

Abstract
When administered subcutaneously HS-599, a new didehydroderivative of buprenorphine (18,19-dehydrobuprenorphine), produced a long-lasting antinociceptive response in rats. Its potency exceeded twice that of buprenorphine. In the tail-flick test it acted as a full agonist but in the plantar test only as a partial agonist. Whereas the μ-opioid antagonists naloxone and naltrexone antagonized HS-599 antinociception the δ-opioid antagonist naltrindole and the κ-opioid antagonist nor-binaltorphimine did not.
Unlike buprenorphine and morphine, HS-599 never induced conditioned place-preference in rats.
In radioligand binding assays, compared with buprenorphine HS-599 had 3 fold higher μ-opioid receptor affinity but lower δ- and κ-opioid receptor affinity.
In isolated guinea-pig ileum preparations, HS-599 only partially inhibited the electrically-stimulated contraction, acting as a partial opioid agonist. When tested against the μ-opioid receptor agonist dermorphin, it behaved as a non-equilibrium antagonist. Conversely, in mouse vas deferens (rich in δ-opioid receptors) and rabbit vas deferens preparations (rich in κ-opioid receptors) HS-599 acted as a pure equilibrium antagonist, shifting the log-concentration-response curves of the δ-opioid agonist deltorphin I and the κ-opioid agonist U-69593 to the right.
In conclusion, HS-599 is a novel buprenorphine derivative with higher affinity, selectivity and potency than the parent compound, for μ-opioid receptors. It produces intense and long-lasting antinociception and does not induce place-preference in rats.
Keywords: HS-599, peripheral administration, opioid activity, antinociception, addicting properties
Introduction
In clinical practice, opioid analgesic drugs are widely used for the relief of severe pain. Unfortunately, their use is limited by their numerous side effects including respiratory depression, constipation and addiction.
In the search for alternative analgesics to morphine, interest has focused on opioids with mixed agonist-antagonist profiles. These compounds commonly exhibit low-to-moderate intrinsic activity at μ-receptors. Hence they produce analgesia with only mild side-effects. Buprenorphine is a potent analgesic drug that has demonstrable agonist and antagonist activity (Cowan et al., 1977a, 1977b; Heel et al., 1979). Whereas buprenorphine is a partial agonist at μ-opioid receptors and an antagonist at δ-opioid receptors, its role at κ-opioid receptors remains less clear (Leander, 1988; Tyers, 1980; Kamei et al., 1995; Pick et al., 1997). Despite strong evidence that buprenorphine acts as a full antagonist at κ receptors (Leander, 1987), it has κ3 and to a lesser extent κ1 agonistic activity (Pick et al., 1997). Although buprenorphine is 20 – 30 times more potent than morphine as an analgesic it induces scarce respiratory depression and is currently indicated as a therapeutic agent with low abuse potential (Pick et al., 1997; Michiteru et al., 1997). Because of these characteristics, buprenorphine is used as an alternative to methadone in maintenance and detoxification treatment of heroin addicts (Reisinger, 1985; Bickel et al., 1988; Kosten & Kleber, 1988; Diamant et al., 1998). Nevertheless, reports from various countries have described buprenorphine abuse (O'connor et al., 1988; Chowdhury & Chowdhury, 1990; San et al., 1993). Buprenorphine also maintains self-administration (Mello et al., 1981; Young et al., 1984) and induces place preference in animals (Gaiardi et al., 1997; Brown et al., 1991; Rowlett et al., 1994; Pchelintsev et al., 1991).
With the aim of developing new and potent analgesics with lower abuse potential than available opiates, we studied the antinociceptive and addicting properties of a derivative of buprenorphine, named HS-599 (Schutz et al., 2001) (Figure 1), after s.c. administration in rats. We also evaluated the receptor affinities and the in vitro biological activity of HS-599 in comparison with those of morphine, buprenorphine, dermorphin (a μ-opioid receptor agonist), deltorphin I and II (δ-opioid receptor agonists) and U69-593 (a κ-opioid receptor agonist).
Figure 1.
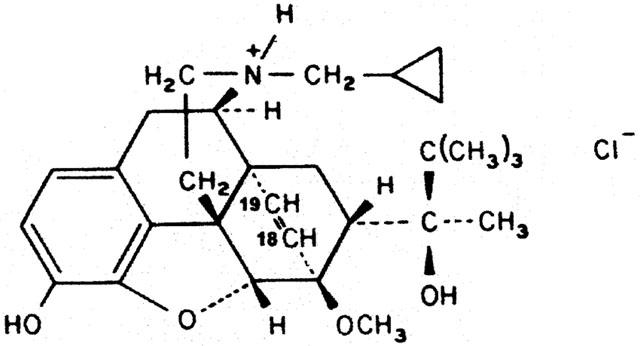
18,19-dehydrobuprenorphine: HS-599.
Methods
In vitro experiments
Binding assays
Binding of HS-599 and buprenorphine to μ, δ and ORL1 opioid receptors was assayed on crude membrane preparations from adult male rat brain (Wistar, 250 – 300 g). Binding to κ-opioid receptors was assayed on crude membrane preparations from adult male guinea-pig brain (450 g) as previously described (Melchiorri et al., 1991). The brains were homogenized in 50 volumes of Tris-HCl buffer (50 mM, pH 7.4, 4°C) using a Kinematica PT 3000 polytron (20 s, speed 16,000 r.p.m.). The homogenate was centrifuged at 41,000×g for 20 min at 4°C, pellets were resuspended in 50 vol of buffer and incubated at 25°C per 30 min to remove endogenous opioids. After centrifugation, pellets were resuspended in buffer, containing 5% glycerol, to give a final w v−1 of 20 mg ml−1 fresh tissue. The affinity of the compounds for μ, δ, κ and ORL1 receptors was determined by competition of unlabelled compounds against the μ-receptor selective ligand [3H]-[D-Ala2, MePhe4, Glyol5]enkephalin ([3H]-DAGO, 0.5 nM; 55.3 Ci mmol−1), the δ-receptor selective ligand [3H]-deltorphin II (0.3 nM; 53 Ci mmol−1), the κ1-receptor preferring ligand [3H]-U-69593 (0.3 nM; 47 Ci mmol−1) and the ORL1-receptor selective ligand [3H]-nociceptin (0.08 nM; 171 Ci mmol−1). Drugs were assayed on 1 mg brain membrane protein, in a final volume of 2 ml Tris-HCl buffer (50 mM, pH 7.4), at 35°C for 90 min. All experiments were performed in triplicate and total binding, nonspecific binding (10 μM naloxone) and 12 inhibitor concentrations were determined. Reagents and membranes were distributed with a robotic sample processor (Tecan RSP 5000-series). Using a Brandel M-24 cell harvester, assays were terminated by filtration through Whatman GF/B filter strips previously soaked in 0.5% polyethylenimine for 1 h. Filters were washed three times with 4 ml of ice-cold buffer and radioactivity was counted in a liquid scintillation spectrometer (Betamatic, Kontron). The inhibition constant (Ki) of the drugs was calculated from competitive binding curves with the computer program Ligand (Munson & Rodbard, 1980). Data are presented as the arithmetic mean±s.e.mean of four independent measurements.
Activity on isolated organ preparations
Preparations of the myenteric plexus-longitudinal muscle obtained from male guinea-pig ileum (GPI, rich in μ-opioid receptors), preparations of mouse vas deferens (MVD, rich in δ-opioid receptors) and rabbit vas deferens (RVD, rich in κ-opioid receptors) were used for field stimulation with bipolar rectangular pulses of supramaximal voltage (Melchiorri et al., 1991). Agonists were evaluated for their ability to inhibit the electrically-evoked twitch. The biological potency of HS-599 was compared with that of the μ-opioid receptor agonist dermorphin in GPI, with that of the δ-opioid receptor agonist deltorphin I in MVD and with that of the κ-opioid receptor agonist U-69593 in RVD. Concentration-response curves were analysed with the Prism computer program.
In vivo experiments
Male Wistar rats (250 – 300 g) were used. All animals were housed at 22±2°C, with food and water ad libitum. A standard light/dark cycle was maintained with a time-regulated light period from 0600 h to 1800 h. The IASP guidelines on ethical standards for investigations of experimental pain in animals were followed. Compounds were dissolved in 10% DMSO and administered to the rats in a volume of 0.2 ml kg−1 of body weight (b.w.). Control rats received vehicle at 0.2 ml kg−1 of b.w. Each animal received one injection only. Every dose of each compound was evaluated in groups of 6 – 8 animals.
Antinociception studies
Analgesia was measured by the tail-flick test (D'amour & Smith, 1941) and the plantar test (Hargreaves et al., 1988). In the tail-flick test a radiant heat stimulus is focused on the blackened area of the tail. In the plantar test the plantar surface of the rat's hindpaw is exposed to a beam of radiant heat applied through the glass floor of a testing chamber. To escape the nociceptive stimulus, the animal flicks its tail or briskly withdraws its paw. Each rat served as its own control. Before drug administration, each rat was tested and the latency to tail-flick or to paw withdrawal recorded (control latency, CL). The latency to tail-flick or to paw withdrawal of each drug-injected rat was defined as the test latency (TL). After drug injections, the test was repeated every 15 min during the first hour and then every 30 min until analgesia disappeared. To avoid tissue damage, rats with a test latency higher than 15 s (cut-off time) were removed from the nociceptive stimuli and assigned a TL of 15. For drawing the dose-response and time-response curves, the antinociceptive response was expressed as percentage of maximum possible effect (%MPE), calculated by the following equation: %MPE=100×(TL − CL)/(15 − CL). Doses that produced peak effects between 20 and 80% MPE were plotted into a log dose-response curve and AD50 values, with 95% confidence intervals (CI), were calculated as doses that produced an analgesic peak effect equal to 50% MPE (Tallarida & Murray, 1986). For each dose at least five rats were checked.
Place-preference
The place-preference apparatus consisted of two boxes (18×18×18 cm): one box had white walls and a smooth white floor whereas the other had black-and-white checkered walls with a wire grid on a black floor. A sliding wall connected the two compartments. A 20×10 cm corridor with a sliding door allowed the rat to go into the centre of the apparatus. An automated timer measured the time rats spent in each compartment. The experiments took place in a small sound insulated and ventilated room illuminated with red lights. On the first day each rat was free to explore the two compartments for 15 min. On the second day, the time each rat spent in the black and the white compartments was recorded for 15 min. Rats had a slight preference for the black compartment; they spent 402±90 s in the black and 380±65 s in the white compartment. All drug injections were therefore paired with the white compartment. From the third day on six groups of rats (n=6) received three pairings of HS-599 (0.003, 0.01, 0.021, 0.06, 0.21, and 0.43 μmol kg−1) with one set of cues and three pairings of saline with the other set of cues on alternate days. The same protocol was used for buprenorphine (0.21 μmol kg−1) and morphine (13.2 μmol kg−1). During a typical session, the rat was injected, then placed for 60 min into its training compartment. On the day after training rats were tested. For testing, animals were placed in the central corridor and allowed to explore both compartments for 15 min; the time (seconds) spent on the drug paired side was recorded.
Drugs
Buprenorphine HCl, morphine HCl, naloxone and naltrexone were bought from S.A.L.A.R.S. (Como, Italy). Dermorphin, deltorphin I and deltorphin II were synthesized as previously described (Negri et al., 1992; Erspamer et al., 1989). Naltrindole was purchased from Sigma (Italy) and nor-binaltorphimine was purchased from Tocris, U.K.
The selective μ-opioid receptor ligand [3H]-DAGO and the selective ORL1-receptor ligand [3H]-nociceptin were purchased from Amersham (U.K.). The selective δ-receptor ligand [3H]-deltorphin II and the κ1-receptor preferring ligand [3H]-U69-593 were obtained from NEN (Italy). HS-599 (18,19-dehydrobuprenorphine) was synthesized as previously described (Schutz et al., 2001).
Results
Receptor binding and GPI-MVD assay
In radioligand binding assays, performed on crude membrane homogenates, HS-599 displayed μ-opioid receptor affinity about three times higher than that of buprenorphine but lower δ- and κ-opioid receptor affinities so that μ/δ and μ/κ selectivity of HS-599 was about one order of magnitude higher than that of buprenorphine. Neither HS-599 nor buprenorphine displayed any affinity for ORL1 receptors (Table 1).
Table 1.
Opioid receptor affinity and selectivity of HS-599 compared with those of buprenorphine and morphine

On GPI preparations, HS-599 inhibited the electrically-evoked twitch concentration-dependently between 2.1 nM and 4.3 μM. HS-599 twitch inhibition was prevented by naloxone (NLX) 200 nM (data not shown). Concentration-response curves (Figure 2) showed that dermorphin produced 100% inhibition of electrically-stimulated contractions of GPI, whereas buprenorphine produced 70% and HS-599 less than 50% of the maximum effect, displaying a significantly lower efficacy than buprenorphine. HS-599 bound irreversibly to GPI: even prolonged, repeated washing left twitch inhibition unchanged. Because of its irreversible binding, each dose of HS-599 was tested on different GPI preparations and compared with dermorphin as an internal standard.
Figure 2.
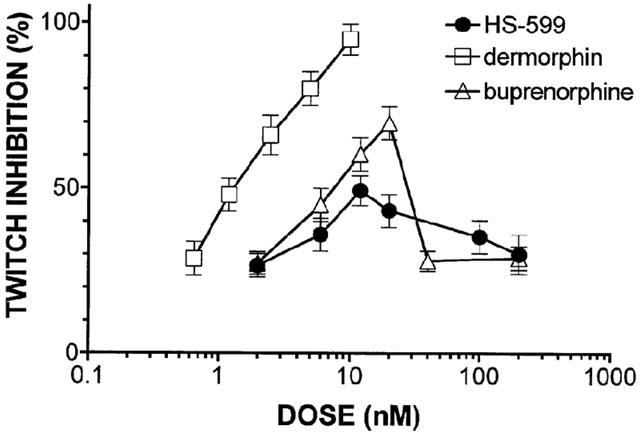
Inhibition of the electrically evoked twitch of guinea-pig ileum preparations. Log-concentration-response curves for the μ-opioid receptor agonist dermorphin, HS-599 and buprenorphine.
HS-599 acted as a non-equilibrium antagonist of the full agonist dermorphin reducing the slope and maximum of the dermorphin log-concentration-response curve (Figure 3). On MVD and RVD, at concentrations up to 3.0 μM HS-599 never inhibited the electrically-stimulated contractions. But it shifted the log-concentration-effect curve of deltorphin I on MVD and U-69593 on RVD to the right (Figures 4 and 5). HS-599 also left the log-concentration-effect slopes of the deltorphin I and U-69593 curves unchanged, indicating that it acted as an equilibrium antagonist of δ- and κ-opioid receptors.
Figure 3.
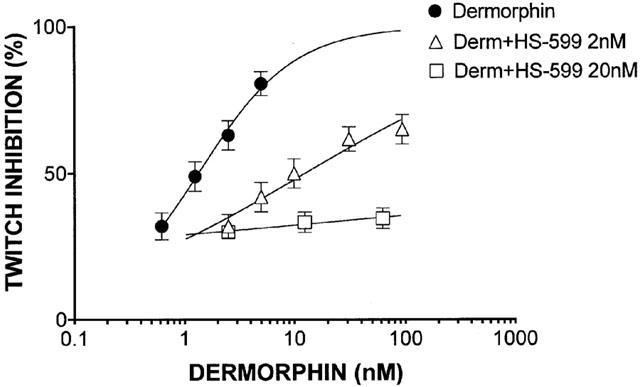
Guinea-pig ileum preparation. Log-concentration-response curves for the μ-opioid receptor selective agonist dermorphin, alone or in the presence of different concentrations of HS-599. Dermorphin, slope=1.002 [0.8123 – 1.092]; dermorphin+HS-599 2 nM, slope=0.3838 [0.2888 – 0.4788], P<0.01; dermorphin+HS-599 20 nM, slope=0.06562 [−0.1417 – 0.1454], P<0.01 (Nonlinear regression analysis, Prism).
Figure 4.
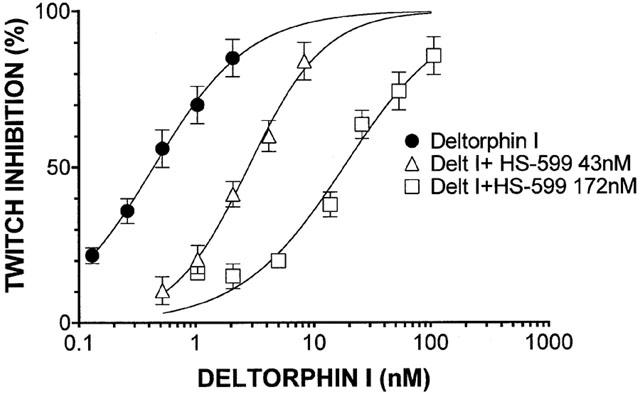
Inhibition of the electrically evoked twitch of mouse vas deferens preparations. Log-concentration response curves for the δ-opioid receptor selective agonist deltorphin I, alone or in the presence of different HS-599 concentrations. HS-599 shifted the concentration-response curve of deltorphin I to the right without changing the slope (nonlinear regression analysis, Prism): Deltorphin I, slope=1.067 [0.9081 – 1.225]; deltorphin I+HS-599 43 nM, slope=1.351 [1.150 – 1.152]; deltorphin I+HS-599 172 nM, slope=0.9758 [0.8176 – 1.134].
Figure 5.
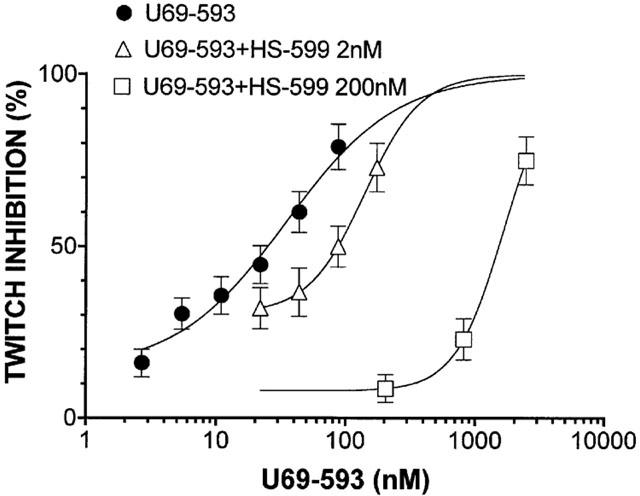
Inhibition of the electrically evoked twitch of rabbit vas deferens preparations. Log-concentration response curves for the κ-opioid receptor selective agonist U-69593, alone or in the presence of different HS-599 concentrations. HS-599 shifted the concentration-response curve of U-69593 to the right without changing the slope (nonlinear regression analysis, Prism): U-69593, slope=1.019 [0.5977 – 1.441]; U69-593+HS-599 2 nM, slope=2.002 [0.7691 – 3.236]; U-69593+HS-599 200 nM, slope=2.387 [1.502 – 3.272].
Antinociception studies
Tail-flick test
Subcutaneous injection of HS-599 in rats produced dose related (0.05 – 0.3 μmol kg−1) antinociceptive effects. The AD50 value was two times lower than that of buprenorphine and 50 times lower than that of morphine (0.110 [0.096 – 0.120] vs 0.219 [0.215 – 0.224] vs 5.01 [1.76 – 15.93] μmol kg−1) (Figure 6). The analgesic effect peaked within 60 – 90 min and remained significant up to 3 – 5 h, depending on the dose. At high doses the antinociceptive effect of HS-599 lasted significantly longer than that of buprenorphine (Figure 7). Pre-treatment with the μ-opioid antagonists NLX (3 mg kg−1 s.c., 20 min before) and NLTX (10 mg kg−1, s.c., 20 min before) abolished antinociception induced by subcutaneous injection of 0.2 μmol kg−1 of HS-599. Conversely, the δ-antagonist naltrindole (NTI, 20 mg kg−1, s.c., 30 min before) and the κ-antagonist nor-binaltorphimine (norBNI, 20 mg kg−1, s.c., 60 min before) left antinociception unaffected (Figure 8). The doses and times used in this study were those known to obtain the best selectivity and antagonist activity in rat.
Figure 6.
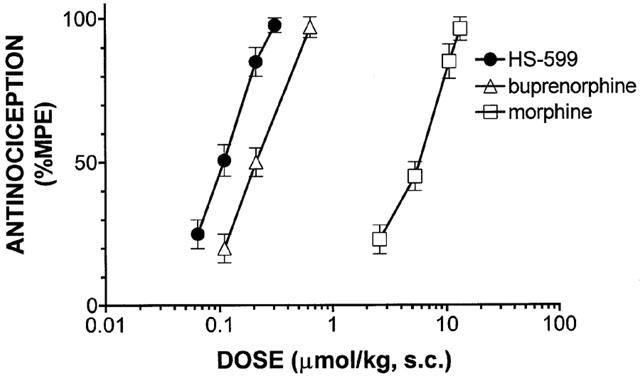
Dose-response relationship of the antinociception produced by s.c. injection of HS-599, buprenorphine and morphine evaluated with tail-flick test in rats. Curves were fitted to data points and analysed by a linear regression computer program (Tallarida & Murray, 1986). HS-599 AD50=0.11 [0.096 – 0.120] μmol kg−1; buprenorphine AD50=0.219 [0.215 – 0.224] μmol kg−1; morphine AD50=5.29 [1.76 – 15.93] μmol kg−1. Each point represents the mean antinociceptive peak effect of 5 – 8 rats±s.e.mean.
Figure 7.
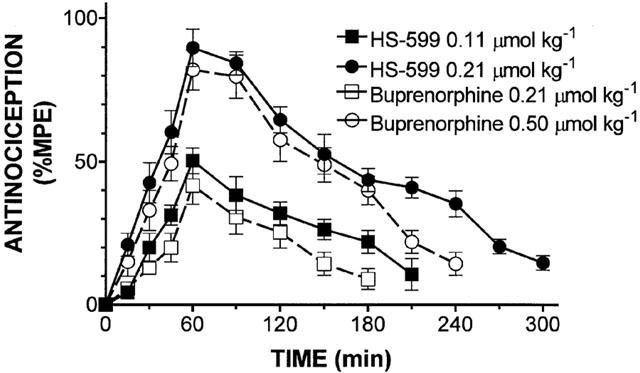
Time course of the antinociception produced by s.c. injection of equianalgesic doses of HS-599 and buprenorphine evaluated by tail-flick test in rats. Each point represents the mean antinociceptive effect of 5 – 8 rats±s.e.mean. Time-response curves have been drawn and analysed by Prism computer program using two-way ANOVA and Bonferroni post-tests.
Figure 8.
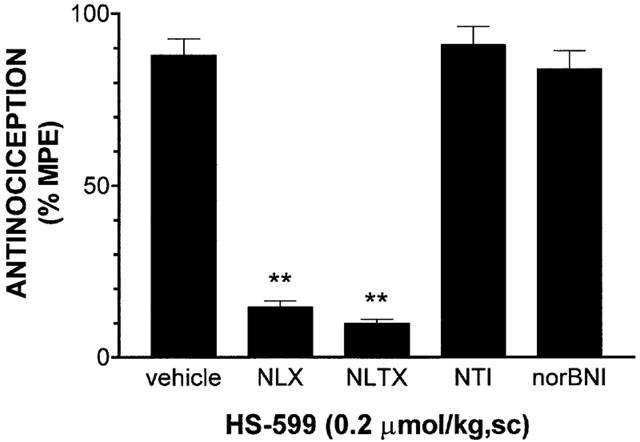
Antagonism by naloxone (NLX, 3 mg kg−1 s.c.), naltrexone (NLTX, 10 mg kg−1 s.c.), naltrindole (NTI, 20 mg kg−1 s.c.) and nor-Binaltorphimine (nor-BNI, 20 mg kg−1 s.c.) of the antinociception induced by HS-599 (0.2 μmol kg−1 s.c.), in rats (tail-flick test). Each column represents the mean±s.e.mean for 5 – 8 rats in each group. **P<0.01 versus the vehicle-pretreated group.
Plantar test
In this test HS-599 displayed poor antinociceptive activity: the 0.25 μmol kg−1 s.c. dose elicited the maximum antinociceptive effect, less than 50% MPE. Higher doses (0.8 – 20 μmol kg−1 s.c.) were always less efficacious. Buprenorphine (0.21 – 1 μmol kg−1 s.c.) and morphine (12 – 50 μmol kg−1 s.c.) both produced dose-related antinociception that reached the maximum analgesic effect (Figure 9). Pre-treatment with NLX 3 mg kg−1 antagonized the HS-599 (0.25 μmol kg−1 s.c.)-induced increased latency to paw withdrawal (data not shown).
Figure 9.
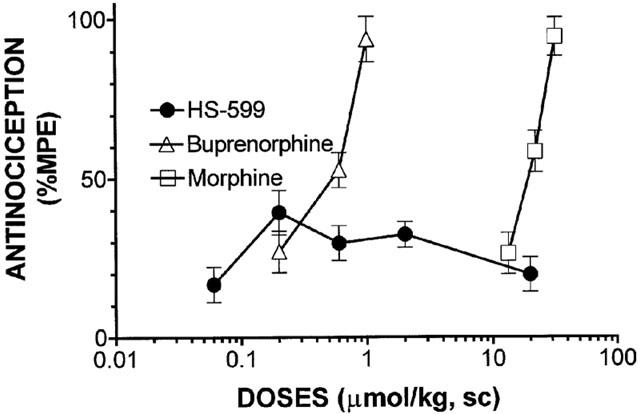
Dose-response curve of the antinociception produced by s.c. administration of HS-599, buprenorphine and morphine in rats (plantar test). Each point represents the mean antinociceptive peak effect of 5 – 8 rats±s.e.mean.
Place-preference
In this test, a standard method for highlighting the rewarding properties of psychoactive, addicting drugs, none of the HS-599 doses tested – from a sub-analgesic dose (0.003 μmol kg−1 s.c.) to a dose (0.43 μmol kg−1 s.c.) twice as high as that inducing the maximum antinociceptive effect in the tail-flick test in rats – induced preference for the drug-paired compartment in rats (for all tested doses: P⩾ 0.0680). Conversely, at doses corresponding to AD50 buprenorphine and morphine induced a significant preference, about 80%, for the all-white compartment (buprenorphine, P=0.0021; morphine, P=0.0003) (Figure 10).
Figure 10.
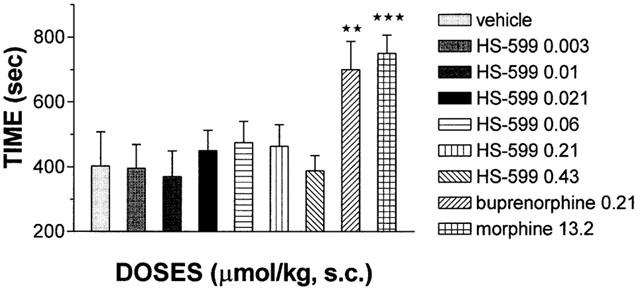
Effect of HS-599, buprenorphine and morphine on conditioned place-preference test in rats. Results are presented as mean±s.e.mean of time (seconds) that rats spent in the drug paired compartment before (control) and after the acquisition of conditioned place-preference (n=6 per group). Results were analysed by a paired two-tailed t-test. All HS-599 values P⩾0.0680 (not significant); buprenorphine, **P=0.0021; morphine, ***P=0.0003.
Discussion
These findings show that like its parent compound, the didehydroderivative of buprenorphine, HS-599 is a potent and long-lasting analgesic in rats. But when tested in the place-conditioning test in rats, unlike buprenorphine, it had no rewarding effects. Our in vitro experiments showed that HS-599 has significantly higher μ-opioid receptor affinity than buprenorphine. Yet it acted as a partial μ-opioid receptor agonist endowed with lower intrinsic activity than buprenorphine. Dissociation from the μ-receptors being very slow in GPI preparations it behaved as a non-equilibrium partial antagonist toward the μ-opioid receptor agonist dermorphin. It bound to δ- and κ-opioid receptors acting as a pure antagonist on MVD and on RVD preparations, reversibly blocking the twitch inhibition produced by the δ-opioid agonist deltorphin I and by the κ-opioid agonist U-69593 respectively.
The in vivo experiments conducted in this study with selective opioid antagonists indicate that HS-599 exerts its antinociceptive activity via μ-opioid receptors and exclude participation of δ- or κ-opioid receptors. Accordingly, the μ antagonists, NLX and NLTX, blocked whereas the δ-selective antagonist, NTI, and the κ-selective antagonist, nor-BNI, failed to reverse HS-599 induced analgesia. Moreover, in the tail-flick test – a test involving spinal reflexes – HS-599 behaved as a full agonist, but in the plantar test – a test involving more complex supraspinal neuronal circuits – analgesia never exceeded 50% MPE. In contrast, at the doses we tested, buprenorphine behaved as a full agonist in both tests. As well as acting as a partial μ-agonist, buprenorphine has been proven to elicit analgesia through an interaction with κ3receptors (Tyers, 1980; Pick et al., 1997). Hence the activity of buprenorphine and HS-599 could have differed in the plantar test owing to the κ3 agonistic activity of buprenorphine, insofar as κ3 analgesia is mediated supraspinally (Paul et al., 1990). Alternatively, owing to its lower intrinsic activity HS-599 was less able than buprenorphine to activate the supraspinal μ-opioid receptors.
As a first approach in assessing the potential reinforcing properties of a test substance the conditioned place preference paradigm is a useful tool. Our place-conditioning data are largely consistent with previous reports indicating that both s.c. (Gaiardi et al., 1997) and i.p. (Brown et al., 1991; Rowlett et al., 1994) injection of buprenorphine has rewarding effects in rats. Under the same conditions and at the doses we tested, HS-599 never caused place-preference.
It is known that the reinforcing and aversive properties of a drug are considered the main attributes that regulate drug-seeking behaviour. In rats, activation of μ-opioid receptors has been predominantly associated with reinforcing properties while activation of κ-opioid receptors has been associated with aversive properties. Buprenophrine is a μ-opioid receptors partial agonist and a κ-opioid receptors antagonist with the same affinity for μ- and κ-opioid receptors. As the aversive properties are thought to work against the positive ones, the dissociation of rewarding and aversive effects, observed with buprenorphine, could facilitate drug-seeking behaviour (Gaiardi et al., 1997). HS-599, thus being a μ-opioid receptors partial agonist and a κ-opioid receptors antagonist like the parent compound, displayed 20 times lower κ-affinity than μ-affinity. Hence, unlike buprenorphine, at doses inducing analgesia through activation of μ-opioid receptors, HS-599 does reasonably not involve κ-opioid receptors. Moreover, to the extent that increased abuse potential is related prevalently to μ-opioid receptors activation (Negus et al., 1990; Riley & Pournaghash, 1995), it is worth noting that HS-599 has significantly lower intrinsic activity than buprenorphine at peripheral (GPI) and supraspinal (plantar test) μ-opioid receptors suggesting that such receptor activity produces stimulus effects insufficient to establish a biological signal necessary to elicit place conditioning.
In conclusion, the higher μ/κ selectivity together with lower μ-intrinsic activity of HS-599 at supraspinal level with respect to buprenorphine could explain why it does not induce place conditioning in rats. HS-599 could be of value as a substitute for methadone and buprenorphine in the weaned substitutive techniques of the drug addicts.
Moreover, at spinal level HS-599 antinociceptive activity is 50 times higher than that of morphine.
Abbreviations
- AD50
dose that induces 50% of antinociceptive effect
- nor-BNI
nor-binaltorphimine
- GPI
guinea-pig ileum
- MPE
maximum possible effect
- MVD
mouse vas deferens
- NLTX
naltrexone
- NLX
naloxone
- NTI
naltrindole
- RVD
rabbit vas deferens
References
- BICKEL W.K., STITZER M.L., BIGELOW G.E., LIEBSON I.A., JANINSKI D.R., JOHNSON R.E. A clinical trial of buprenorphine: comparison with methadone in the detoxification of heroin addicts. Clin. Pharmacol. Ther. 1988;43:72–78. doi: 10.1038/clpt.1988.13. [DOI] [PubMed] [Google Scholar]
- BROWN E.E., FINLAY J.M., WONG J.T.F., DAMSMA G., FIBIGER H.C. Behavioral and neurochemical interactions between cocaine and buprenorphine: implications for the pharmacotherapy of cocaine abuse. J. Pharmacol. Exp. Ther. 1991;256:119–126. [PubMed] [Google Scholar]
- CHOWDHURY A.N., CHOWDHURY S. Buprenorphine abuse: report from India. Br. J. Addict. 1990;85:1349–1350. doi: 10.1111/j.1360-0443.1990.tb01612.x. [DOI] [PubMed] [Google Scholar]
- COWAN A., DOXEI J.C., HARRY E.J.R. The animal pharmacology of buprenorphine, an oripavine analgesic agent. Br. J. Pharmacol. 1977b;60:547–554. doi: 10.1111/j.1476-5381.1977.tb07533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COWAN A., LEWIS J.W., MACFARLANE I.R. Agonist and antagonist properties of buprenorphine, a new antinociceptive agent. Br. J. Pharmacol. 1977a;60:537–545. doi: 10.1111/j.1476-5381.1977.tb07532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'AMOUR F.E., SMITH D.L. Method for determining loss of pain sensation. J. Pharmacol. Exp. Ther. 1941;72:74–79. [Google Scholar]
- DIAMANT K., FISCHER G., SCHNEIDER C., LENZINGER E., PEZAWAS L., SCHINDLER S., EDER H. Outpatient opiate detoxification treatment with buprenorphine. Preliminary investigation. Eur. Addict. Res. 1998;4:198–202. doi: 10.1159/000018953. [DOI] [PubMed] [Google Scholar]
- ERSPAMER V., MELCHIORRI P., FALCONIERI ERSPAMER G., NEGRI L., CORSI R., SEVERINI C., BARRA D., SIMMACO M., KREIL G. Deltorphins: a family of naturally occurring peptides with high affinity and selectivity for δ-opioid binding sites. Proc. Natl. Acad. Sci. U.S.A. 1989;86:5188–5192. doi: 10.1073/pnas.86.13.5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAIARDI M., BARTOLETTI M., BACCHI A., GUBELLINI C., BABBINI M. Motivational properties of buprenorphine as assessed by place preference and taste conditioning in rats. Psychopharmacology. 1997;130:104–108. doi: 10.1007/s002130050216. [DOI] [PubMed] [Google Scholar]
- HARGREAVES F., DUBNER R., BROWN F., FLORES C., JORIS J. A new sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- HEEL R.C., BROGDEN R.N., SPEIGHT T.M., AVERY G.S. Buprenorphine: a review of its pharmacological properties and therapeutic efficacy. Drugs. 1979;17:81–110. doi: 10.2165/00003495-197917020-00001. [DOI] [PubMed] [Google Scholar]
- KAMEI J., SAITOH A., SUZUKI T., MISAWA M., NAGASE H., KASUYA Y. Buprenorphine exerts its antinociceptive activity via μ1-opioid receptors. Life Science. 1995;56:285–290. doi: 10.1016/0024-3205(95)00078-x. [DOI] [PubMed] [Google Scholar]
- KOSTEN T.R., KLEBER H.D. Buprenorphine detoxification from opioid dependence: a pilot study. Life Sci. 1988;42:635–641. doi: 10.1016/0024-3205(88)90454-7. [DOI] [PubMed] [Google Scholar]
- LEANDER J.D. Buprenorphine has a potent kappa opioid receptor antagonistic activity. Neuropharmacology. 1987;26:1445–1447. doi: 10.1016/0028-3908(87)90112-2. [DOI] [PubMed] [Google Scholar]
- LEANDER J.D. Buprenorphine is a potent κ-opioid receptor antagonist in pigeons and mice. Eur. J. Pharmacol. 1988;151:457–461. doi: 10.1016/0014-2999(88)90543-2. [DOI] [PubMed] [Google Scholar]
- MELCHIORRI P., NEGRI L., FALCONIERI ERSPAMER G., SEVERINI C., CORSI R., SOAJE M., ERSPAMER V., BARRA D. Structure-activity relationships of the δ-opioid selective agonists, deltorphins. Eur. J. Pharmacol. 1991;195:201–207. doi: 10.1016/0014-2999(91)90536-y. [DOI] [PubMed] [Google Scholar]
- MELLO N.K., BREE M.P., MENDELSON J.H. Buprenorphine self-administration by rhesus monkey. Pharmacol. Biochem. Behav. 1981;15:215–225. doi: 10.1016/0091-3057(81)90180-5. [DOI] [PubMed] [Google Scholar]
- MICHITERU O., HAJIME K., KENJIN N., YASUFUMI S., TATSUJI I. Kinetics of respiratory depression in rats induced by buprenorphine and its metabolite, norbuprenorphine. J. Pharmacol. Exp. Ther. 1997;281:428–433. [PubMed] [Google Scholar]
- MUNSON P.J., RODBARD D. Ligand: a versatile computerized approach for the characterization of ligand binding systems. Anal. Biochem. 1980;107:220–224. doi: 10.1016/0003-2697(80)90515-1. [DOI] [PubMed] [Google Scholar]
- NEGRI L., FALCONIERI ERSPAMER G., SEVERINI C., POTENZA R.L., MELCHIORRI P., ERSPAMER V. Dermorphin related peptides from the skin of Phyllomedusa bicolor and their amidated analogs activate two μ-opioid receptor subtypes which modulate antinociception and catalepsy, in the rat. Proc. Natl. Acad. Sci. U.S.A. 1992;89:7203–7207. doi: 10.1073/pnas.89.15.7203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEGUS S.S., PICKER M.J., DYKSTRA L.A. Interactions between mu and kappa opioid agonists in the rat drug discrimination procedure. Psychopharmacology (Berlin) 1990;102:465–473. doi: 10.1007/BF02247126. [DOI] [PubMed] [Google Scholar]
- O'CONNOR J.J., MOLONEY E., TRAVERS R., CAMPBELL A. Buprenorphine abuse among opiate addicts. Br. J. Addict. 1988;83:1085–1087. doi: 10.1111/j.1360-0443.1988.tb00536.x. [DOI] [PubMed] [Google Scholar]
- PAUL D., LEVISON J.A., HOWARD D., PICK C.G., HAHN E.F., PASTERNAK G.W. Naloxone benzoylhydrazone (NalBzoH) analgesia. J. Pharmacol. Exp. Ther. 1990;255:769–774. [PubMed] [Google Scholar]
- PCHELINTSEV M.V., GORBACHEVA E.N., ZVARTAU E.E. Simple methodology of assessment of analgesics' addictive potential in mice. Pharmacol. Biochem. Behav. 1991;39:873–876. doi: 10.1016/0091-3057(91)90046-5. [DOI] [PubMed] [Google Scholar]
- PICK C.G., PETER Y., SCHREIBER S., WEIZMAN R. Pharmacological characterization of buprenorphine, a mixed agonist-antagonist with κ3 analgesia. Brain Res. 1997;744:41–46. doi: 10.1016/s0006-8993(96)01069-4. [DOI] [PubMed] [Google Scholar]
- REISINGER M. Buprenorphine as new treatment for heroin dependence. Drug Alcohol Depend. 1985;16:257–262. doi: 10.1016/0376-8716(85)90050-x. [DOI] [PubMed] [Google Scholar]
- RILEY A.L., POURNAGHASH S. The effects of chronic morphine on the generalization of buprenorphine stimulus control: an assessment of kappa antagonist activity. Pharmacol. Biochem. Behav. 1995;52:779–787. doi: 10.1016/0091-3057(95)00180-5. [DOI] [PubMed] [Google Scholar]
- ROWLETT J.K., GIBSON T.R., BARDO M.T. Dissociation of buprenorphine-induced locomotor sensitization and conditioned place preference. Pharmacol. Biochem. Behav. 1994;49:241–245. doi: 10.1016/0091-3057(94)90484-7. [DOI] [PubMed] [Google Scholar]
- SAN L., TORRENS M., CASTILLO C., PORTA M., DE LA TORRE R. Consumption of buprenorphine and other drugs among heroin addicts under ambulatory treatment: results from cross-sectional studies in 1988 and 1990. Addiction. 1993;88:1341–1349. doi: 10.1111/j.1360-0443.1993.tb02020.x. [DOI] [PubMed] [Google Scholar]
- SCHUTZ J., KRASSNIG R., SCHMIDHAMMER H., WURST K., LATTANZI R., NEGRI L. Synthesis and pharmacological evaluation of 18,19-dehydrobuprenorphine. Heterocycles. 2001;54:989–998. [Google Scholar]
- TALLARIDA R.J., MURRAY R.B. Manual of Pharmacological calculation 1986New York: Springer-Verlag; 2nd ed [Google Scholar]
- TYERS M.B. A classification of opiate receptors that mediate antinociception in animals. Br. J. Pharmacol. 1980;69:503–512. doi: 10.1111/j.1476-5381.1980.tb07041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOUNG A.M., STEPHENS K.R., HEIN D.W., WOODS J.H. Reinforcing and discriminative stimulus properties of mixed agonist-antagonist opioids. J. Pharmacol. Exp. Ther. 1984;229:118–126. [PubMed] [Google Scholar]