

Abstract
To investigate the effect of the hydrophilic Asn amino acid at position 230 of the human μ-opioid receptor (hMOR230) on the potency of various agonists, we mutated this residue to Thr and Leu (hMORN230T and hMORN230L respectively).
Taking advantage of the functional coupling of the opioid receptor with the heteromultimeric G-protein-coupled inwardly rectifying K+ (GIRK1/GIRK2) channel, either the wild type hMOR or one of the mutated receptors (hMORN230L or hMORN230T) were functionally coexpressed with GIRK1/GIRK2 channels and a regulator of G-protein signalling (RGS4) in Xenopus laevis oocytes.
The two-microelectrode voltage clamp technique was used to measure the opioid receptor-activated GIRK1/GIRK2 channel responses. The potency of [D-Ala2,N-MePhe4,Gly5-ol]-enkephalin (DAMGO), remained unaffected as measured via hMORN230T and hMORN230L, while the potency of fentanyl and morphine significantly increased via these mutated receptors.
Our results are indicative for the existence of hydrophobic interactions between a methyl-group of the side chain of Thr or Leu on the one hand and the piperidine-ring of fentanyl and the hexene-ring of morphine on the other. The mutations also had no influence on the potency of morphine-6-glucuronide (M6G) and morphine-3-glucuronide (M3G).
We conclude that the hydrophilic side chain of Asn in position 230 is not involved in the formation of a H-bond with the aliphatic alcohol of morphine and that an enhancement of the potency of morphine and fentanyl can be explained by mutating this residue towards more hydrophobic amino acids.
Keywords: Cloned opioid receptors, Xenopus oocytes, morphine, DAMGO, fentanyl, site-directed mutagenesis, voltage clamp, opioid agonists, RGS
Introduction
Opiate and opioid compounds act on opioid receptors to produce their pharmacological action (Xu et al., 1999). Molecular cloning studies have identified cDNAs and genes encoding μ-, δ- and κ-opiate receptors from several species (Kieffer et al., 1992; Knapp et al., 1995; Wang et al., 1993), providing a unique opportunity to examine individual opioid receptor types with regard to pharmacological profile, structure-function analysis, cellular effector coupling, anatomical distribution, regulation and expression.
Deduced amino acid sequences of these clones display the motif of putative seven α-helical lipophilic transmembrane helices (TM's) connected by alternating intracellular and extracellular hydrophilic loops, that is characteristic of G protein-coupled receptors (Xu et al., 1999). A family of 20 endogenous opioid receptor ligands were generated from three precursor proteins, prodynorphin, proenkephalin and proopiomelanocortin. In addition to these endogenous peptide ligands, a large series of alkaloid opiate drugs interact with these receptors. Although the opioid peptides belong to a class of molecules distinct from opiate alkaloids, they all share common structural features including a positive charge juxtaposed with an aromatic ring (Evans et al., 1992).
Opioid receptor activation produces a wide array of cellular responses like inhibition of adenylyl cyclase, inhibition of voltage-dependent calcium channels (N, P, Q and R type) and activation of an inwardly rectifying potassium channel (Ikeda et al., 1995). The μ-opioid receptor serves as the principal physiological target for most clinically important opioid analgesics, such as morphine and fentanyl. δ-agonists are poor analgesic compounds, but exhibit less addictive potential, while κ-agonists should be restricted to the periphery because of the strong dysphoric properties of these compounds (Kieffer, 1999).
In general, two approaches are used to examine issues of selectivity and affinity. First, constructing opioid chimeric receptors provides a powerful tool for mapping the regions involved in drug selectivity. Using receptor chimeras, several groups have reported that the docking sites for the opioid peptides and alkaloids are different (Li et al., 1996; Meng et al., 1995; Onogi et al., 1995; reviewed in Law et al., 1999). The second approach involves computational modelling (Filizola et al., 1999; Pogozheva et al., 1998) which predicts possible specific amino acid residues that may be critical for receptor binding and selectivity. Nevertheless, caution must be taken in interpreting the data because these structural modifications must retain the conformation of the parent receptors to a large extent (Mansour et al., 1997).
Structural models and experimental data have indicated Asp147 and His297, which are located in the bottom of the binding pocket, as key anchor residues because they could serve as attachment points for the quaternary nitrogen and hydroxyl-group, respectively, of the tyramine moiety of the opiate ligand (Befort et al., 1996; Li et al., 1999; Spivak et al., 1997). Using this alignment Pogozheva et al. (1998) suggested that the preferred binding of morphinans to the μ receptor could be explained by the presence of an additional H-bond between Asn230 and the 6-OH group of morphine. Figure 1 shows a schematic representation of the human μ-opioid receptor with the localization of residue Asn230 together with other residues shown to interact with opiate ligand binding.
Figure 1.

Schematic drawing of the human μ-opioid receptor. Solid black horizontal lines represent the approximate membrane boundaries. The bold circle indicates the position of Asn230. Other residues shown to interact with opiate ligand binding are illustrated and encircled, in their one letter code. The transmembrane helices (TM), the different extracellular loops (EL) and intracellular loops (IL) are denoted by roman numerals.
The goal of this work was to examine the importance of Asn230 in the μ-opioid receptor. Therefore we mutated Asn230 to Thr and Leu, which are neutral and hydrophobic residues, respectively, and determined the EC50-values for GIRK1/GIRK2 channel activation through consecutive activation of wild type and mutant opioid receptors coupled to G proteins and expressed in Xenopus laevis oocytes.
To achieve this, we coexpressed GIRK1- and GIRK2-channels, as the neuronal G-protein-gated inwardly rectifying K+ channels function as heteromultimers (Kofuji et al., 1995) together with RGS4, a regulator of G-protein signalling. RGS proteins accelerate the GTPase rate of heterotrimeric Gα proteins (de vries et al., 2000; Doupnik et al., 1997). Coexpression of RGS4, the brain-expressed isoform of RGS proteins, reconstitutes the native gating kinetics by accelerating GIRK1/GIRK2 channel deactivation (Ulens et al., 2000a).
Overall, this experimentally created model (Ulens et al., 2000b) provides a defined population of functionally active opioid receptors (wild type receptor or one of the mutant receptors) and an excellent tool for measuring the efficacy and potency of a ligand for a certain receptor.
Methods
Subcloning and in vitro transcription of cDNA clones encoding GIRK1/2 channels, human μ opioid receptors and RGS4
Plasmids containing the entire coding sequence for the mouse GIRK1 and the mouse GIRK2 channel were subcloned into the vector pSP35T and pBScMXT, respectively and designated as pSP/GIRK1 (Kobayashi et al., 1995) and pBScMXT/GIRK2 (Kofuji et al., 1995). The polylinker in each of these vectors is flanked by Xenopus globin 5′ and 3′ untranslated regions, resulting in an enhanced protein expression after injection of in vitro transcribed cRNA (Krieg & Melton, 1984). For in vitro transcription, plasmids were first linearized either with EcoRI (for pSP/GIRK1), or with SalI (for pBScMXT/GIRK2). Next the cRNAs were synthesized from the linearized plasmids using the large-scale SP6 mMessage mMachine (for pSP/GIRK1) or T3 mMessage mMachine (for pBScMXT/GIRK2) transcription kit (Ambion, U.S.A.).
The human MOR and rat RGS4 were subcloned into pGEMHE by Ulens et al. (2000b) as described. For in vitro transcription, each clone was linearized with NheI. Next the capped cRNAs were synthesized from the linearized plasmids using the large-scale T7 mMessage mMachine transcription kit (Ambion, U.S.A.).
Construction of mutant human μ-opioid receptors
Asn230 in hMOR was mutated to Leu230 and Thr230 using the Quickchange site-directed mutagenesis kit (Stratagene, U.S.A.). The primers we used were 5′-CCAACTGGTACTGGGAGMYCCTGCTGAAGATCTGTG-3′ and 5′-CACAGATCTTCAGCAGGRKCTCCCAGTACCAGGTTGG-3′, with M=A or C, Y=C or T, K=T or G, R=G or A, (codon and complementary codon are underlined). Cycling parameters were set according to the manufacturer's guidelines. A 299 basepair fragment containing the desired mutation was isolated by a double restriction digest with MscI and BglII. The mutant cDNA was then loaded on an agarose gel, the fragment of interest was cut out, genecleaned (QIAQUICK, QIAGEN, U.S.A.) and ligated with T4 DNA ligase (Promega, U.S.A.) into the corresponding sites of the WT hMOR/pGEMHE. The same mutant fragment was subcloned into pGEM7Zf(+) (Promega, U.S.A.) for DNA sequencing (Eurogentec, Belgium). For in vitro transcription, each mutant, hMORN230L/pGEMHE and hMORN230T/pGEMHE was linearized with NheI. Next the capped cRNAs were synthesized from the linearized plasmids using the large-scale T7 mMessage mMachine transcription kit (Ambion, U.S.A.).
Experimental model
Xenopus laevis oocytes were prepared for injection as described (Liman et al., 1992). Oocytes were co-injected with 0.5 ng 50 nl−1 GIRK1, 0.5 ng 50 nl−1 GIRK2 and 10 ng 50 nl−1 RGS4 cRNA, with the addition of 10 ng 50 nl−1 of either hMOR, hMORN230L or hMORN230T cRNA. Injected oocytes were maintained in ND-96 solution (composition in mM: KCl 2, NaCl 96, MgCl2 1, CaCl2 1.8, HEPES 5, pH 7.5) supplemented with 50 μg ml−1 gentamicin sulphate and incubated at 18°C.
Electrophysiological recordings
Whole-cell currents from oocytes were recorded 1 day after injection using the two-microelectrode voltage clamp technique (Geneclamp 500, Axon Instruments, U.S.A.). Resistances of voltage and current electrodes were kept as low as possible (approximately 200 kΩ) and were filled with 3 M KCl. To eliminate the effect of voltage drop across the bath-grounding electrode, the bath potential was actively controlled. All experiments were performed at room temperature (19 – 23°C). At the start and the end of each experiment, oocytes were superfused with low-potassium (ND-96) solution (composition in mM: KCl 2, NaCl 96, MgCl2 1, CaCl2 1.8, HEPES 5; pH 7.5). During application of increasing concentrations of ligands, oocytes were superfused with high-potassium (HK) solution (composition in mM: KCl 96, NaCl 2, MgCl2 1, CaCl2 1.8, HEPES 5; pH 7.5). In HK solution, the K+ equilibrium potential is close to 0 mV and enables K+ inward currents to flow through inwardly rectifying K+ channels at negative holding potentials. A gravity controlled fast perfusion system (Warner Instruments, U.S.A.) was used to ensure rapid solution exchanges. Application of opioid ligands did not evoke an increase of the conductance in uninjected oocytes. In each experiment, oocytes were clamped at a holding potential of −70 mV for approximately 10 min and superfused with ND-96 solution. Next, the superfusion was switched from ND-96 to HK solution, after which increasing concentrations of an opioid receptor agonist were applied. Each concentration was applied for as long as needed to achieve a steady state GIRK1/GIRK2 current activation. Each ligand concentration was washed out by superfusing with HK solution. During this washout period, the channels return to the control current level as a result of the deactivation process that is dramatically accelerated in the presence of RGS4, as described (Ulens et al., 2000a). At the end of each experiment, the oocyte was superfused with HK solution containing 300 μM BaCl2, causing block of the net GIRK1/GIRK2-gated inward current. Finally, the superfusion was switched back to ND-96 solution to confirm complete reversibility. To avoid the receptor expression level affecting the EC50-values of the investigated agonists in the study, the expression system was standardized as previously described (Ulens et al., 2000b).
Data analysis
The pCLAMP program was used for data acquisition and data files (Axon Instruments, U.S.A.) were imported in Microsoft Excel. The percentage activated current was calculated using the equation:
 |
and 0% was taken as the control current level. Current percentages were used for the calculation of the EC50-value, using the Hill equation:
where I represents the current percentage, Imax the maximal current percentage, EC50 the concentration of the agonist that evokes the half-maximal response, A the concentration of agonist, and nH the Hill coefficient. Averaged data are indicated as mean±s.e.mean and were calculated using n experiments, where n indicates the number of oocytes tested. Statistical analysis of differences between groups was carried out with Student's t-test and a probability of 0.05 was taken as the level of statistical significance.
Compounds
[D-Ala2,N-MePhe4,Gly5-ol]-enkephalin (DAMGO: Sigma, U.S.A.), fentanyl HCl (kindly provided by NIDA, U.S.A.), morphine HCl (Federa, Belgium), morphine-6-glucuronide (M6G) (Sigma, U.S.A.) and morphine-3-glucuronide (M3G) (Sigma, U.S.A.) were dissolved in high potassium (HK) solution, stored at 4°C, and diluted appropriately for the experiments.
Results
To investigate the role of interactions of opioid agonists with the hydrophilic residue Asn230 in hMOR, we mutated Asn230 to threonine (hMORN230T) and to leucine (hMORN230L). Using the functional coupling of the opioid receptors to the heteromultimeric GIRK1/GIRK2 channels in Xenopus oocytes, we examined the effects of various opioid agonists on the wild type human μ-opioid receptor and on the mutant human receptors, hMORN230T and hMORN230L. GIRK1/GIRK2 channels were coexpressed with RGS4, mimicking the native neuronal G-protein-mediated pathway of K+-channel activation, together with either the wild type μ-opioid receptor hMORwt or one of the mutant receptors, hMORN230T or hMORN230L. We used the two-microelectrode voltage clamp technique to measure the opioid receptor-activated GIRK1/GIRK2 channel response as the increase of the inward K+-current at −70 mV, evoked by the application of increasing concentrations of opioid ligands. In our study we examined the potency of DAMGO, fentanyl, morphine, M6G and M3G on hMORwt, and on the mutant receptors, hMORN230T and hMORN230L.
Figure 2 shows representative current traces of agonist-gated currents evoked from oocytes expressing either hMOR wild type (Figure 2A), hMORN230T (Figure 2B) or hMORN230L (Figure 2C) by fentanyl. Analogously, Figures 3 and 4 show representative traces of currents evoked by fentanyl and M6G, respectively. Current traces evoked by DAMGO and M3G are not shown.
Figure 2.
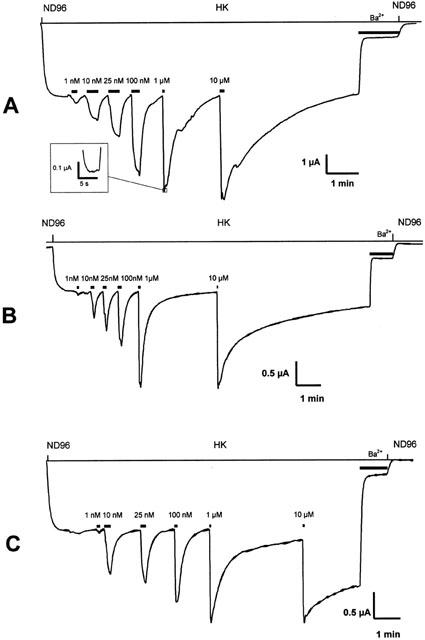
Representative current traces evoked from Xenopus laevis oocytes coexpressing GIRK1/GIRK2 channels and RGS4 with hMOR (A), hMORN230T (B) or hMORN230L (C). Agonist gated currents were evoked at −70 mV by application of increasing concentrations of fentanyl. The inset in A shows a trace on an expanded time scale to illustrate that steady state was reached.
Figure 3.
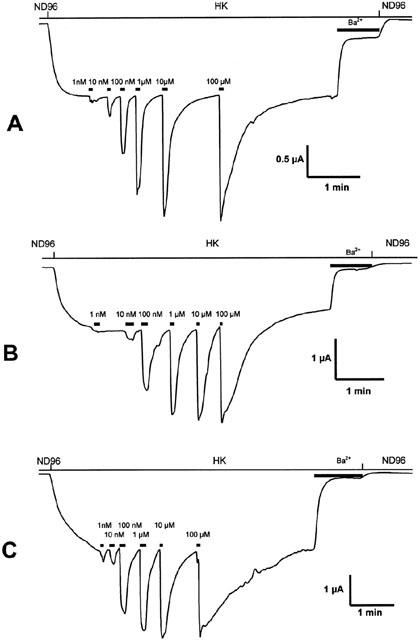
Representative current traces evoked from Xenopus laevis oocytes coexpressing GIRK1/GIRK2 channels and RGS4 with hMOR (A), hMORN230T (B) or hMORN230L (C). Agonist gated currents were evoked at −70 mV by application of increasing concentrations of morphine.
Figure 4.
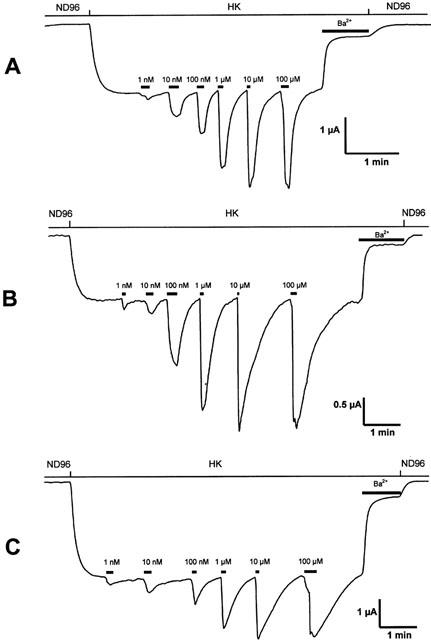
Representative current traces evoked from Xenopus laevis oocytes coexpressing GIRK1/GIRK2 channels and RGS4 with hMOR (A), hMORN230T (B) or hMORN230L (C). Agonist gated currents were evoked at −70 mV by application of increasing concentrations of morphine-6-glucuronide.
Concentration-response relationships (Figure 5A – E) are shown for DAMGO, fentanyl, morphine, M6G and M3G, respectively.
Figure 5.
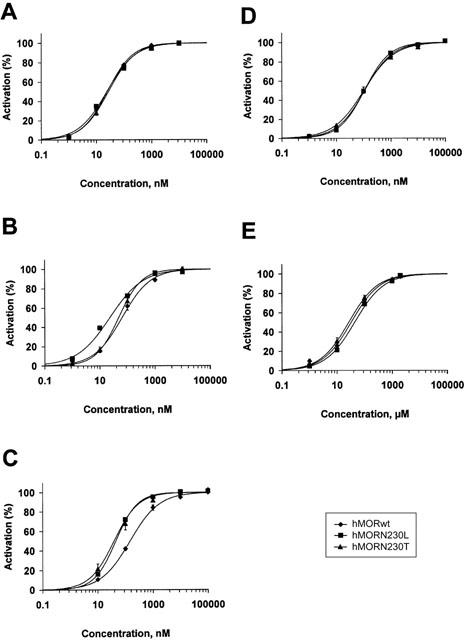
Concentration-response curves for GIRK1/GIRK2 channel activation by increasing concentrations of DAMGO (A), fentanyl (B), morphine (C), M3G (D) and M6G (E). Agonist-gated currents were evoked from Xenopus laevis oocytes coexpressing GIRK1/GIRK2 and RGS4 with hMORwt, hMORN230L or hMORN230T. The agonist-gated increase of GIRK current at each concentration was normalized to a maximal response of 100%. Each point represents the average current activation evoked from 5 to 8 oocytes (mean±s.e.mean).
Table 1 summarizes EC50-values calculated for these agonists via the receptors and also shows the structure of the amino acid at position 230. Averaged data are indicated as mean±s.e.mean and were calculated using n experiments (n=5 – 8).
Table 1.
EC50 values calculated for GIRK1/GIRK2 channel activation via hMORwt, hMORN230L or hMORN230T, coexpressed with RGS4 in Xenopus oocytes

DAMGO, a μ-opioid-selective ligand showed no difference in potency between the wild type receptor (EC50: 25.6±5.0 nM) and the mutant opioid receptors (25.8±4.7 nM for hMORN230T and 24.7±2.1 nM for hMORN230L).
Fentanyl was approximately two times less potent than DAMGO via the wild type receptor. The EC50-value of fentanyl via hMORN230T (47.9±3.1 nM) was significantly lower as compared to hMORwt (64.9±7.8 nM). A 3 fold increase in potency was seen when fentanyl was applied to the hMORN230L-receptor (EC50: 20.2±1.9 nM).
Morphine also showed a higher affinity for the mutated receptors than for the wild type receptor (EC50: 148.6±16.3 nM), with no statistical difference between hMORN230T (EC50: 41.2±10.7 nM) and hMORN230L (EC50: 46.3±1.3 nM).
The EC50-value of morphine-6-glucuronide on the wild type receptor (107.4±10.8 nM) was only slightly, albeit significantly, lower as compared to the EC50-value of morphine on the wild type receptor (148.6±16.3 nM). M6G also displayed the same potency via hMORN230T (EC50: 97.1±12.9 nM) as via hMORN230L (EC50: 106.3±6.3 nM), and this potency was not higher as compared to the wild type receptor.
Table 1 also shows that morphine-3-glucuronide is more than 200 times less potent as compared to morphine and morphine-6-glucuronide. Statistical analysis revealed that there was no difference in potency of morphine-3-glucuronide on hMORwt (EC50: 34.4±8.1 μM), hMORN230T (EC50: 23.6±6.3 μM) and hMORN230T (EC50: 43.1±15.9 μM).
Discussion
In the present study we demonstrate the importance of the hydrophilic amino acid Asn230 in hMOR in the potency of various agonists on the cloned hMORwt, hMORN230T and hMORN230L receptors co-expressed with the GIRK1/GIRK2 channels and RGS4 in Xenopus oocytes. Therefore, Asn230 was mutated to Thr (hMORN230T) and to Leu (hMORN230L), because these residues are neutral and hydrophobic, respectively. The scaled hydrophobicity values of these amino acids are 0.236 for Asn, 0.450 for Thr and 0.943 for Leu (Black & Mould, 1991). These particular mutants were preferred because a Thr (position 211) and Leu (position 224) are present at the corresponding positions in the δ- and κ-opioid receptor, respectively (Lomize et al., 1999). Position Asn230 is situated at the end of the second extracellular loop, near the fifth transmembrane helix (Figure 1) (Xu et al., 2000).
The ligand-binding cavity is partially covered by the extracellular loops (EL). These loops, which have been reported to participate in the binding of opioid ligands, create an almost continuous surface made originating from the β-hairpin formed by EL-2 and the smaller EL-1 and EL-3. These loops appear to act as filters for the ligands, regulating the ability to interact within the core of the receptor (Law et al., 1999). EL-2 also appears to be an absolute requirement for nociceptin-induced receptor activation (Mollereau et al., 1999).
The significance of polar interactions involving Asp147 and His297 residues in the bottom of the binding pocket has been shown by site-directed mutagenesis by different groups (Befort et al., 1996; Li et al., 1999; Spivak et al., 1997). These residues serve as ‘attachment points' for the quaternary nitrogen and hydroxyl group, respectively, of the tyramine moiety common to most opioid ligands (Lomize et al., 1999).
DAMGO is a μ-opioid selective peptidic ligand. Onogi et al. (1995) showed the importance of the first extracellular loop (EL-1) for the binding of DAMGO, by the use of μ/δ chimeras. Chimeras that include the first extracellular loop from the δ-opioid receptor lost the high-affinity binding for DAMGO (Wang et al., 1995). A later mutational study suggested that Lys108 in EL-1 of the δ-opioid receptor prevents DAMGO from binding to the δ-opioid receptor (Fukuda et al., 1995; Minami et al., 1996). Studies with μ/κ chimeric receptors revealed the importance of the region around the third extracellular loop to distinguish between μ- and κ-opioid receptors for the high affinity binding of the μ-opioid receptor selective peptide ligand, DAMGO (Minami et al., 1995). Seki et al. (1998) demonstrated that Lys303, Val316, Trp318 and His319 around the third extracellular loop of the μ-opioid receptor act as determinants for the discrimination between μ- and κ-opioid receptors by DAMGO. Results from point mutations relatively deep within the binding cavity (e.g. Tyr326), suggest that DAMGO not only interacts with the extracellular loops of the receptor, but likely extends within the binding cavity itself that is formed by the seven TM-helices (Mansour et al., 1997).
Our results show that mutation of Asn230 in the second extracellular loop of the μ-opioid receptor did not affect the EC50-value, neither on hMORN230T mutant nor on hMORN230L. Obviously, this residue is not critical in the affinity of DAMGO towards the μ-opioid receptor. This result is in line with a chimeric opiate receptor study which showed that replacing the second extracellular loop of the μ-opioid receptor by the corresponding loop of the κ-opioid receptor did not affect the affinity of DAMGO (Wang et al., 1994).
Nevertheless, these results can serve as a control for the functionality of the mutant receptors. It is therefore very likely that the mutations did not markedly alter the secondary and tertiary structures of the resulting mutants.
Fentanyl, a 4-anilidopiperidine, is a highly selective μ-opioid agonist. Substitution of Asn230 to Thr or Leu significantly decreased the EC50-value of fentanyl. A gradual increase in affinity was seen as the amino acid in position 230 became more lipophilic, suggesting the importance of hydrophobic interactions between the receptor and fentanyl. Pogozheva et al. (1998) suggested in their receptor structure function study that the COOCH3 group in position 4 of the piperidine ring of lofentanyl forms a H-bond with Lys233 and Asn230. In the case of fentanyl, its piperidine ring could possibly benefit from hydrophobic interactions with the mutant receptors hMORN230T and even more with the hMORN230L receptor, resulting in an increased affinity.
Much to our surprise, the same tendency was seen with morphine, a rather small alkaloid agonist. The potency of this primary μ-agonist was increased 3.5 fold by mutating the wild type hMOR receptor to hMORN230T or hMORN230L, although a decreased potency was expected. The EC50-values for both mutated receptors did not differ statistically. These findings are not in line with the models of Lomize et al. (1999) and Pogozheva et al. (1998), who emphasized the importance of the formation of a hydrogen bond between Asn230 and the 6-α-OH group of morphine. The recent alignment of morphine by McFadyen et al. (2000) is very similar to previous models, but places the 6-hydroxyl group of morphine outside the binding pocket and postulates that the hexene ring of morphine is surrounded with Lys233, Tyr148, Asn230 and Trp318. Lipophilic methylgroups in the mutated receptors could interact via hydrophobic interactions with the hexene ring of morphine and enhance the potency. The finding that Lys233 is involved in covalent bond formation with the fumarate group of β-funaltrexamine provides additional information to determine the site of interaction with Asn230 (Chen et al., 1996).
Based on computer modelling manipulations, yet another model was hypothesized in which Tyr326, in TM-VII, would lie adjacent to the aliphatic alcohol of morphine (Mansour et al., 1997). Furthermore it was suggested in this model that the change of Tyr326 to Phe, thereby removing the possible hydrogen bonding site, decreased the affinity for morphine, as well as for DAMGO and fentanyl. According to the models of Pogozheva et al. (1998) and Subramanian et al. (2000) this residue lies in the vicinity of residues that interact with the piperidine ring of the tyramine moiety, like the conserved Asp147.
We suggest that our mutated receptors form hydrophobic interactions with the hexene ring of morphine, which is oriented approximately perpendicular to the tyramine and directed toward the β-hairpin formed by EL-2 (Lomize et al., 1999), and consequently can explain the increase in potency for morphine. In this view, it is highly unlikely that Asn230 forms a hydrogen bond with the 6α-OH group of morphine and that this residue serves as a determinant for the preferred binding of morphinans to the μ receptor as proposed by Pogozheva et al. (1998).
Morphine-6-glucuronide activates GIRK1/GIRK2 channels through the wild type μ-opioid receptor with a significantly higher potency than morphine. Analogous results for both ligands were obtained by Ulens et al. (2001) who also showed that the potency profile changed from μ>δ=κ for morphine to μ>κ>δ for morphine-6-glucuronide. According to McFadyen et al. (2000) the 6-hydroxyl group lies outside the binding pocket, so the glucuronide in morphine-6-glucuronide can also lie outside the binding pocket. The enhanced potency could be due to favourable interactions between the hydrophilic glucuronide moiety and residues in the EL-2, EL-3 or in the aminoterminus of the receptor, which was shown to contribute in the affinity for fentanyl, methadone, morphine and other ligands (Chaturvedi et al., 2000). We observed no difference in EC50-value for morphine-6-glucuronide between the mutated receptors and the wild type μ-opioid receptor. Taken together, glucuronidation of the 6-hydroxyl group enhances the potency via the wild type hMOR receptor but abolishes the increased potency of morphine via the mutated hMORN230T and hMORN230L receptor. The precise reasons for these disparate findings are not clear at present and further investigations are needed to elucidate the responsible residues and interactions.
Our electrophysiological EC50-values for morphine-3-glucuronide, another metabolite of morphine, are situated in the micromolar range, resulting in a 200 fold reduction in potency as compared with morphine on the wild type receptor. This rather big decrease in potency can be explained using the classical alignment of morphine that places the quaternary nitrogen of morphine close enough to Asp147 and the phenolic hydroxyl (3-hydroxyl) group close to His297, rather deep in the binding pocket (McFadyen et al., 2000; Lomize et al., 1999). Substitution of the hydroxyl group at C3 of morphine with a glucuronide moiety disturbs the interaction with these key interaction points. Recently, a revised pharmacophore model was presented by McFadyen et al. (2000) where the only required moieties are a quaternary amine nitrogen and a relatively planar, electron-rich region, commonly a phenyl ring. A para-hydroxyl substituent contributes to high-affinity binding but is not critical. The design of 3-deoxyclocinnamox, a high affinity μ-opioid antagonist lacking a phenolic hydroxyl group, confirmed this proposition (Derrick et al., 2000). Morphine-3-glucuronide showed no difference in the affinity for μ-, κ- and δ-receptors (Ulens et al., 2001). Our mutated receptors also displayed the same potency for morphine-3-glucuronide.
In conclusion, the present study sheds new light on the role of Asn230 in the potency of various agonists. Mutation of this residue to the more hydrophobic threonine and leucine resulted in an increased potency for fentanyl and morphine, while the potency of DAMGO, M6G and M3G remained unaffected. Possible hydrophobic interactions between this residue and the piperidine and hexene ring of fentanyl and morphine, respectively, can explain these findings.
Acknowledgments
We are grateful to Chris Ulens for subcloning hMOR and RGS4 and fruitful comments. GIRK1 cDNA was a gift from Kazutaka Ikeda (The Institute of Physical and Chemical Research, RIKEN, Wako, Japan). GIRK2 and RGS4 were kindly donated by Henry Lester (California Institute of Technology, Pasadena, U.S.A.). The human μ-opioid receptor was a gift from Lei Yu (University of Cincinnati, Cincinnati, U.S.A.). Fentanyl was a gift from the National Institute on Drug Abuse (Bethesda, MD, U.S.A.) and M6G and M3G was a gift from Anna Ratka (Idaho State University, Idaho, U.S.A.).
Abbreviations
- DAMGO
[D-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin
- EL
extracellular loop
- GIRK channels
G-protein-coupled inwardly rectifying K+ channels
- HK
high potassium solution
- hMOR
human μ-opioid receptor
- IL
intracellular loop
- M3G
morphine-3-glucuronide
- M6G
morphine-6-glucuronide
- ND-96
low potassium solution
- RGS
regulators of G-protein signalling
- TM
transmembrane domain
References
- BEFORT K., TABBARA L., BAUSCH S., CHAVKIN C., EVANS C., KIEFFER B. The conserved asparate residue in the third putative transmembrane domain of the delta-opioid receptor is not the anionic counterpart for cationic opiate binding but is a constituent of the receptor binding site. Mol. Pharmacol. 1996;49:216–223. [PubMed] [Google Scholar]
- BLACK S.D., MOULD D.R. Development of hydrophobicity parameters to analyze proteins which bear post- or cotranslational modifications. Anal. Biochem. 1991;193:72–82. doi: 10.1016/0003-2697(91)90045-u. [DOI] [PubMed] [Google Scholar]
- CHATURVEDI K., SHAHRESTANIFAR M., HOWELLS R.D. mu Opioid receptor: role for the amino terminus as a determinant of ligand binding affinity. Brain Res. Mol. Brain Res. 2000;76:64–72. doi: 10.1016/s0169-328x(99)00332-0. [DOI] [PubMed] [Google Scholar]
- CHEN C., YIN J., RIEL J.K., DESJARLAIS R.L., RAVEGLIA L.F., ZHU J., LIU-CHEN L.Y. Determination of the amino acid residue involved in [3H]beta-funaltrexamine covalent binding in the cloned rat mu-opioid receptor. J. Biol. Chem. 1996;271:21422–21429. doi: 10.1074/jbc.271.35.21422. [DOI] [PubMed] [Google Scholar]
- DERRICK I., NEILAN C.L., ANDES J., HUSBANDS S.M., WOODS J.H., TRAYNOR J.R., LEWIS J.W. 3-Deoxyclocinnamox: the first high-affinity nonpeptide mu-opioid antagonist lacking a phenolic hydroxyl group. J. Med. Chem. 2000;43:3348–3350. doi: 10.1021/jm0009641. [DOI] [PubMed] [Google Scholar]
- DE VRIES L., ZHENG B., FISCHER T., ELENKO E., FARQUHAR M.G. The regulator of G protein signaling family. Annu. Rev. Pharmacol. Toxicol. 2000;40:235–271. doi: 10.1146/annurev.pharmtox.40.1.235. [DOI] [PubMed] [Google Scholar]
- DOUPNIK C.A., DAVIDSON N., LESTER H.A., KOFUJI P. RGS proteins reconstitute the rapid gating kinetics of gbetagamma-activated inwardly rectifying K+ channels. Proc. Natl. Acad. Sci. U.S.A. 1997;94:10461–10466. doi: 10.1073/pnas.94.19.10461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EVANS C.J., KEITH D.E., JR, MORRISON H., MAGENDZO K., EDWARDS R.H. Cloning of a delta opioid receptor by functional expression [see comments] Science. 1992;258:1952–1955. doi: 10.1126/science.1335167. [DOI] [PubMed] [Google Scholar]
- FILIZOLA M., LAAKKONEN L., LOEW G.H. 3D modeling, ligand binding and activation studies of the cloned mouse delta, mu; and kappa opioid receptors. Protein Eng. 1999;12:927–942. doi: 10.1093/protein/12.11.927. [DOI] [PubMed] [Google Scholar]
- FUKUDA K., TERASAKO K., KATO S., MORI K. Identification of the amino acid residues involved in selective agonist binding in the first extracellular loop of the delta- and mu-opioid receptors. FEBS Lett. 1995;373:177–181. doi: 10.1016/0014-5793(95)01034-c. [DOI] [PubMed] [Google Scholar]
- IKEDA K., KOBAYASHI T., ICHIKAWA T., USUI H., KUMANISHI T. Functional couplings of the delta- and the kappa-opioid receptors with the G-protein-activated K+ channel. Biochem. Biophys. Res. Commun. 1995;208:302–308. doi: 10.1006/bbrc.1995.1338. [DOI] [PubMed] [Google Scholar]
- KIEFFER B.L. Opioids: first lessons from knockout mice. Trends Pharmacol. Sci. 1999;20:19–26. doi: 10.1016/s0165-6147(98)01279-6. [DOI] [PubMed] [Google Scholar]
- KIEFFER B.L., BEFORT K., GAVERIAUX-RUFF C., HIRTH C.G. The delta-opioid receptor: isolation of a cDNA by expression cloning and pharmacological characterization [published erratum appears in Proc. Natl. Acad. Sci. U.S.A. 1994 Feb 1;91(3):1193] Proc. Natl. Acad. Sci. U.S.A. 1992;89:12048–12052. doi: 10.1073/pnas.89.24.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KNAPP R.J., MALATYNSKA E., COLLINS N., FANG L., WANG J.Y., HRUBY V.J., ROESKE W.R., YAMAMURA H.I. Molecular biology and pharmacology of cloned opioid receptors. FASEB J. 1995;9:516–525. doi: 10.1096/fasebj.9.7.7737460. [DOI] [PubMed] [Google Scholar]
- KOBAYASHI T., IKEDA K., ICHIKAWA T., ABE S., TOGASHI S., KUMANISHI T. Molecular cloning of a mouse G-protein-activated K+ channel (mGIRK1) and distinct distributions of three GIRK (GIRK1, 2 and 3) mRNAs in mouse brain. Biochem. Biophys. Res. Commun. 1995;208:1166–1173. doi: 10.1006/bbrc.1995.1456. [DOI] [PubMed] [Google Scholar]
- KOFUJI P., DAVIDSON N., LESTER H.A. Evidence that neuronal G-protein-gated inwardly rectifying K+ channels are activated by G beta gamma subunits and function as heteromultimers. Proc. Natl. Acad. Sci. U.S.A. 1995;92:6542–6546. doi: 10.1073/pnas.92.14.6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRIEG P.A., MELTON D.A. Functional messenger RNAs are produced by SP6 in vitro transcription of cloned cDNAs. Nucleic Acids Res. 1984;12:7057–7070. doi: 10.1093/nar/12.18.7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAW P.Y., WONG Y.H., LOH H.H. Mutational analysis of the structure and function of opioid receptors. Biopolymers. 1999;51:440–455. doi: 10.1002/(SICI)1097-0282(1999)51:6<440::AID-BIP6>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- LI J.G., CHEN C., YIN J., RICE K., ZHANG Y., MATECKA D., DE RIEL J.K., DESJARLAIS R.L., LIU-CHEN L.Y. ASP147 in the third transmembrane helix of the rat mu opioid receptor forms ion-pairing with morphine and naltrexone. Life Sci. 1999;65:175–185. doi: 10.1016/s0024-3205(99)00234-9. [DOI] [PubMed] [Google Scholar]
- LI X., VARGA E.V., STROPOVA D., ZALEWSKA T., MALATYNSKA E., KNAPP R.J., ROESKE W.R., YAMAMURA H.I. delta-Opioid receptor: the third extracellular loop determines naltrindole selectivity. Eur. J. Pharmacol. 1996;300:R1–R2. doi: 10.1016/0014-2999(96)00098-2. [DOI] [PubMed] [Google Scholar]
- LIMAN E.R., TYTGAT J., HESS P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron. 1992;9:861–871. doi: 10.1016/0896-6273(92)90239-a. [DOI] [PubMed] [Google Scholar]
- LOMIZE A.L., POGOZHEVA I.D., MOSBERG H.I. Structural organization of G-protein-coupled receptors. J. Comput. Aided Mol. Des. 1999;13:325–353. doi: 10.1023/a:1008050821744. [DOI] [PubMed] [Google Scholar]
- MANSOUR A., TAYLOR L.P., FINE J.L., THOMPSON R.C., HOVERSTEN M.T., MOSBERG H.I., WATSON S.J., AKIL H. Key residues defining the mu-opioid receptor binding pocket: a site-directed mutagenesis study. J. Neurochem. 1997;68:344–353. doi: 10.1046/j.1471-4159.1997.68010344.x. [DOI] [PubMed] [Google Scholar]
- MCFADYEN I.J., HOUSHYAR H., LIU-CHEN L.Y., WOODS J.H., TRAYNOR J.R. The steroid 17alpha-Acetoxy-6-dimethylaminomethyl -21 -fluoro -3 -ethoxy -pregna -3,5-dien-20-one (SC17599) is a selective mu-opioid agonist: implications for the mu-opioid pharmacophore. Mol. Pharmacol. 2000;58:669–676. doi: 10.1124/mol.58.4.669. [DOI] [PubMed] [Google Scholar]
- MENG F., HOVERSTEN M.T., THOMPSON R.C., TAYLOR L., WATSON S.J., AKIL H. A chimeric study of the molecular basis of affinity and selectivity of the kappa and the delta opioid receptors. Potential role of extracellular domains. J. Biol. Chem. 1995;270:12730–12736. doi: 10.1074/jbc.270.21.12730. [DOI] [PubMed] [Google Scholar]
- MINAMI M., NAKAGAWA T., SEKI T., ONOGI T., AOKI Y., KATAO Y., KATSUMATA S., SATOH M. A single residue, Lys108, of the delta-opioid receptor prevents the mu-opioid selective ligand [D-Ala2,N-MePhe4,Gly-ol5]enkephalin from binding to the delta-opioid receptor. Mol. Pharmacol. 1996;50:1413–1422. [PubMed] [Google Scholar]
- MINAMI M., ONOGI T., NAKAGAWA T., KATAO Y., AOKI Y., KATSUMATA S., SATOH M. DAMGO, a mu-opioid receptor selective ligand, distinguishes between mu- and kappa-opioid receptors at a different region from that for the distinction between mu- and delta-opioid receptors. FEBS Lett. 1995;364:23–27. doi: 10.1016/0014-5793(95)00340-f. [DOI] [PubMed] [Google Scholar]
- MOLLEREAU C., MOULEDOUS L., LAPALU S., CAMBOIS G., MOISAND C., BUTOUR J.L., MEUNIER J.C. Distinct mechanisms for activation of the opioid receptor-like 1 and kappa-opioid receptors by nociceptin and dynorphin A. Mol. Pharmacol. 1999;55:324–331. doi: 10.1124/mol.55.2.324. [DOI] [PubMed] [Google Scholar]
- ONOGI T., MINAMI M., KATAO Y., NAKAGAWA T., OAKI Y., TOYA T., KATSUMATA S., SATOH M. DAMGO, a mu-opioid receptor selective agonist, distinguishes between mu- and delta-opioid receptors around their first extracellular loops. FEBS Lett. 1995;357:93–97. doi: 10.1016/0014-5793(94)01341-w. [DOI] [PubMed] [Google Scholar]
- POGOZHEVA I.D., LOMIZE A.L., MOSBERG H.I. Opioid receptor three-dimensional structures from distance geometry calculations with hydrogen bonding constraints. Biophys. J. 1998;75:612–634. doi: 10.1016/S0006-3495(98)77552-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEKI T., MINAMI M., NAKAGAWA T., IENAGA Y., MORISADA A., SATOH M. DAMGO recognizes four residues in the third extracellular loop to discriminate between mu- and kappa-opioid receptors. Eur. J. Pharmacol. 1998;350:301–310. doi: 10.1016/s0014-2999(98)00240-4. [DOI] [PubMed] [Google Scholar]
- SPIVAK C.E., BEGLAN C.L., SEIDLECK B.K., HIRSHBEIN L.D., BLASCHAK C.J., UHL G.R., SURRATT C.K. Naloxone activation of mu-opioid receptors mutated at a histidine residue lining the opioid binding cavity. Mol. Pharmacol. 1997;52:983–992. doi: 10.1124/mol.52.6.983. [DOI] [PubMed] [Google Scholar]
- SUBRAMANIAN G., PATERLINI M.G., PORTOGHESE P.S., FERGUSON D.M. Molecular docking reveals a novel binding site model for fentanyl at the mu-opioid receptor. J. Med. Chem. 2000;43:381–391. doi: 10.1021/jm9903702. [DOI] [PubMed] [Google Scholar]
- ULENS C., BAKER L., RATKA A., WAUMANS D., TYTGAT J.Morphine-6-glucuronide and morphine-3-glucuronide, opioid receptor agonists with different potency Biochem. Pharmacol. 2001. in press [DOI] [PubMed]
- ULENS C., DAENENS P., TYTGAT J. Changes in GIRK1/GIRK2 deactivation kinetics and basal activity in the presence and absence of RGS4. Life Sci. 2000a;67:2305–2317. doi: 10.1016/s0024-3205(00)00820-1. [DOI] [PubMed] [Google Scholar]
- ULENS C., VAN BOVEN M., DAENENS P., TYTGAT J. Interaction of p-fluorofentanyl on cloned human opioid receptors and exploration of the role of Trp-318 and His-319 in mu-opioid receptor selectivity. J. Pharmacol. Exp. Ther. 2000b;294:1024–1033. [PubMed] [Google Scholar]
- WANG J.B., IMAI Y., EPPLER C.M., GREGOR P., SPIVAK C.E., UHL G.R. mu opiate receptor: cDNA cloning and expression. Proc. Natl. Acad. Sci. U.S.A. 1993;90:10230–10234. doi: 10.1073/pnas.90.21.10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG J.B., JOHNSON P.S., WU J.M., WANG W.F., UHL G.R. Human kappa opiate receptor second extracellular loop elevates dynorphin's affinity for human mu/kappa chimeras. J. Biol. Chem. 1994;269:25966–25969. [PubMed] [Google Scholar]
- WANG W.W., SHAHRESTANIFAR M., JIN J., HOWELLS R.D. Studies on mu and delta opioid receptor selectivity utilizing chimeric and site-mutagenized receptors. Proc. Natl. Acad. Sci. U.S.A. 1995;92:12436–12440. doi: 10.1073/pnas.92.26.12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XU W., CHEN C., HUANG P., LI J., DE RIEL J.K., JAVITCH J.A., LIU-CHEN L.Y. The conserved cysteine 7.38 residue is differentially accessible in the binding-site crevices of the mu, delta, and kappa opioid receptors. Biochemistry. 2000;39:13904–13915. doi: 10.1021/bi001099p. [DOI] [PubMed] [Google Scholar]
- XU W., OZDENER F., LI J.G., CHEN C., DE RIEL J.K., WEINSTEIN H., LIU-CHEN L.Y. Functional role of the spatial proximity of Asp114(2.50) in TMH2 and Asn332(7.49) in TMH 7 of the mu opioid receptor. FEBS Lett. 1999;447:318–324. doi: 10.1016/s0014-5793(99)00316-6. [DOI] [PubMed] [Google Scholar]