 50 μM 2APB caused a marked rightward shift in the phenylephrine concentration response curve and prevented the acute myogenic response to a step change in pressure while having comparatively little effect on steady-state arteriolar tone at 70 mmHg. This could conceivably indicate that once steady-state contraction is achieved in arteriolar smooth muscle, the requirement for IP3 receptor-mediated mechanisms of Ca2+ handling is reduced compared to during the initiation of the contraction. Although the IP3 receptor may be linked to capacitative Ca2+ entry it must be remembered that under physiological conditions these vessels would be expected to have a membrane potential of approximately −35 mV favouring Ca2+ entry via voltage-gated Ca2+ channels. Further, calcium channel antagonists such as nifedipine and verapamil inhibit steady-state myogenic tone and induce passive mechanical behaviour to subsequent changes in intraluminal pressure (Wesselman et al., 1996; Knot et al., 1998; Potocnik et al., 2000).
An interesting observation was that under some circumstances 2APB exposure resulted in dissociation between [Ca2+]i and contractile activity. For example, during the acute pressure step [Ca2+]i increased to comparable levels (Figure 7) both in the absence and presence of 2APB (50 μM) yet in the presence of this compound, contraction was almost totally inhibited. Similarly, the higher concentrations of 2APB (100 and 300 μM) led to a reduction in steady-state tone without a concomitant decrease in [Ca2+]i. These data may be explained by the relative insensitivity of global [Ca2+]i measurements for estimating regional or focal changes of Ca2+ that are of functional significance.
Evidence from a variety of cell types suggests that Ca2+ changes within cellular compartments may activate distinct intracellular responses. For example Fagan et al. (1998) have shown that Ca2+ entering C6-2B rat glioma cells by a store depletion or capacitative mechanism can regulate Ca2+ sensitive isoforms of adenylyl cyclase while a global change in intracellular Ca2+ (as caused by ionomycin) is ineffective in regulating the same adenylyl cyclase subtype. In arteriolar smooth muscle it is likely that Ca2+ regulates functions other than contraction, such as ion channel activity. For example a localized increase in Ca2+ between the SR and plasma membrane may conceivably activate KCa channels and Na+-Ca2+ exchange while not directly affecting contraction. Such regional differences in Ca2+ may in fact limit rises in bulk cytoplasmic Ca2+ and thereby attenuate contractile activity (van breemen et al., 1995). Similarly we have previously found that in the presence of verapamil (10 μM) arterioles exposed to phenylephrine (0.1 μM) or a 50 – 120 mmHg pressure step exhibit a similar change in intracellular Ca2+ yet contraction only occurs in response to the alpha agonist (Zou et al., 2000). Thus it is conceivable that changes in [Ca2+]i underlie multiple functions in myogenically active vessels and therefore highlight the need for an understanding of the spatio-temporal aspects of Ca2+ control.
A further novel finding of these studies is the existence of what appears to be a component of capacitative Ca2+ entry in cannulated and mechanically functional arterioles. In this context it is defined as Ca2+ entry that follows a period of extracellular Ca2+ deprivation when voltage-gated Ca2+ influx is blocked with nifedipine (1 or 10 μM). Interestingly there was not an observable increase in the Ca2+ influx by this non-voltage gated mechanism when intraluminal pressure was raised to higher levels. Paradoxically there was evidence for an inverse relationship despite the fact that our previous studies have shown global [Ca2+]i to increase with increasing intraluminal pressure (Zou et al., 1995, 2000). Thus it appears that capacitative Ca2+ entry per se is not directly related to myogenic vasoconstriction but perhaps responds to the pressure-induced increase in [Ca2+]i or other signalling molecules. Given this the question then becomes what is the stimulus for capacitative Ca2+ entry in this preparation? Presumably the influx on re-addition of Ca2+ to the superfusate reflects a loss of intracellular Ca2+ and, in particular, SR Ca2+ during the exposure to a 0 mM Ca2+ buffer containing 0.5 mM EGTA and nifedipine. Under similar conditions previous studies have shown that arterioles do tend to lose Ca2+ from intracellular stores as a function of time (Hynes & Duling, 1991).
That 2APB was having an effect on Ca2+ entry independent of voltage-gated Ca2+ entry as opposed to Ca2+ release from the sarcoplasmic reticulum was further confirmed using the Mn2+ quench technique. Mn2+ quenches Fura 2 fluorescence at a rate determined by factors controlling cation influx (Sage et al., 1989). As voltage-gated Ca2+ entry was first inhibited by exposing arterioles to nifedipine it is likely that quenching of the Fura 2 signal, in part, reflects Mn2+ entry via a capacitative Ca2+ entry mechanism. The observation that 2APB decreased the rate of quenching of the fluorescent signal is consistent with this agent impairing a non-voltage dependent Ca2+ entry mechanism.
Although early studies in vascular smooth muscle provided impetus to the idea of capacitative Ca2+ entry (Casteels & Droogmans, 1981) characterization of such mechanisms has largely been undertaken in non-excitable cells (Sage et al., 1989). In such studies evidence has been provided for Ca2+ selective entry channels including calcium release-activated (CRAC) and TRP channels (Putney & McKay, 1999; Ma et al., 2000). Similar channels, specific for Ca2+ entry, have not been identified in smooth muscle although evidence has been provided for non-selective cation channels the activity of which is modulated by the filling state of intracellular Ca2+ stores (Arnon et al., 2000; Trepakova et al., 2001). Under physiological conditions such channels would likely carry Na+ which could lead to membrane depolarization and opening of voltage-dependent Ca2+ channels. In the present studies, however, capacitative Ca2+ entry was still evident in the presence of nifedipine suggesting the existence of at least a component of Ca2+ entry that was not related to voltage dependency.
Two general hypotheses have been proposed to explain the coupling between store-depletion and Ca2+ entry through non-voltage dependent mechanisms. The first of these is that store depletion leads to the production of a diffusable factor which modulates Ca2+ entry. One such factor, termed calcium influx factor, was suggested to be a small molecular weight phosphorus containing compound (Randriamampita & Tsien, 1991) while other studies have implicated a role for arachidonic acid metabolites (Rzigalinski et al., 1999). The second hypothesis suggests that store depletion leads to a conformational change which results in a physical coupling between the endoplasmic reticulum and cell membrane Ca2+ entry channels. Support for this has been provided by studies demonstrating that capacitative calcium entry is inhibited by agents condensing (calyneurin) or disrupting (cytochalasin D) the actin cytoskeleton (Holda & Blatter, 1997; Ma et al., 2000; Bishara et al., unpublished observations). With respect to the latter, in related studies we have found 1 μM cytochalasin (as used in the current study) markedly inhibits capacitative calcium entry in cultured endothelial cells. In apparent contrast this concentration of cytochalasin did not markedly inhibit capacitative calcium entry in arterioles. This may reflect differences in the underlying Ca2+ entry mechanisms in the two cell types or suggest that higher concentrations of cytochalasin are required in vascular smooth muscle to disrupt a physical relationship between the sarcoplasmic reticulum and Ca2+ entry. In regard to the latter, although higher concentrations are required to cause gross changes in cell morphology (Tseng et al., 1997), a concentration of 1 μM cytochalasin does inhibit force production in tracheal smooth muscle (Mehta et al., 2000; Youn et al., 1998). Alternatively, capacitative calcium entry in endothelial cells may be dependent on dynamic properties of the cytoskeleton as opposed to purely structural features. Regardless of the underlying reason it appears that vascular smooth muscle and endothelial cells differ with respect to sensitivity to cytochalasin D for inhibiting capacitative calcium entry.
In summary, the present studies demonstrate capacitative Ca2+ entry in functional arterioles and provide support for IP3 receptor-related mechanisms in myogenic tone and reactivity. Capacitative Ca2+ entry while being evident in the presence of blockade of dihydropyridine-sensitive Ca2+ entry mechanisms does not appear to play a direct role in supplying Ca2+ for myogenic tone as its efficacy does not increase with increasing intraluminal pressures. Inhibition of capacitative Ca2+ entry at higher intraluminal pressures may indicate that steady-state levels of [Ca2+]i, or another pressure-induced signalling molecule, modulates non-voltage gated mechanisms of Ca2+ entry in arteriolar smooth muscle. The effectiveness of 2APB in inhibiting both non-voltage gated Ca2+ entry and responsiveness to an acute pressure step (in the face of an unchanged global cytosolic Ca2+ level) is consistent with it inhibiting an element of an axis involving IP3-mediated and or capacitative Ca2+ entry mechanisms in myogenic reactivity. It is suggested that such mechanisms participate on a localized intracellular level to couple the myogenic stimulus to contraction.
Acknowledgments
Work described in these studies was supported by grants from the National Health and Medical Research Council of Australia and the National Heart Foundation.
Abbreviations
- 2APB
2 amino ethoxy diphenyl borate
- [Ca2+]i
intracellular Ca2+
- IP3
inositol trisphosphate
- PLC
phospholipase C
References
- ARNON A., HAMLYN J.M., BLAUSTEIN M.P. Na+ entry via store-operated channels modulates Ca2+ signaling in arterial myocytes. Am. J. Physiol. 2000;278:C163–C173. doi: 10.1152/ajpcell.2000.278.1.C163. [DOI] [PubMed] [Google Scholar]
- ASCHER-LANDSBERG J., SAUNDERS T., ELOVITZ M., PHILLIPPE M. The effects of 2-aminoethoxydiphenyl borate, a novel inositol 1,4,5-trisphosphate receptor modulator on myometrial contractions. Biochem. Biophys. Res. Commun. 1999;264:979–982. doi: 10.1006/bbrc.1999.1602. [DOI] [PubMed] [Google Scholar]
- BROAD L.M., BRAUN F.J., LIEVREMONT J.P., BIRD G.S., KUROSAKI T., PUTNEY J.W., JR Role of the phospholipase C-inositol 1,4,5-trisphosphate pathway in calcium release-activated calcium current (Icrac) and capacitative calcium entry. J. Biol. Chem. 2001;276:15945–15952. doi: 10.1074/jbc.M011571200. [DOI] [PubMed] [Google Scholar]
- CASTEELS R., DROOGMANS G. Exchange characteristics of the noradrenaline-sensitive calcium store in vascular smooth muscle cells of the rabbit ear artery. J. Physiol. 1981;317:263–279. doi: 10.1113/jphysiol.1981.sp013824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVIS M.J., HILL M.A. Signaling mechanisms underlying the vascular myogenic response. Physiol. Rev. 1999;79:387–423. doi: 10.1152/physrev.1999.79.2.387. [DOI] [PubMed] [Google Scholar]
- DOBRYDNEVA Y., WILLIAMS R.L., BLACKMORE P.F. 2-Aminoethoxydiphenylborate (2APB): a direct inhibitor of Ca2+ influx channels in human platelets. FASEB J. 2001;15:A296. [Google Scholar]
- DULING B.R., GORE R.W., DACEY R.G., JR, DAMON D.N. Methods for isolation, cannulation, and in vitro study of single microvessels. Am. J. Physiol. 1981;241:H108–H116. doi: 10.1152/ajpheart.1981.241.1.H108. [DOI] [PubMed] [Google Scholar]
- FELLNER S.K., ARENDSHORST W.J. Capacitative calcium entry in smooth muscle cells from preglomerular vessels. Am. J. Physiol. 1999;277:F533–F542. doi: 10.1152/ajprenal.1999.277.4.F533. [DOI] [PubMed] [Google Scholar]
- FELLNER S.K., ARENDSHORST W.J. Ryanodine receptor and capacitative Ca2+ entry in fresh preglomerular vascular smooth muscle cells. Kidney Int. 2000;58:1686–1694. doi: 10.1046/j.1523-1755.2000.00329.x. [DOI] [PubMed] [Google Scholar]
- FAGAN K.A., MONS N., COOPER D.M.F. Dependence of the Ca2+-inhibitable adenylyl cyclase of C6-2B glioma cells on capacitative Ca2+ entry. J. Biol. Chem. 1998;273:9297–9305. doi: 10.1074/jbc.273.15.9297. [DOI] [PubMed] [Google Scholar]
- FRIEL D.D. TRP: It's role in phototransduction and store-operated Ca2+ entry. Cell. 1996;85:617–619. doi: 10.1016/s0092-8674(00)81021-1. [DOI] [PubMed] [Google Scholar]
- HILL M.A., ZOU H., DAVIS M.J., POTOCNIK S.J., PRICE S. Transient increases in diameter and Ca2+i following acute pressure increases are not obligatory for myogenic constriction. Am. J. Physiol. 2000;278:H345–H352. doi: 10.1152/ajpheart.2000.278.2.H345. [DOI] [PubMed] [Google Scholar]
- HILL M.A., ZOU H., POTOCNIK S.J., MEININGER G.A., DAVIS J.M. Arteriolar smooth muscle mechanotransduction; Ca2+ signaling pathways underlying myogenic reactivity. J. App. Physiol. 2001;91:973–983. doi: 10.1152/jappl.2001.91.2.973. [DOI] [PubMed] [Google Scholar]
- HOLDA J.R., BLATTER L.A. Capacitative calcium entry is inhibited in vascular endothelial cells by disruption of cytoskeletal microfilaments. FEBS Lett. 1997;403:191–196. doi: 10.1016/s0014-5793(97)00051-3. [DOI] [PubMed] [Google Scholar]
- HYNES M.R., DULING B.R. Ca2+sensitivity of isolated arterioles from the hamster cheek pouch. Am. J. Physiol. Heart Circ. Physiol. 1991;260:H355–H316. doi: 10.1152/ajpheart.1991.260.2.H355. [DOI] [PubMed] [Google Scholar]
- INSCHO E.W., COOK A.K., MUI V., IMIG J.D. Calcium mobilization contributes to pressure-mediated afferent arteriolar vasoconstriction. Hypertension. 1998;31:421–428. doi: 10.1161/01.hyp.31.1.421. [DOI] [PubMed] [Google Scholar]
- JAGGAR J.H., WELLMAN G.C., HEPPNER T.J., PORTER V.A., PEREZ G.J., GOLLASCH M., KLEPPISCH T., RUBART M., STEVENSON A.S., LEDERER W.J., KNOT H.J., BONEV A.D., NELSON M.T. Ca2+ channels, ryanodine receptors and Ca2+-activated K+ channels: a functional unit for regulating arterial tone. Acta Physiol. Scand. 1998;164:577–587. doi: 10.1046/j.1365-201X.1998.00462.x. [DOI] [PubMed] [Google Scholar]
- KNOT H.J., STANDEN N.B., NELSON M.T. Ryanodine receptors regulate arterial diameter and [Ca2+] in cerebral arteries of rat via Ca2+-dependent K+ channels. J. Physiol. 1998;508:211–221. doi: 10.1111/j.1469-7793.1998.211br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAPORTE R., LAHER I. Sarcoplasmic reticulum-sarcolemma interactions and vascular smooth muscle tone. J. Vasc. Res. 1997;34:325–343. doi: 10.1159/000159242. [DOI] [PubMed] [Google Scholar]
- MA H-T., PATTERSON R.L., VAN ROSSUM D.B., BIRNBAUMER L., MIKOSHIBA K., GILL D.L. Requirement of the inositol trisphosphate receptor for activation of store-operated Ca2+ channels. Science. 2000;287:1647–1651. doi: 10.1126/science.287.5458.1647. [DOI] [PubMed] [Google Scholar]
- MARUYAMA T., KANAJI T., NAKADE S., KANNO T., MIKOSHIBA K. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of ins(1,4,5)P3-induced Ca2+ release. J. Biochem. (Tokyo) 1997;122:498–505. doi: 10.1093/oxfordjournals.jbchem.a021780. [DOI] [PubMed] [Google Scholar]
- MEHTA D., TANG D.D., WU M.F., ATKINSON S., GUNST S.J. Role of Rho in Ca2+-insensitive contraction and paxillin tyrosine phosphorylation in smooth muscle. Am. J. Physiol. 2000;279:C308–C318. doi: 10.1152/ajpcell.2000.279.2.C308. [DOI] [PubMed] [Google Scholar]
- MEININGER G.A., ZAWEIJA D.C., FALCONE J.C., HILL M.A., DAVEY J.P. Calcium measurement in isolated arterioles during myogenic and agonist stimulation. Am. J. Physiol. 1991;261:H950–H959. doi: 10.1152/ajpheart.1991.261.3.H950. [DOI] [PubMed] [Google Scholar]
- MIRIEL V.A., MAUBAN J.R., BLAUSTEIN M.P., WIER W.G. Local and cellular Ca2+ transients in smooth muscle of pressurized rat resistance arteries during myogenic and agonist stimulation. J. Physiol. 1999;518:815–824. doi: 10.1111/j.1469-7793.1999.0815p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NARAYANAN K., IMIG M., ROMAN R.J., HARDER D.R. Pressurization of isolated renal arteries increases inositol trisphosphate and diacylglycerol. Am. J. Physiol. 1994;266:H1840–H1845. doi: 10.1152/ajpheart.1994.266.5.H1840. [DOI] [PubMed] [Google Scholar]
- OSOL G., LAHER I., KELLEY M. Myogenic tone is coupled to phospholipase C and G protein activation in small cerebral arteries. Am. J. Physiol. 1993;265:H415–H420. doi: 10.1152/ajpheart.1993.265.1.H415. [DOI] [PubMed] [Google Scholar]
- POTOCNIK S.J., MURPHY T.V., KOTECHA N., HILL M.A. Effects of mibefradil and nifedipine on arteriolar myogenic responsiveness and intracellular Ca2+ Br. J. Pharmacol. 2000;131:1065–1072. doi: 10.1038/sj.bjp.0703650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PUTNEY J.W., MCKAY R.R. Capacitative calcium entry channels. Bioessays. 1999;21:38–46. doi: 10.1002/(SICI)1521-1878(199901)21:1<38::AID-BIES5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- RANDRIAMANPITA C., TSEIN R.Y. Emptying of intracellular Ca2+ stores releases a novel small messenger that simulates Ca2+ influx. Nature. 1991;364:809–814. doi: 10.1038/364809a0. [DOI] [PubMed] [Google Scholar]
- RZIGALINSKI B.A., WILLOUGHBY K.A., HOFFMAN J.W., FALCK J.R., ELLIS E.F. Calcium influx factor, further evidence it is 5,6,-epoxyeicosatrienoic acid. J. Biol. Chem. 1999;274:175–182. doi: 10.1074/jbc.274.1.175. [DOI] [PubMed] [Google Scholar]
- SAGE S.O., MERRITT J.E., HALLAM T.J., RINK T.J. Receptor-mediated calcium entry in fura-2-loaded human platelets stimulated with ADP and thrombin. Biochem. J. 1989;258:923–926. doi: 10.1042/bj2580923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SKUTELLA M., RUEGG U.T. Studies on capacitative calcium entry in vascular smooth muscle cells by measuring 45 Ca2+ influx. J. Recept. Signal Tranduct. Res. 1997;17:163–175. doi: 10.3109/10799899709036601. [DOI] [PubMed] [Google Scholar]
- TREPAKOVA E.S., GERICKE M., HIRAKAWA Y., WEISBROD R.M., COHEN R.A., BOLOTINA V.M. Properties of a native cation channel activated by Ca2+ store depletion in vascular smooth muscle cells. J. Biol. Chem. 2001;276:7782–7790. doi: 10.1074/jbc.M010104200. [DOI] [PubMed] [Google Scholar]
- TSENG S., KIM R., KIM T., MORGAN K.G., HAI C.M. F-actin disruption attenuates agonist-induced [Ca2+], myosin phosphorylation, and force in smooth muscle. Am. J. Physiol. 1997;27:C1960–C1967. doi: 10.1152/ajpcell.1997.272.6.C1960. [DOI] [PubMed] [Google Scholar]
- VAN BREEMEN C., CHEN Q., LAHER I. Superficial buffer barrier function of smooth muscle sarcoplasmic reticulum. Trends Pharmacol. Sci. 1997;16:98–105. doi: 10.1016/s0165-6147(00)88990-7. [DOI] [PubMed] [Google Scholar]
- WANG X., VAN BREEMEN C. Multiple mechanisms of activating Ca2+ entry in freshly isolated rabbit aortic endothelial cells. J. Vasc. Res. 1997;34:196–207. doi: 10.1159/000159223. [DOI] [PubMed] [Google Scholar]
- WESSELMAN J.P., VANBAVEL E., PFAFFENDORF M., SPAAN J.A. Voltage-operated calcium channels are essential for the myogenic responsiveness of cannulated rat mesenteric small arteries. J. Vasc. Res. 1996;33:32–41. doi: 10.1159/000159129. [DOI] [PubMed] [Google Scholar]
- WU J., KAMIMURA N., TAKEO T., SUGA S., WAKUI M., MARUYAMA T., MIKOSHIBA K. 2-Aminoethoxydiphenyl borate modulates kinetics of intracellular Ca(2+) signals mediated by inositol 1,4,5-trisphosphate-sensitive Ca(2+) stores in single pancreatic acinar cells of mouse. Mol. Pharmacol. 2000;58:1368–1374. doi: 10.1124/mol.58.6.1368. [DOI] [PubMed] [Google Scholar]
- YOUN T., KIM S.A., HAI C.M. Length-dependent modulation of smooth muscle activation: effects of agonist, cytochalasin, and temperature. Am. J. Physiol. 1998;274:C1601–C1607. doi: 10.1152/ajpcell.1998.274.6.C1601. [DOI] [PubMed] [Google Scholar]
- ZOU H., RATZ P.H, HILL M.A. Role of myosin phosphorylation and [Ca2+]i in myogenic reactivity and arteriolar tone. Am. J. Physiol. 1995;269:H1590–H1596. doi: 10.1152/ajpheart.1995.269.5.H1590. [DOI] [PubMed] [Google Scholar]
- ZOU H., RATZ P.H, HILL M.A. Temporal aspects of [Ca2+]i and myosin phosphorylation during myogenic and agonist-induced arteriolar constriction. J. Vasc. Res. 2000;37:556–567. doi: 10.1159/000054089. [DOI] [PubMed] [Google Scholar]