

Abstract
The accumulation of amyloid β protein (Aβ) in the brain is a characteristic feature of Alzheimer's disease (AD). Clinical trials of AD patients with nonsteroidal anti-inflammatory drugs (NSAIDs) indicate a clinical benefit. NSAIDs are presumed to act by suppressing inhibiting chronic inflammation in the brain of AD patients.
In the present study, we investigated effects of S-2474 on Aβ-induced cell death in primary cultures of rat cortical neurons. S-2474 is a novel NSAID, which inhibits cyclo-oxygenase-2 (COX-2) and contains the di-tert-butylphenol antioxidant moiety.
S-2474 significantly prevented neurons from Aβ(25 – 35)- and Aβ(1 – 40)-induced cell death. S-2474 ameliorated Aβ-induced apoptotic features such as the condensation of chromatin and the fragmentation of DNA completely.
Prior to cell death, Aβ(25 – 35) generated prostaglandin D2 (PGD2) and free radicals from neurons. PGD2 is a product of cyclo-oxygenase (COX), and caused neuronal cell death.
S-2474 significantly inhibited the Aβ(25 – 35)-induced generation of PGD2 and free radicals.
The present cortical cultures contained little non-neuronal cells, indicating that S-2474 affected neuronal survival directly, but not indirectly via non-neuronal cells. Both an inhibitory effect of COX-2 and an antioxidant effect might contribute to the neuroprotective effects of S-2474.
In conclusion, S-2474 exhibits protective effects against neurotoxicity of Aβ. Furthermore, the present study suggests that S-2474 may possess therapeutic potential for AD via ameliorating degeneration in neurons as well as suppressing chronic inflammation in non-neuronal cells.
Keywords: Alzheimer's disease, amyloid β protein, apoptosis, nonsteroidal anti-inflammatory drug, S-2474, cyclo-oxygenase-2, free radical, neuroprotection
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) are commonly prescribed in drug therapy for inflammatory diseases such as Alzheimer's disease (AD) (McGeer et al., 1996; Stewart et al., 1997). Currently available NSAIDs have dual inhibitory activities against cyclo-oxygenase (COX) and 5-lipoxygenase (5-LO) (Vane & Botting, 1998; Hidaka et al., 1984; Ikuta et al., 1987). COX is classified into two distinct isoforms, a constitutive form, COX-1, and a mitogen-inducible form, COX-2. By COX and 5-LO, arachidonic acid (AA) is metabolized to prostaglandins (PGs) and leukotrienes (LTs), respectively. S-2474 is a novel NSAID containing the γ-sultam skeleton and the di-tert-butylphenol antioxidant moiety (Inagaki et al., 2000). This compound very strongly suppresses the COX-2 pathway (IC50=0.2 – 3.6 nM), and inhibits production of the multiple inflammatory mediators such as LTs, interleukin (IL)-1, IL-6, IL-8 and NO (IC50=0.5 – 30 μM) (Matsumoto et al., 1997). On the other hand, it does not affect the COX-1 pathway (IC50>10 μM) (Inagaki et al., 2000). COX-1 is responsible for the synthesis of cytoprotective prostaglandins (PGs) in the gastrointestinal tract and its inhibition of COX-1 leads to adverse effects, i.e. gastrointestinal ulceration (Miller, 1983). Oral S-2474 exerts anti-inflammatory and analgestic action without side effects, gastrointestinal ulceration (Jyoyama et al., 1997). Thus, S-2474 is thought to offer anti-inflammatory therapeutic effects via inhibition of COX-2, 5-LO, IL production and radical generation.
Alzheimer's disease (AD) is characterized clinically by progressive dementia and pathologically by cortical atrophy, neuronal loss, neurofibrillary tangles, senile plaques, and deposits of amyloid β protein (Aβ) in the various region of brain such as cerebral cortex and hippocampus (Selkoe, 1991). Aggregated deposits of Aβ are generally assumed to have a causative role in neurodegeneration and development of AD (Selkoe, 1994; Cummings & Cotman, 1995). Aβ is a 39 to 43-amino-acid hydrophobic peptide, and causes neuronal cell death in primary cultures (Pike et al., 1991; Ueda et al., 1994). Aβ-induced neuronal cell death is typified by several characteristic features of apoptosis, such as formation of cell surface blebs, chromatin condensation, and DNA fragmentation (Forloni et al., 1993; Ueda et al., 1996). Therefore, neuroprotective compounds against Aβ toxicity are drug candidates for the clinical management of AD.
Aβ causes peroxidation of plasma membrane (Behl et al., 1994; Ueda et al., 1997a). Aβ neurotoxicity is attenuated by a number of antioxidants, e.g., vitamin E (Behl et al., 1994; Ueda et al., 1997a). Aβ-induced lipid peroxidation impairs plasma membrane ion-motive ATPases, leading to depolarization and the activation of L-type voltage-dependent calcium channels (Weiss et al., 1994; Ueda et al., 1997a). Subsequently, intracellular Ca2+ levels ([Ca2+]i) are elevated (Mattson et al., 1992; Ueda et al., 1997b). The resulting increase in [Ca2+]i activates phospholipase A2 (PLA2), which releases AA from the membrane. Cytosolic PLA2 immunoreactivity is increased in AD brain (Stephenson et al., 1996). However, it has not yet been clearly demonstrated how AA metabolites are involved in Aβ neurotoxicity and AD.
A clinical trial of AD patients with a COX inhibitor, indomethacin, indicated a beneficial effect (Rogers et al., 1993; McGeer, 2000). In the brain, both COX-1 and COX-2 are expressed (Yasojima et al., 1999). COX-2 is up-regulated in AD brain and in Aβ-treated SH-SY5Y neuroblastoma cells (Pasinetti & Aisen, 1998), suggesting the involvement of COX-2 in AD. Among PGs, the formation of PGD2 is significantly increased in AD brain (Iwamoto et al., 1989). On the other hand, 5-LO inhibitors protect hippocampal neurons against Aβ-toxicity (Goodman et al., 1994), suggesting the association of LTs with AD. Recently, NSAIDs are reported to reduce apoptotic neuronal cell death via activation of peroxysome proliferator-activated receptor γ (PPAR γ) in rat cerebella granule cells. (Heneka et al., 2000). However, an endogenous ligand for PPAR γ, 15-deoxy-Δ12,14-prostaglandin J2 (15d-Δ12,14-PGJ2) induces apoptosis in rat cortical neurons (Rohn et al., 2001). Thus, it is not yet understood how NSAIDs exhibit clinically beneficial effects on AD.
Aβ-induced neuronal cell death is established as in vitro model of AD (Pike et al., 1991). Using this model, we found neuroprotective effects of S-2474 against Aβ neurotoxicity in the primary culture of rat cortical neurons. In the present study, we examined how S-2474 exhibits neuroprotective effects in this model.
Methods
Materials
S-2474 ((E)-(5)-(3,5-di-tert-butyl-4-hydroxybenzylidene)-2-ethyl-1,2-isothiazolidine-1,1-dioxide) and U-46619 (1,5,5-hydroxy-11α,9α-(epoxymethano)prosta-5Z,13E-dienoic acid), a stable agonist for thromboxane A2 (TXA2) receptor, were synthesized in our laboratory, and its stock solution was prepared by solution at 10 mM in DMSO (Inagaki et al., 2000). Aβ(25 – 35) and Aβ(1 – 40) were purchased from Bachem AG (Bubendorf, Switzerland). Their scrambled and reversed forms were obtained from Takara (Shiga, Japan). A stock solution of Aβ was prepared by solution of the peptide at 1 mM in deionized water and was incubated at 37°C for 2 days to aggregate the peptide (Ueda et al., 1994). AA, PGD2, PGE2, 9α-11β-PGF2, PGF2α, PGI2, 15d-Δ12,14-PGJ2, LTB4, LTC4, LTD4 were purchased from Cayman Chemical (Ann Arbor, MI, U.S.A.). Their solvents are evaporated under a gentle stream of nitrogen and the residues are redissolved in ethanol. They are stored at −20°C. Hoechst 33258 fluorescent dye was purchased from Molecular Probes (Eugene, OR, U.S.A.). 2′,7′-Dichlorofluorescin diacetate (DCFDA) was purchased from Kodak (Tokyo, Japan). Anti-microtuble-associated protein 2 (anti-MAP2) and anti-glial fibrillary acidic protein (anti-GFAP) were obtained from Sigma (St. Louis, MO, U.S.A.). Anti-microglial antigen (OX-42) was purchased from BMA biomedicals AG (Augst, Switzerland). Reversed phase (C18) Sep-Pack cartridges was purchased from Waters (Bedford, MA., U.S.A.). PGD2[3H]-assay system was purchased from Amersham (Buckinghamshire, U.K.). PGE2 [125I]-RIA kit and LTB4 [3H]-RIA kit were purchased from DuPont NEN (Boston, MA, U.S.A.).
Animals
All experiments were carried out according to the guidelines of the European Community's Council for Animal Experiments. The following experimental procedures were approved by the Institutional Animal Care and Use Committee at the Discovery Research Laboratories of Shionogi & Co., Ltd., and all efforts were made to minimize the number of animals used and their suffering. Pregnant Sprague-Dawley rats were used. The rats were individually housed in macrolon cages with free access to food and water and maintained on a 12-h light/dark cycle, at 25°C room temperature.
Tissue culture
Neuronal cell cultures were prepared from cerebral cortices of day-19 Sprague-Dawley rat embryos as previously reported (Mattson et al., 1995; Ueda et al., 1996). Cells were plated at a density of 2.5×105 cells cm−2 on poly-L-lysine-coated dishes in conditioning medium, Leibovitz's L-15 medium supplemented with 5% foetal bovine serum and 5% horse serum at 37°C. Cultures were treated with 0.1 μM arabinosylcytosine C only on day 1 and used for experiments on day 2 after plating. The compositions of neurons, astrocytes and microglias in cortical cultures were determined by use of antibodies for MAP2, GFAP and microglial antigen, which are specific for neurons, astrocytes and microglias, respectively. Most of the cells (more than 95%) were stained by anti-MAP2 antibody, whereas there were few anti-GFAP-positive cells (less than 4%) and anti-microglial antigen positive cells (less than 3%). Thus, the present culture contained primarily neurons and few non-neuronal cells.
Analysis of neuronal survival
Experiments were principally performed in the two conditions as follows. (i) Neurons (2.5×105 cells cm−2) were treated with 10 μM Aβ(25 – 35) or Aβ(1 – 40) in the presence or absence of S-2474 at 37°C. Vehicle controls were treated with culture medium containing 1% deionized water and 0.1% DMSO. Aβ controls were treated with culture medium containing 10 μM Aβ(25 – 35) and 0.1% DMSO. (ii) Neurons (2.5×105 cells cm−2) were treated with eicosanoids at 37°C. Vehicle controls were treated with culture medium containing 0.1% ethanol.
Two different methods were employed for assessment of neurotoxicity of Aβ, as previously reported (Ueda et al., 1996). First, the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide dye (MTT) reduction assay reflecting mitochondrial succinate dehydrogenase activity was employed. Second, residual cells were counted according to morphologic criteria; neurons with intact neurites and a smooth, round soma were considered viable, whereas those with degenerated neurites and an irregular soma were considered nonviable.
Fluorescent microscopy
Assessment of condensation of chromatin, an index of apoptosis (Deckwerth & Jonson, 1993), was performed as previously described (Ueda et al., 1996). Neurons (2.5×105 cells cm−2) were treated with 10 μM Aβ(25 – 35) in the presence or absence of 10 μM S-2474 at 37°C for 48 h. Vehicle controls were treated with culture medium containing 1% deionized water and 0.1% DMSO. Aβ controls were treated with culture medium containing 10 μM Aβ(25 – 35) and 0.1% DMSO. At this time, culture medium was exchanged with PBS containing 10 μM Hoechst 33258 fluorescent dye (Molecular Probes, Eugene, OR, U.S.A.). Cells were incubated for 10 min at 37°C in the dark and washed with PBS. Stained nuclei were categorized as follows: (i) normal nuclei, homogeneously stained chromatin; (ii) intact nuclei with condensed chromatin, crescent-shaped areas of condensed chromatin often located near the periphery of the nucleus; and (iii) fragmented nuclei, more than two condensed micronuclei within the area of a neuron.
In situ labelling of nuclear DNA fragments
Neurons (2.5×105 cells cm−2) were treated with 10 μM Aβ(25 – 35) in the presence or absence of 10 μM S-2474 at 37°C for 48 h. Vehicle controls were treated with culture medium containing 1% deionized water and 0.1% DMSO. Aβ controls were treated with culture medium containing 10 μM Aβ(25 – 35) and 0.1% DMSO. Cortical cell cultures were stained by the TUNEL technique (TdT-mediated dUTP-biotin nick end-labelling), as described (Gavrieli et al., 1992). Apoptotic cells could be distinguished morphologically from necrotic cells by the presence of condensed brown nuclei.
Measurement of PGD2, PGE2 and LTB4
Neurons (2.5×105 cells cm−2) were treated with 10 μM Aβ(25 – 35) in the presence or absence of 10 μM S-2474 at 37°C. Vehicle controls were treated with culture medium containing 1% deionized water and 0.1% DMSO. Aβ controls were treated with culture medium containing 10 μM Aβ(25 – 35) and 0.1% DMSO. At the indicated times in Figure 4 or 5, eicosanoids were extracted according to the method of previous report (Powell, 1980). Supernatants of culture medium (1 ml) was mixed homogenously with cold ethanol (4 ml). The mixture was centrifuged at 1500×g at 4°C for 10 min to remove the particulate matter. Supernatants were diluted with an appropriate volume of distilled water to yield a final concentration of 10% ethanol, and the pH was adjusted to 3.5 – 4.0. Samples were loaded onto reversed phase (C18) Sep-Pack cartridges, which had been prepared by washing with ethanol, followed by distilled water. Samples were washed onto the Sep-Pak with 15 ml of 10% aqueous ethanol, followed by 15 ml of petroleum ether. Samples were extracted with 5 ml of methyl formate. The methyl formate effluents were pooled and evaporated with a heating module and dissolved in RIA buffer (50 mM phosphate buffer, pH 7.3, with 0.1% gelatin and 0.1% azide). The samples were stored frozen until RIA analysis for PG D2, PGE2 and LTB4. PG D2, PGE2 and LTB4 were measured with their RIA kits (duplicate/sample). The cross reactivity of PGJ2 with the PGD2 RIA kit is 7%, whereas that of other PGs such as PGA2, PGE1, PGE2, PGF1α, PGF2α, 6-keto PGE1, 6-keto PGF1α and TXB2 is less than 1%. The cross-reactivity of PGE1 with the PGE2 RIA kit was 30%, and that of other PGs such as AA, PGA1, PGA2, PGB2, PGD2, DHKPGE2, PGF1α, 6-keto PGF1α, PGF2α, DHKPGF2α and 6-keto linoleic acid was less than 1%. The cross reactivity of 20-OH-LTB4, 6-trans LTB4, 5,12-DiHETE with the LTB4 RIA kit are 1.3%, 1.0% and 3.6%, respectively. On the other hand, that of other eicosanoides such as AA, ETYA, 5S,6S-DiHETE, 5S,6R-DiHETE, 5S,12S-DiHETE, 5-HETE, 5,12-HETE, 12-HETE, 20-COOH-LTB4, 6-trans, 12-epi-LTB4, 12-epi-LTB4, 20-COOH-LTB4, LTC4, PGA1, PGA2, PGD2, PGE1, PGE2, PGF1α, PGF2α, 13,14 dihydro, 15-keto PGF2α, NDGA and TXB2 was less than 1%.
Figure 4.
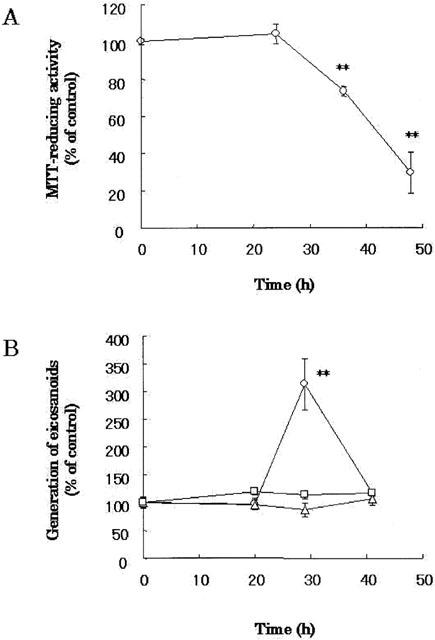
Aβ(25 – 35)-induced neuronal cell death and generation of eicosanoids from Neurons. Cortical neurons were treated with 10 μM Aβ(25 – 35). MTT-reducing activity (A) and eicosanoids (B) were measured at the indicated time points after Aβ treatment. PGD2 (circles), PG E2 (triangles) and LBT4 (squares) were measured with their RIA kits. The control level of PGD2, PGE2 or LTB4 was 73±3, 198±20 or 752±38 pg/ml, respectively. Data are expressed as mean±s.e.mean values (n=4). **P<0.01, compared with vehicle control by ANOVA followed by Dunnett's test. Vehicle control is treated with culture medium containing 1% deionized water and 0.1% DMSO.
Figure 5.

Effect of S-2474 on Aβ(25 – 35)-induced generation of PGD2 from Neurons. (A) PGD2-induced neuronal cell death: Cortical neurons were treated with 10 μM PGD2. MTT-reducing activity was determined at the indicated times after the PG treatment. *P<0.05, **P<0.01, compared with vehicle control by ANOVA followed by Dunnett's test. Vehicle control is treated with culture medium containing 0.1% ethanol. (B) Effect of S-2474 on Aβ(25 – 35)-induced generation of PGD2 from Neurons: Cortical neurons were treated with S-2474 at the indicated concentration in the presence of 10 μM Aβ(25 – 35). Generation of PGD2 was determined 29 h later. Data are expressed as mean±s.e.mean values (n=4). **P<0.01, compared with Aβ control by ANOVA followed by Dunnett's test. Aβ control is treated with culture medium containing 10 μM Aβ (25 – 35) and 0.1% DMSO.
Measurement of reactive oxygen species
Neurons (2.5×105 cells cm−2) were treated with 10 μM Aβ(25 – 35) in the presence or absence of 10 μM S-2474 at 37°C for 24 h later. Vehicle controls were treated with culture medium containing 1% deionized water and 0.1% DMSO. Aβ controls were treated with culture medium containing 10 μM Aβ(25 – 35) and 0.1% DMSO. Intracellular reactive oxygen species were measured by the 2′,7′-dichlorofluorescin diacetate (DCFDA) assay (Chacon & Acosta, 1991). In brief, cultures were loaded with 1 μM DCFDA for 20 min, and then washed twice with 1 ml of PBS. One ml of deoxycholate (1%) was added to lyse the cells. The lysates were transferred to a new tube and chilled on ice. The dishes were washed with 1 ml of distilled water, and the liquid used for washing was added to the lysate. The fluorescent intensity of the lysate was determined with a spectrometer using excitation and emission wavelengths of 488 and 525 nm, respectively. Data are given as percentage of DCFDA fluorescence of corresponding vehicle-treated values.
Statistical analysis
Data are given as means±s.e.mean (n=numbers of observations). We performed two experiments at least on different days, and confirmed their reproductivity. We analysed observations obtained on the same day, and presented the typical experimental results among independent ones on different days to minimize experimental errors. Comparison tests were performed by two-way ANOVA followed by Dunnett's test. IC50 values were calculated by Microsoft Excel Fit as previously reported (Asakura et al., 1999).
Results
Effects of S-2474 on Aβ(25 – 35)- and Aβ(1 – 40)- induced neuronal cell death
Primary cultures of dissociated cortical neurons were exposed to Aβ-related peptides for 48 h, and their toxicity was quantified by the MTT reduction assay (Table 1). Aβ(25 – 35), the toxic fragment of Aβ (Yanker et al., 1990), showed neurotoxicity in a concentration-dependent manner (LD50=3.4±0.4 μM). At 10 μM, Aβ(25 – 35) decreased 70% of MTT-reducing activity markedly [F(3,36)=8.797, P<0.001]. The application of 10 μM Aβ(1 – 40), a native form of Aβ, resulted in a 34% decrement of the MTT-reducing activity [F(3,36)=4.154, P=0.001]. There was no statistical difference in MTT-reducing activity between vehicle-treated and scrambled Aβ(25 – 35) [F(3,36)=0.778, P<0.389] or Aβ(35-25), the reversed sequence of Aβ(25 – 35) [F(3,36)=0.156, P=0.197].
Table 1.
Effects of amyloid-related peptides on neuronal cell survival

Next, we investigated effects of S-2474, a novel NSAID, on the Aβ(25 – 35)-induced neuronal cell death (Figure 1 and Table 1). S-2474 prevented neurons from Aβ-induced cell death significantly in a concentration-dependent manner (IC50=26±12 nM). At 10 μM, S-2474 completely inhibited Aβ(25 – 35)-induced neuronal cell death [F(3,36)=11.276, P<0.001]. Moreover, S-2474 also showed neuroprotective effects in the Aβ(1 – 40)-induced neuronal cell death (Table 1) [F(3,36)=6.356, P<0.001]. As compared to vehicle-treated neurons, there was a slight but statistical difference in S-2474-treated [F(3,36)=2.310, P=0.014], 10 μM S-2474-added Aβ(25 – 35)-treated [F(3,36)=2.147, P=0.015] and S-2474-added Aβ(1 – 40)-treated neurons [F(3,36)=2.066, P=0.017].
Figure 1.
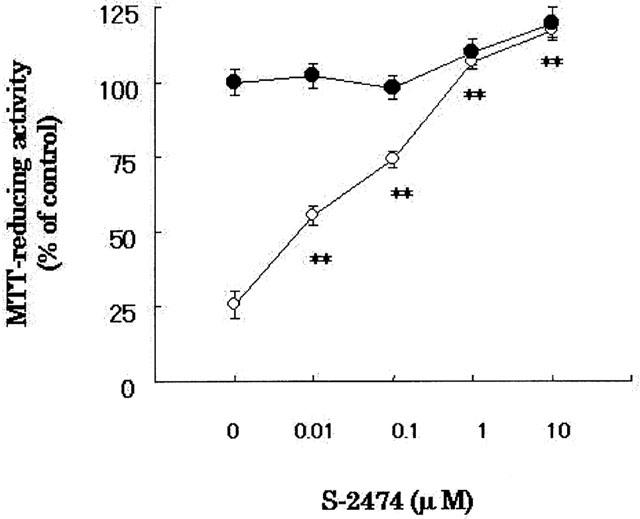
Effects of S-2474 on Aβ(25 – 35)-induced neuronal cell death. Cortical neurons were treated with S-2474 at the indicated concentrations in the presence (open circles) or absence (closed circles) of 10 μM Aβ(25 – 35). MTT reducing activity was determined 48 h after Aβ(25 – 35) treatment. Data are expressed as mean±s.e.mean values (n=4). **P<0.01, compared with Aβ(25 – 35) control by ANOVA followed by Dunnett's test. Vehicle control is treated with culture medium containing 1% deionized water and 0.1% DMSO. Aβ control is treated with culture medium containing 10 μM Aβ (25 – 35) and 0.1% DMSO.
Effects of S-2474 on Aβ(25 – 35)-induced morphological changes of neurons
The compositions of neurons, astrocytes and microglias in cortical cultures were determined by use of antibodies for MAP2, GFAP and OX-42, which are specific for neurons, astrocytes and microglias, respectively. Approximately 95% of the cells were stained by anti-MAP2 antibody, whereas there were few anti-GFAP-positive cells (less than 4%) and anti-microglial antigen-positive ones (less than 3%) (Figure 2A). Examination of cultures treated with Aβ(25 – 35) by light microscopy showed disruption of neurites at 48 h. Some cell bodies shrank and lost their bright phase-contrast appearance. There were markedly fewer cells, and extensive debris was seen attached to the substratum. The morphologic disruption in Aβ(25 – 35)-treated neurons was suppressed completely by S-2474 (Figure 2B).
Figure 2.
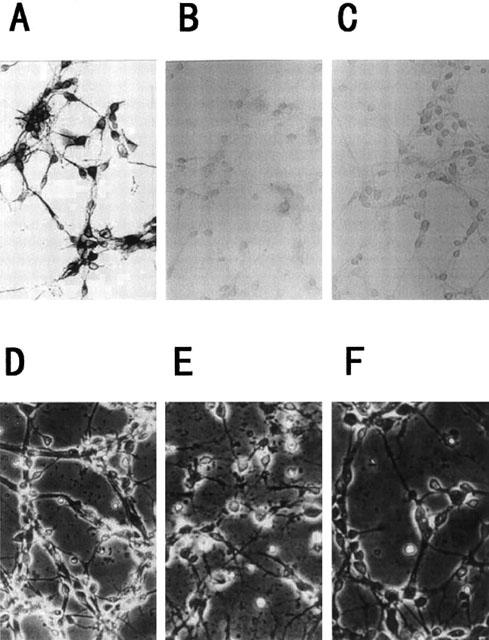
Morphologic changes in cortical neurons by Aβ(25 – 35). Immunocytochemical analysis for anti-MAP2 (A), anti-GFAP (B) or OX-42 (C) was performed on the present cortical cultures. Magnification, ×95. Cortical neurons were treated with vehicle control (D), 10 μM Aβ(25 – 35) (E), or 10 μM Aβ(25 – 35)+10 μM S-2474 (F). Neurons were examined by phase-contrast microscopy 48 h later. Magnification, ×95. Vehicle control is treated with culture medium containing 1% deionized water and 0.1% DMSO. Aβ control is treated with culture medium containing 10 μM Aβ (25 – 35) and 0.1% DMSO.
Effects of S-2474 on Aβ(25 – 35)-induced apoptotic features of neurons
Previously, we reported that Aβ(25 – 35)-induced neuronal cell death was accompanied with characteristic features of apoptosis, such as chromatin condensation and DNA fragmentation (Ueda et al., 1996). Therefore, chromatin condensation was examined with Hoechst 33258 fluorescent dye (Figure 3A and Table 2). In vehicle-treated cultures, cells showed little fluorescence in the nucleus. On the other hand, condensed and fragmented chromatin was clearly observed in Aβ(25 – 35)-treated cultures [F(3,36)=8.550, P=0.001]. The amount of condensed chromatin in Aβ(25 – 35)-treated neurons was decreased significantly by S-2474 [F(3,36)=8.382, P<0.01]. There was no statistical difference in MTT-reducing activity between vehicle-treated and 10 μM S-2474-added Aβ(25 – 35)-treated neurons [F(3,36)=0.168, P=0.293].
Figure 3.
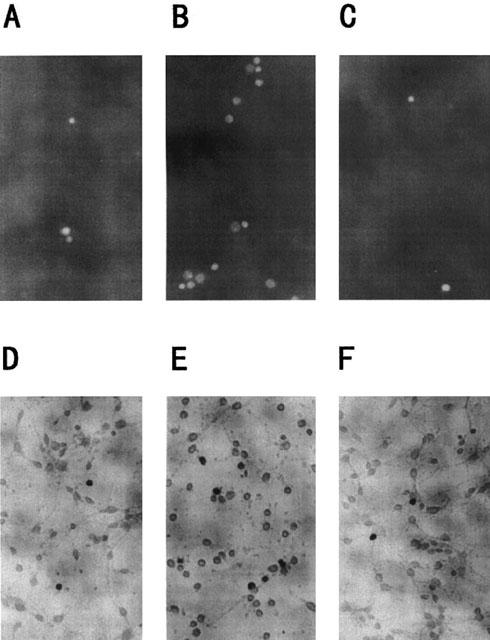
Apoptotic features of cortical neurons induced by Aβ(25 – 35). Cortical neurons were treated with vehicle control (A and D), 10 μM Aβ(25 – 35) control (B and E) or 10 μM Aβ(25 – 35)+10 μM S-2474 (C and F). Neurons were stained with 1 μM Hoechst 33258 for 10 min 48 h later (A, B, and C). Neurons were fixed with 4% paraformaldehyde, washed twice with PBS, and stained by the TUNEL technique 48 h later (D, E, and F). Magnification, ×95. Vehicle control is treated with culture medium containing 1% deionized water and 0.1% DMSO. Aβ control is treated with culture medium containing 10 μM Aβ (25 – 35) and 0.1% DMSO.
Table 2.
Effects of S-2474 on Aβ-induced Hoechst 33258 and TUNEL-positive neurons (cells/cm2)

Fragmentation of DNA was also estimated with the TUNEL technique (Figure 3B and Table 2). After neurons were incubated with or without Aβ(25 – 35) for 48 h, the number of TUNEL-positive nuclei was increased significantly in Aβ(25 – 35)-treated neurons as compared to vehicle-treated ones [F(3,36)=3.949, P=0.004]. The number of TUNEL-positive nuclei in Aβ(25 – 35)-treated neurons was decreased significantly by S-2474 [F(3,36)=4.584, P=0.003]. There was no statistical difference in MTT-reducing activity between vehicle-treated and 10 μM S-2474-added Aβ(25 – 35)-treated neurons [F(3,36)=0.635, P=0.107]. Thus, S-2474 ameliorated Aβ-induced apoptotic features.
Aβ(25 – 35)-induced generation of AA metabolites
Activation of PLA2 contributes to the neurotoxicity of Aβ(25 – 35) (Stephenson et al., 1996), and level of PGD2 among AA metabolites is significantly elevated in AD (Iwamoto et al., 1989). We examined the association of eicosanoids generation with Aβ(25 – 35)-induced neuronal cell death (Figure 4). There was no statistical difference in MTT-reducing activity between vehicle-treated and Aβ(25 – 35)-treated neurons at 24 h [F(3,36)=0.354, P=0.483]. After 24 h, 10 μM Aβ(25 – 35) decreased MTT-reducing activity in a time-dependent manner, and killed 70% of neurons at 48 h later [F(3,36)=9.009, P=0.001] (Figure 4A).
Next, we examined generation of eicosanoids from neurons after Aβ-treatment. The level of PGD2 in vehicle controls was not altered throughout the culture. When neurons were exposed to Aβ(25 – 35), there was no statistical difference in generation of PGD2 between vehicle-treated and Aβ(25 – 35)-treated neurons at 20 h [F(3,36)=0.073, P=0.810]. Then, PGD2 was increased transiently at 29 h [F(3,36)=5.803, P=0.004] and decreased thereafter [F(3,36)=0.444, P=0.019] (Figure 4B). On the other hand, there was no statistical difference in generation of PGE2 [F(3,36)=0.908, P=0.409] and LTB4 [F(3,12)=1.490, P=0.187] between vehicle-treated and Aβ(25 – 35)-treated neurons at 29 h (Figure 4B). These results indicated that Aβ significantly generated PGD2, but not PGE2 nor LTB4, from neurons prior to neuronal cell death.
Effects of PGD2 on neuronal cell survival
To ascertain whether PGD2 possesses neurotoxicity, we examined effect of PGD2 on neuronal survival (Figure 5 and Table 3). As shown in Table 3, PGD2 caused neuronal cell death in a concentration-dependent manner (LD50=7.9±0.8 μM). MTT reducing activity was decreased to 34% of control neurons by 10 μM PGD2 [F(3,56)=6.531, P<0.001] at 24 h. At 6 h, there was no statistical difference in MTT-reducing activity between vehicle-treated and 10 μM PGD2-treated neurons [F(3,56)=0.633, P=0.072]. Then, PGD2 displayed significant neurotoxicity at 9 h [F(3,56)=2.897, P=0.001] and killed 80% of neurons at 24 h [F(3,52)=7.250, P<0.001] (Figure 5A). Moreover, 10 μM 15d-Δ12,14-PGJ2 caused neuronal cell death more potently than PGD2 [F(3,56)=8.197, P<0.001] at 24 h (Table 3). As compared to vehicle controls, however, no statistical difference was detected in other eicosanoids such as AA [F(3,56)=1.207, P=0.388], PGE2[F(3,56)=0.268, P=0.739], 9α, 11β-PGF2[F(3,56)=0.525, P=0.328], PGF2α[F(3,56)=0.746, P=0.132], PGI2[F(3,56)=0.418, P=0.416], U-46619[F(3,56)=0.363, P=0.588], LTB4 [F(3,56)=1.003, P=0.083], LTC4 [F(3,56)=0.490, P=0.437] and LTD4 [F(3,56)=0.200, P=0.661].
Table 3.
Effects of eicosanoids on neuronal cell survival

Next, we examined effect of S-2474, a COX-2 inhibitor, on Aβ-induced PGD2 generation. As shown in Figure 5B, S-2474 inhibited the PGD2 generation in a concentration dependent manner (IC50=69.8±21.9 nM). At 10 μM, S-2474 lowered the elevated level of PGD2 significantly [F(3,36)=4.285, P=0.012]. There was a slight but statistical difference in PGD2 generation between vehicle-treated and 10 μM S-2474-added Aβ(25 – 35)-treated neurons [F(3,36)=1.286, P=0.015]. Thus, S-2474 suppressed generation of PGD2 from Aβ-treated neurons significantly, although not completely.
Aβ(25 – 35)-induced generation of free radicals
S-2474 contains the di-tert-butylphenol antioxidant moiety (Inagaki et al., 2000). To examine effects of S-2474 on Aβ(25 – 35)-induced generation of free radicals, we measured the intracellular reactive oxygen species by the DCFDA assay (Figure 6). Application of Aβ(25 – 35) for 24 h markedly increased the intracellular reactive oxygen species [F(3,24)=8.599, P<0.001]. The radical scavenger, vitamin E (100 μM), potently reduced accumulation of reactive oxygen species [F(3,24)=8.378, P<0.001]. There was no statistical difference in generation of free radicals between vehicle-treated and 100 μM vitamin E-added Aβ(25 – 35)-treated neurons [F(3,24)=0.384, P=0.488]. As well as vitamine E, S-2474 significantly decreased the elevated level of free radicals in a concentration-dependent manner (IC50=601±75 nM) (Figure 6A). At 10 μM, S-2474 significantly reduced radicals from Aβ(25 – 35)-treated neurons [F(3,24)=5.883, P=0.003]. There was no statistical difference in generation of free radicals between vehicle-treated and 10 μM S-2474-added Aβ(25 – 35)-treated neurons [F(3,24)=2.291, P=0.051].
Figure 6.
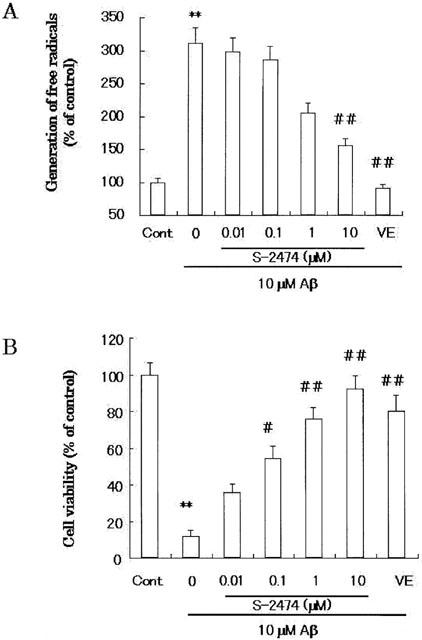
Involvement of free radicals in Aβ(25 – 35) neurotoxicity. (A) Level of free radicals measured by DCFDA assay: Cortical neurons were treated with 10 μM Aβ(25 – 35) in the presence or absence of S-2474 or 100 μM vitamin E, and the DCFDA assay was performed 24 h later. Data are expressed as mean±s.e.mean (n=4), per cent of control cultures, which were incubated for the same amount of time as experimental cultures (n=4). **P<0.01, compared with vehicle control by ANOVA followed by Dunnett's test. ##P<0.01, compared with Aβ control by ANOVA followed by Dunnett's test. Vehicle control is treated with culture medium containing 1% deionized water and 0.1% DMSO. Aβ control is treated with culture medium containing 10 μM Aβ (25 – 35) and 0.1% DMSO. (B) Effect of S-2474 on Aβ(25 – 35)-induced neuronal cell death. Cortical neurons were treated with S-2474 at the indicated concentration or 100 μM vitamin E in the presence of 10 μM Aβ(25 – 35). Forty-eight hours later, cell viability was measured according to the morphological criteria. Data are expressed as mean±s.e.mean values (n=4). **P<0.01, compared with vehicle control by ANOVA followed by Dunnett's test. #P<0.05, ##P<0.01, compared with Aβ control by ANOVA followed by Dunnett's test. Vehicle control is treated with culture medium containing 1% deionized water and 0.1% DMSO. Aβ control is treated with culture medium containing 10 μM Aβ (25 – 35) and 0.1% DMSO.
To examine the effects of anti-oxidants on Aβ neurotoxicity, we performed morphometric cell counting to quantify the magnitude of cell death according to the previous report (Ueda et al., 1997a). The presence of anti-oxidants may affect the MTT reduction assay (Behl et al., 1992). Therefore, neurotoxicity was measured morphologically (Figure 2). S-2474 or vitamin E was added to cortical cultures simultaneously with Aβ(25 – 35). Morphometric cell counting was then performed 48 h later (Figure 6B). In accordance with the results using MTT-reducing activity, Aβ(25 – 35) induced morphologic neurodegeneration [F(3,24)=19.403, P<0.001]. Vitamin E significantly ameliorated Aβ(25 – 35)-induced neurodegeneration [F(3,24)=16.818, P<0.001]. S-2474 also exhibited neuroprotective effects in a concentration-dependent manner (IC50=43±19 nM). At 10 μM, S-2474 rescued neurons from Aβ(25 – 35)-induced neurodegeneration [F(3,24)=16.594, P<0.001]. There was no statistical difference in molphometric cell viability between vehicle-treated and vitamin E-added Aβ(25 – 35)-treated neurons [F(3,24)=2.585, P=0.048]. There was a slight but statistical difference in molphometric cell viability between vehicle-treated and 10 μM S-2474-added Aβ(25 – 35)-treated neurons [F(3,24)=2.810, P=0.030]. Thus, S-2474 ameliorated Aβ-induced molphologic neurodegeneration significantly, although not completely.
Discussion
In the present study, we demonstrated that S-2474 prevented neurons from Aβ-induced cell death completely. S-2474 also ameliorated Aβ-induced morphological changes and apoptotic features such as the condensation of chromatin and the fragmentation of DNA significantly. The present cortical cultures contained little non-neuronal cells, indicating that S-2474 exhibited neuroprotective effect directly, but not indirectly via non-neuronal cells.
S-2474 is reported to inhibit COX-2 and 5-LO (Inagaki et al., 2000). COX-2 is up-regulated in AD brain and in Aβ-treated SH-SY5Y neuroblastoma cells (Pasinetti & Aisen, 1998). Among PGs, the formation of PGD2 is significantly increased in AD brain (Iwamoto et al., 1989). In the present culture of rat cortical neurons, changes in COX-2 expression could not be detected after Aβ-treatment (data not shown). However, PGD2 was transiently increased prior to Aβ-induced neuronal cell death. PGD2 induced neuronal cell death in a concentration- and time-dependent manner. Neuronal cell survival was not affected by other eicosanoids such as PGE2, 9α-11β-PGF2, PGF2α, PGI2 and U-46619, a stable agonist for thromboxane A2 receptor. Among eicosanoids tested, only PGD2 showed neurotoxicity. Furthermore, S-2474 decreased significantly the PGD2. On the other hand, LTs including LTB4, C4, and D4 were not altered, and did not kill neurons. Thus, S-2474 appears to rescue neurons from Aβ-induced cell death via inhibiting generation of PGD2.
PGD2 is the major PG in the brain of the rat and other mammalian species (Abdel-Halim et al., 1977), and PGD2 levels were significantly increased in AD brain (Iwamoto et al., 1989), suggesting involvement in neurodegeneration. Indeed, PGD2 caused neuronal cell death in our cortical cultures, indicating a possible involvement of PGD2 in the apoptosis of neurons induced by Aβ. Interpretation of PGD2 as an apoptosis inducer requires circumspection for several reasons, however. First, the LD50 value of PGD2 is high as compared to the affinity for its receptors. Second, there is a latent period for PGD2 to induce neuronal cell death. Third, it has not been clarified sufficiently how PGD2 is associated with inflammatory diseases including AD. Therefore, further studies are necessary for determining the pathologic role of PGD2 in AD.
There was a close correlation between MTT-reducing activity and morphologic viability. A comparison of the former activity with the latter viability in detail gave rise to a small question, however. In the presence of 10 μM S-2474, the former activity of Aβ-treated neurons was significantly higher than that of vehicle controls. On the other hand, in the presence of 10 μM S-2474, the latter viability of Aβ-treated neurons was lower than that of control neurons. S-2474 contains the di-tert-butylphenol antioxidant moiety (Inagaki et al., 2000), and scavenged free radicals from Aβ-treated neurons. The presence of anti-oxidants may affect the MTT reduction assay (Behl et al., 1992), and vitamin E increased the MTT-reducing activity regardless of the presence of Aβ (data not shown). Thus, S-2474 appears to increase MTT-reducing activity via an antioxidant effect at high concentrations.
Besides COX-2, expression of peroxysome proliferator-activated receptor-γ (PPARγ) is also increased in AD brains (Kitamura et al., 1999). PPARγ is activated by NSAIDs such as indomethacine (Kitamura et al., 1999; Klegeris et al., 1999). Recent studies have demonstrated that NSAIDs with PPARγ agonist effects reduce apoptosis in cerebellar granule cells (Heneka et al., 2000). However, an endogenous PPARγ agonist, 15d-Δ12,14-PGJ2, induces apoptosis in rat cortical neurons (Rohn et al., 2001). In the present study, we confirmed the latter report. Thus, it is unlikely that PPARγ contributes to the neuroprotective effect of S-2474.
The combination of the present study with our previous reports gave rise to therapeutic potentials of S-2474 for AD. First, S-2474 causes little adverse effect such as gastrointestinal ulceration (Jyoyama et al., 1997), because it is a weak inhibitor for COX-1 (Inagaki et al., 2000). Second, S-2474 possesses inhibitory activities against the production of inflammatory factors (Inagaki et al., 2000), including IL-1, IL-6 and NO, which are elevated in the AD brain (Minster et al., 2000). It suggests a suppressive effect of S-2474 on the inflammation in AD brain. Third, S-2474 appears to exhibit neuroprotective effect as a COX-2 inhibitor and an anti-oxydant. Thus, we conclude that S-2474 has neuroprotective effect against Aβ toxicity. Furthermore, we demonstrate that S-2474 possesses therapeutic potentials for AD via a direct neuroprotective effect as well as an indirect anti-inflammatory effect.
Abbreviations
- AA
arachidonic acid
- Aβ
amyloid β protein
- AD
Alzheimer's disease
- ANOVA
analysis of variance
- [Ca2+]i
intracellular Ca2+ levels
- COX
cyclo-oxygenase
- DCFDA
2′,7′-dichlorofluorescin diacetate
- 15d-Δ12,14-PGJ2
15-deoxy-Δ12,14-prostaglandin J2
- GFAP
glial fibrillary acidic protein
- IL
interleukin
- 5-LO
5-lipoxygenase
- LT
leukotriene
- MAP-2
microtuble-associated protein 2
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide dye
- NSAID
nonsteroidal anti-inflammatory drug
- PG
prostaglandin
- PLA2
phospholipase A2
- PPAR γ
peroxysome proliferator-activated receptor γ
- TUNEL
TdT-mediated dUTP-biotin nick end-labelling
- TXA2
thromboxane A2
References
- ABDEL-HALIM M.S., HAMBERG M., SJÖQUIST, ÄNGGÅRD E. Identification of prostaglandin in homogenates of rat brain. Prostaglandin. 1977;14:633–643. doi: 10.1016/0090-6980(77)90190-3. [DOI] [PubMed] [Google Scholar]
- ASAKURA K., KANEMASA T., MINAGAWA K., KAGAWA K., NINOMIYA M. The nonpeptide α-eudesmol from Juniperus virginiana Linn. (Cupressaceae) inhibits ω-agatoxin IVA-sensitive Ca2+ currents and synaptosomal 45Ca2+ uptake. Brain Res. 1999;823:169–176. doi: 10.1016/s0006-8993(99)01165-8. [DOI] [PubMed] [Google Scholar]
- BEHL C., DAVIS J.B., COLE G.M., SCHUBERT D. Vitamin E protects nerve cells from amyloid β protein toxicity. Biochem. Biophys. Res. Commun. 1992;186:944–950. doi: 10.1016/0006-291x(92)90837-b. [DOI] [PubMed] [Google Scholar]
- BEHL C., DAVIS J.B., LESLEY R., SCHUBERT D. Hydrogen peroxide mediates amyloid β protein toxicity. Cell. 1994;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- CHACON E., ACOSTA D. Mitochondrial regulation of superoxide by Ca2+: an alternate mechanism for the cardiotoxicity of doxorubicin. Toxicol. Appl. Pharmacol. 1991;107:117–128. doi: 10.1016/0041-008x(91)90336-d. [DOI] [PubMed] [Google Scholar]
- CUMMINGS B.J., COTMAN C.W. Image analysis and beta-amyloid load in Alzheimer's disease and relation to dementia severity. Lancet. 1995;346:1524–1528. doi: 10.1016/s0140-6736(95)92053-6. [DOI] [PubMed] [Google Scholar]
- DECKWERTH T.L., JONSON E.M., JR Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J. Cell. Biol. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FORLONI G., CHISA R., SMIROLDO S., VERGA L., SALMONA M., TAGLIVINI F., ANGERETTI N. Apoptosis mediated neurotoxicity induced by chronic application of β amyloid fragment 25–35. Neuro. Report. 1993;4:523–526. doi: 10.1097/00001756-199305000-00015. [DOI] [PubMed] [Google Scholar]
- GAVRIELI Y., SHERMAN Y., BEN-SASSON S.A. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOODMAN Y., STEINER M.R., STEINER S.M., MATTSON M.P. Nordihydroguaiaretic acid protects hippocampal neurons against amyloid β-peptide toxicity, and attenuates free radical and calcium accumulation. Brain Res. 1994;654:171–176. doi: 10.1016/0006-8993(94)91586-5. [DOI] [PubMed] [Google Scholar]
- HENEKA M.T., KLOCKGETHER T., FEINSTEIN D.L. Peroxisome proliferator-activated receptor-γ ligands reduce neuronal inducible nitric oxide synthase expression and cell death in vivo. J. Neurosci. 2000;20:6862–6867. doi: 10.1523/JNEUROSCI.20-18-06862.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HIDAKA T., HOSOE K., ARIKI Y., TAKEO K., YAMASHITA T., KATSUMI I., KONDO H., YAMASHITA K., WATANABE K. Pharmacological properties of a new anti-inflammatory compound, α-(3,5-Di-tert-butyl-4-hydroxy-benzylidene)-γ-butyrolactone(KME-4), and its inhibitory effects on prostaglandin synthase and 5-lipooxygenase. Jpn. J. Pharmacol. 1984;36:77–85. doi: 10.1254/jjp.36.77. [DOI] [PubMed] [Google Scholar]
- IKUTA H., SHIROTA H., KOBAYASHI S., YAMAGISHI Y., YAMADA K., YAMATSU I., KATAYAMA K. Synthesis and anti-inflammatory activities of 3-(3,5-Di-tert-butyl-4-hydroxy-benzylidene)pyrrolidin-2-ones. J. Med. Chem. 1987;30:1995–1998. doi: 10.1021/jm00394a011. [DOI] [PubMed] [Google Scholar]
- INAGAKI M., TSURI T., JYOYAMA H., ONO T., YAMADA K., KOBAYASHI M., HORI Y., ARIMURA A., YASUI K., OHNO K., KAKUDO S., KOIZUMI K., SUZUKI R., KATO M., KAWAI S., MATSUMOTO S. Novel antiarthritic agents with 1,2-isothiazolidine-1,1-dioxide (γ-sultam) skeleton: cytokine suppressive dual inhibitors of cyclooxygenase-2 and 5-lipoxygenase. J. Med. Chem. 2000;43:2040–2048. doi: 10.1021/jm9906015. [DOI] [PubMed] [Google Scholar]
- JYOYAMA H., HORI Y., YASUI K., OHNO K., MATSUMOTO S., NAKAMURA K. A novel antiarthritic agent S-2474 with dual profile of NSAID plus DMARD: In vivo characterization. Inflamm. Res. 1997;46:S–257. [Google Scholar]
- IWAMOTO N., KOBAYASHI K., KOSAKA K. The formation of prostaglandins in the postmortem cerebral cortex of Alzheimer-type dementia patients. J. Neurol. 1989;236:80–84. doi: 10.1007/BF00314401. [DOI] [PubMed] [Google Scholar]
- KITAMURA Y., SHIMOHAMA S., KOIKE H., KAKIMURA J., MATSUOKA Y., NOMURA Y., GEBICKE-HAERTER P.J., TANIGUCHI T. Increased expression of cyclooxygenase and peroxisome proliferator-activated receptor-γ in Alzheimer's disease brains. Biochem. Biophys. Res. Commun. 1999;254:582–586. doi: 10.1006/bbrc.1998.9981. [DOI] [PubMed] [Google Scholar]
- KLEGERIS A., WALKER D.G., MCGEER P.L. Toxicity of human THP-monocytic cells towards neuron-like cells is reduced by non-steroidal anti-inflammatory drugs (NSAIDs) Neuropharmacol. 1999;38:1017–1025. doi: 10.1016/s0028-3908(99)00014-3. [DOI] [PubMed] [Google Scholar]
- MATSUMOTO S., JYOYAMA H., YAMADA K., ONO T., KOIZUMI K., KAKUDO S., ARIMURA A., SUZUKI R. A novel antiarthritic agent S-2474 with dual profile of NSAID plus DMARD: In vitro characterizations. Inflamm. Res. 1997;46:S257. [Google Scholar]
- MATTSON M.P., BARGER S.W., BEGLEY J.G., MARK R.J. Calcium, free radicals, and excitotoxic neuronal death in primary cell culture. Methods Cell. Biol. 1995;46:187–216. doi: 10.1016/s0091-679x(08)61930-5. [DOI] [PubMed] [Google Scholar]
- MATTSON M.P., CHENG B., DSAVIS D., BRYANT K., LIEBERBURG I., RYDLE R.E. β-amyloid peptide destabilized calcium homeostatis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCGEER P.L. Cyclo-oxygenase-2 inhibitors. Relational and therapeutic potential for Alzheimer's disease. Drugs and Aging. 2000;17:1–11. doi: 10.2165/00002512-200017010-00001. [DOI] [PubMed] [Google Scholar]
- MCGEER P.L., SCHULZER M., MCGEER E.G. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer's disease: A review of 17 epidemiologic studies. Neurology. 1996;47:425–432. doi: 10.1212/wnl.47.2.425. [DOI] [PubMed] [Google Scholar]
- MILLER T.A. Protective effects of prostaglandins against gastric mucosal damage: Current knowledge and proposed mechanisms. Am. J. Physiol. 1983;245:G601–G623. doi: 10.1152/ajpgi.1983.245.5.G601. [DOI] [PubMed] [Google Scholar]
- MINSTER R.L., DEKOSKY S.T., GANGLI M., BELLE S., KAMBOH M.I. Genetic association studies of interleukin-1 (IL-1A and IL-1B) and interleukin-1 receptor antagonist genes and the risk of Alzheimer's disease. Ann. Neurol. 2000;48:817–819. [PubMed] [Google Scholar]
- PASINETTI G.M., AISEN P.S. Cyclooxygenase-2 expression is increased in frontal cortex of Alzheimer's disease brain. Neurosci. 1998;87:319–324. doi: 10.1016/s0306-4522(98)00218-8. [DOI] [PubMed] [Google Scholar]
- PIKE C.J., WALENCEWICZ A.J., GLABE C.G., COTMAN C.W. In vitro aging of β-amyloid protein causes peptide aggregation and neurotoxicity. Brain Res. 1991;621:279–282. doi: 10.1016/0006-8993(91)91553-d. [DOI] [PubMed] [Google Scholar]
- POWELL W.S. Rapid extraction of oxygenated metabolites of arachidonic acid from biological samples using octadecylsilyl silica. Prostaglandins. 1980;20:947–957. doi: 10.1016/0090-6980(80)90144-6. [DOI] [PubMed] [Google Scholar]
- ROGERS J., KIRBY L.C., HEMPELMAN S.R., BERRY D.L., MCGEER P.L., KASZNIAK A.W., ZALINSKI J., COFIELD M., MANSUKHANI L., WILLSON P., KOGAN F. Clinical trial of indomethacin in Alzheimer's disease. Neurology. 1993;43:1609–1611. doi: 10.1212/wnl.43.8.1609. [DOI] [PubMed] [Google Scholar]
- ROHN T.T., WONG S.M., COTMAN C.W., CRIBBS D.H. 15-deoxy-Δ12,14- prostaglandin J2, a specific ligand for peroxisome proliferator-activated receptor-γ, induces neuronal apoptosis. Neuroreport. 2001;12:839–843. doi: 10.1097/00001756-200103260-00043. [DOI] [PubMed] [Google Scholar]
- SELKOE D.J. The molecular pathology of Alzheimer's disease. Neuron. 1991;6:487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- SELKOE D.J. Alzheimer's disease: a central role for amyloid. J. Neuropathol. Exp. Neurol. 1994;53:438–447. doi: 10.1097/00005072-199409000-00003. [DOI] [PubMed] [Google Scholar]
- STEPHENSON D.T., LEMERE C.A., SELKOE D.J., CLEMENS J.A. Cytosolic phospholipase A2 (cPLA2) immunoreactivity is elevated in Alzheimer's disease brain. Neurobil. Dis. 1996;3:51–63. doi: 10.1006/nbdi.1996.0005. [DOI] [PubMed] [Google Scholar]
- STEWART W.F., KAWAS C., CORRDA M., METTER J. Risk of Alzheimer's disease and duration of NSAIDs use. Neurology. 1997;48:626–632. doi: 10.1212/wnl.48.3.626. [DOI] [PubMed] [Google Scholar]
- UEDA K., FUKUI Y., KAGEYAMA H. Amyloid β protein-induced neuronal cell death: neurotoxic properties of aggregated amyloid β protein. Brain Res. 1994;639:240–244. doi: 10.1016/0006-8993(94)91736-1. [DOI] [PubMed] [Google Scholar]
- UEDA K., SHINOHARA S., YAGAMI T., ASAKURA K., KAWASAKI K. Amyloid β protein potentiates Ca2+ influx through L-type voltage-sensitive Ca2+ channels: a possible involvement of free radicals. J. Neurochem. 1997a;68:265–271. doi: 10.1046/j.1471-4159.1997.68010265.x. [DOI] [PubMed] [Google Scholar]
- UEDA K., YAGAMI T., ASAKURA K., KAWASAKI K. Chlorplomazine reduces toxicity and Ca2+ uptake induced by amyloid β protein (25–35) in vitro. Brain Res. 1997b;748:184–188. doi: 10.1016/s0006-8993(96)01300-5. [DOI] [PubMed] [Google Scholar]
- UEDA K., YAGAMI T., KAGEYAMA H., KAWASAKI K. Protein kinase inhibitor attenuates apoptotic cell death induced by amyloid β protein in culture of the rat cerebral cortex. Neurosci. Lett. 1996;203:175–178. doi: 10.1016/0304-3940(95)12288-5. [DOI] [PubMed] [Google Scholar]
- VANE J.R., BOTTING R.M. Antiinflammatory drugs and their mechanism of action. Inflammation Res. 1998;47:S78–S87. doi: 10.1007/s000110050284. [DOI] [PubMed] [Google Scholar]
- WEISS J.H., PIKE C.J., COTMAN C.W. Ca2+ channel blockers attenuates β-amyloid peptide toxicity to cortical neurons in culture. J. Neurochem. 1994;62:372–375. doi: 10.1046/j.1471-4159.1994.62010372.x. [DOI] [PubMed] [Google Scholar]
- YANKER B.A., DUFFY L.K., KIRSCHNER D.A. Neurotrophic and neurotoxic effects of amyloid β protein: reversal by tachykinin neuropeptides. Science. 1990;250:279–282. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]
- YASOJIMA K., SCHWAB C., MCGEER E.G., MCGEER P.L. Distribution of cyclooxygenase-1 and cyclooxygenase-2 mRNAs and proteins in human brain and peripheral organs. Brain Res. 1999;830:226–236. doi: 10.1016/s0006-8993(99)01389-x. [DOI] [PubMed] [Google Scholar]