

Abstract
Human mast cell tryptase appears to display considerable variation in activating proteinase-activated receptor 2 (PAR2). We found tryptase to be an inefficient activator of wild-type rat-PAR2 (wt-rPAR2) and therefore decided to explore the factors that may influence tryptase activation of PAR2.
Using a 20 mer peptide (P20) corresponding to the cleavage/activation sequence of wt-rPAR2, tryptase was as efficient as trypsin in releasing the receptor-activating sequence (SLIGRL…). However, in the presence of either human-PAR2 or wt-r PAR2 expressing cells, tryptase could only activate PAR2 by releasing SLIGRL from the P20 peptide, suggesting that PAR2 expressed on the cells was protected from tryptase activation.
Three approaches were employed to test the hypothesis that PAR2 receptor glycosylation restricts tryptase activation. (a) pretreatment of wt-rPAR2 expressing cells or human embryonic kidney cells (HEK293) with vibrio cholerae neuraminidase to remove oligosaccharide sialic acid, unmasked tryptase-mediated PAR2 activation. (b) Inhibiting receptor glycosylation in HEK293 cells with tunicamycin enabled tryptase-mediated PAR2 activation. (c) Wt-rPAR2 devoid of the N-terminal glycosylation sequon (PAR2T25−), but not rPAR2 devoid of the glycosylation sequon located on extracellular loop-2 (PAR2T224A), was selectively and substantially (>30 fold) more sensitive to tryptase compared with the wt-rPAR2.
Immunocytochemistry using antisera that specifically recognized the N-terminal precleavage sequence of PAR2 demonstrated that tryptase released the precleavage domain from PAR2T25− but not from wt-rPAR2.
Heparin : tryptase molar ratios of greater than 2 : 1 abrogated tryptase activation of PAR2T25−.
Our results indicate that glycosylation of PAR2 and heparin-inhibition of PAR2 activation by tryptase could provide novel mechanisms for regulating receptor activation by tryptase and possibly other proteases.
Keywords: Tryptase, PAR2, glycosylation
Introduction
Mast cell tryptase is the major secretory product of the human mast cell (Schwartz et al., 1987). Within the mast cell granules, tryptase is found associated with heparin proteoglycan that serves to stabilize the enzyme and regulate its biological function upon release (Schwartz & Bradford, 1986). This 134 kDa tetrameric serine protease adopts a square flat ring structure composed of four monomers, whose active sites face a central pore (Pereira et al., 1998), and consequently restrict access to potential macromolecular substrates and proteinase inhibitors. Nevertheless, tryptase has profound actions on cells, such as causing cytokine release from epithelial and endothelial cells (Cairns & Walls, 1996; Compton et al., 1998), proliferation of fibroblasts, epithelial and smooth muscle cells (Brown et al., 1995; Cairns & Walls, 1996; Ruoss et al., 1991), and fibroblast collagen synthesis (Cairns & Walls, 1997). Furthermore, injection of tryptase into the peritoneum of mice induces considerable inflammatory cell recruitment (He et al., 1997), and injection into the skin of guinea-pigs induces oedema (He & Walls, 1997). These findings have prompted suggestions that tryptase maybe implicated in inflammatory disease (Compton & Walls, 1999). However, the mechanism whereby tryptase activates cells remains unclear. Although it has recently been suggested that tryptase can affect target cells via activation of protease-activated receptor 2 (PAR2) (Corvera et al., 1997; Mirza et al., 1997; Molino et al., 1997), others using recombinant tryptase have failed to detect PAR2 activation by tryptase (Huang et al., 2001).
PAR2 is a member of the novel family of G-protein coupled receptors activated by proteolytic cleavage (Dery et al., 1998). Activation occurs by proteolysis of the amino-terminal exodomain resulting in the exposure of a tethered ligand, which presumably binds to, and activates the receptor. There are currently three active members of the PAR family, PAR 1, 2 and 4 (Bohm et al., 1996; Ishihara et al., 1997; Vu et al., 1991; Kahn et al., 1998; Xu et al., 1998). Family member, PAR3, has been found to be a co-factor for PAR4 and as such may not be considered as an active receptor in its own right (Nakanishi-Matsui et al., 2000). Interestingly, peptides corresponding to the first five to six amino acids of the tethered ligand will activate these receptors independently of proteolysis. Consequently these PAR-activating peptides (PAR-APs) have become useful tools for investigating the biological roles of PARs (Vergnolle, 1999; Vergnolle et al., 1999). For example, the administration of PAR2-APs into animal tissues generates all the hallmarks of an inflammatory response (Vergnolle et al., 1999). Furthermore, inflammatory cytokines upregulate the expression of PAR2 on endothelial cells for up to 96 h (Nystedt et al., 1996).
Tryptase activation of PAR2 has been demonstrated in a number of cell types including cultured human endothelial cells (HUVEC), rat colonic myocytes, keratinocytes and guinea-pig myenteric neurons (Corvera et al., 1997; 1999; Molino et al., 1997; Schechter et al., 1998). However, some reports (Alm et al., 2000; Corvera et al., 1999; Molino et al., 1997; Schechter et al., 1998), but not others (Corvera et al., 1997; Steinhoff et al., 2000), have indicated that tryptase appears to behave as a partial agonist compared to trypsin for activating PAR2, implying that PAR2 activation by tryptase may be influenced by other factors. Interestingly, Huang et al. (2001) demonstrated that recombinant βI-tryptase was unable to activate any of the PARs cloned to date, including PAR2 expressed on HEK293 cells. Furthermore, it has been reported that the ability of tryptase to activate PAR2 on keratinocytes is abolished by the addition of heparin (Schwartz & Bradford, 1986), whilst others have indicated that tryptase in the presence of heparin can stimulate activation of PAR2 (Corvera et al., 1997; Steinhoff et al., 2000). Therefore, we sought to investigate the factors that might regulate the ability of tryptase to activate PAR2. We decided to (1) determine whether tryptase could recognize the cleavage/activation site of wt-rPAR2 represented by a synthetic 20 mer peptide (P20), (2) investigate the factors that influence the efficacy of tryptase in activating endogenous PAR2 present at the cell membrane, and finally (3) determine the role of heparin in regulating the ability of tryptase in activating PAR2.
Methods
Reagents
Heparin agarose, Sephacryl S200, Nα-Benzoyl-DL-Arginine 4-Nitro-anilide (BAPNA), porcine pancreatic type IX trypsin (13 – 20,000 units mg−1), sulphinpyrazone, calcium ionophore (A23187), porcine intestinal mucosa heparin (MW 16 – 17,000 kDa), soya trypsin inhibitor and leupeptin were from Sigma (St Louis, MO, U.S.A.); foetal calf serum (FCS), Dulbecco's modified Eagle's medium, nonenzymatic cell dissociation fluid, penicillin, streptomycin, amphotericin, sodium pyruvate, and phosphate buffered saline (PBS) (without calcium and magnesium) were from Life Technologies, Inc. All oligonucleotides were synthesized by the University Core DNA & Protein Services, University of Calgary, Calgary, AB, Canada. All peptides were synthesized by the Peptide Synthesis Facility, University of Calgary, Calgary, AB, Canada. Stock solutions of peptides in 25 mM HEPES, pH 7.4 were standardized by quantitative amino acid analysis to confirm peptide concentration and purity. Vibrio cholerae neuraminidase (Cat N° 480717) was purchased from Calbiochem; Fraxiparine® (Sanofi, ON, Canada) was a gift from Dr Graham Pino, University of Calgary; Pluronic acid from Molecular Probes, Eugene, OR, U.S.A. and tunicamycin from ICN Biomedicals-Inc, Aurora, Ohio, U.S.A.
Purification of tryptase
Human lung tryptase was purified essentially as described previously and stored in 2 M NaCl, 20 mM MES buffer, pH 6.1 at −80°C (Compton et al., 1998). Human lung was obtained according to procedures approved by the University of Calgary, Faculty of Medicine Ethics Committee. To determine tryptase activity, an aliquot of 90 μl tryptase assay buffer (100 mM Tris base, 1 M glycerol, pH 8.0) containing 1 mM BAPNA was added to 10 μl tryptase sample, incubated for 5 min at room temperature and the reaction monitored at 450 nm on an ELISA plate reader (MR5000; Dynatech). One unit (U) of tryptase activity was defined as the amount of tryptase required to hydrolyse 1 μmol of BAPNA per min at 25°C. The purity of the isolated tryptase was assessed by specific activity and SDS – PAGE on a 12% gel wherein tryptase was identified by Western blot analysis using the tryptase specific monoclonal antibody, AA5 (a kind gift from Dr Andrew Walls, University of Southampton, U.K.) (Walls et al., 1990). The identity of the tryptase was verified further by amino-acid sequence analysis of protein recovered by Western blot transfer (Alberta Peptide Institute, University of Alberta, Edmonton, AB, Canada). Tryptase was used in the absence of heparin unless stated otherwise and concentrations (nM) used in all experiments were calculated on the basis of the molecular weight (134 kDa) of the tryptase tetramer (Schwartz et al., 1981).
Analysis of peptide cleavage by HPLC and N-terminal sequence analysis
To test whether tryptase recognized the cleavage site of wt-rPAR2, tryptase or trypsin were incubated with a peptide (P20) which corresponds to the cleavage/activation site (residues 30 to 45) of the wt-rPAR2 N-terminus (30GPNSKGR↓SLIGRLDT45P-YGGC (↓trypsin cleavage site, -YGGC added for affinity column coupling)). Tryptase (1 – 10 nM) or trypsin (1 – 10 nM) was incubated with P20 peptide (10 μM) in isotonic phosphate buffered saline pH 7.4 (PBS) for 10 min at room temperature and the hydrolysis products were subjected to HPLC analysis. For inhibitor studies, soya trypsin inhibitor (STI, 1 mg ml−1) was incubated with either tryptase or trypsin for 30 min prior to addition to the P20-containing solution. In separate tubes, tryptase was also incubated with leupeptin (50 μg ml−1) for 30 min prior to addition to the P20 solution. Proteolysis products were applied to an HPLC column (Vydac, 5 μm×25 cm, C4 column) and eluted with a gradient of 0.1% trifluoro acetic acid in acetonitrite (0 – 45%) with a flow rate of 1 ml min−1. Peak fractions (E210) eluting from the column were collected and freeze dried. N-terminal sequencing (performed by the Alberta Peptide Institute, University of Alberta, Edmonton, AB, Canada) was utilized in order to identify the peptide fragments so isolated.
Generation of cDNAs encoding wild-type and mutant PAR2
The wt-rPAR2 and wt-hPAR2 cell lines used in this study have been described in detail previously (Compton et al., 2000). Three mutant rPAR2 clones devoid of glycosylation sequons were generated using the QuikChange site directed mutagenesis kit (Stratagene, CA, U.S.A.) according to the manufacturer's instructions. Two clones were deficient in the N-terminus glycosylation sequon (rPAR2T25−: Threonine25 was deleted to eliminate the glycosylation sequon NXS/T (where X=any amino acid except proline) and rPAR2N23A where asparagine23 was substituted by alanine). The third mutant receptor was deficient in the glycosylation sequon on extracellular loop-2 (rPAR2T224A; Threonine224 substituted by alanine). One mutant hPAR2 clone devoid of an N-terminal glycosylation sequon was also generated (hPAR2N30A; asparagine30 substituted for alanine). The mutant PAR2 clones were subsequently sequenced to confirm the engineered mutations using fluorescence based automated cycle sequencing by the University Core DNA & Protein Services, University of Calgary, Calgary, AB, Canada.
Transfection
Semi-confluent Kirsten virus sarcoma transformed rat kidney epithelial cells (KNRK, American Tissue Type Culture Collection, Bethesda, MD, U.S.A.) in 60 mm petri dishes were transfected using the LipofectAMINE® method according to the manufacturer's protocol (Gibco/BRL). Although, a weak endogenous PAR2 signal has been detected by RT – PCR in non-transfected KNRK cells, no immunoreactivity is detected on the cell surface of these cells using a PAR2 specific antibody; nor is a calcium signal generated in non-transfected KNRK cells by high concentrations of the PAR2-AP, SLIGRL-NH2 (Al Ani et al., 1999). Transfected cells were subcloned in geneticin (0.6 mg ml−1) containing medium (DMEM, 5% FCS, 100 μM sodium pyruvate, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin, and 250 ng ml−1 amphotericin B). To obtain permanent mutant PAR2 cell lines, cells expressing high levels of PAR2 were isolated by FACS® using the B5 anti-PAR2 rabbit polyclonal antibody (Al Ani et al., 1999; Kong et al., 1997). For the human PAR1 cell line, cotransfection was performed using the pcDNA3 vector (Invitrogen, CA, U.S.A.) in conjunction with the pBJ1 human PAR1 containing vector (a kind gift from Dr Shaun Coughlin, University of California, San Francisco, U.S.A. (Ishii et al., 1993)), to allow for antibiotic selection.
Cell culture
Permanently expressing PAR2 and PAR1 cell lines were propagated in DMEM containing 5% FCS, 0.6 mg ml−1 geneticin, 100 μM sodium pyruvate, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin, and 250 ng ml−1 amphotericin B. Human Embryonic Kidney cells (HEK293, T-antigen positive, kindly provided by Dr Johnathan Lytton, University of Calgary, Faculty of Medicine, Calgary, AB, Canada) were grown in DMEM containing FCS 10%, sodium pyruvate 100 μM, penicillin 100 U ml−1, streptomycin 100 μg ml−1, and amphotericin B 250 ng ml−1. All cell lines were propagated without the use of trypsin in 95% air, 5% CO2, at 37°C.
Production of antiserum to the precleavage sequence of rPAR2
A polyclonal antiserum (SLAW) was raised in rabbits (exactly as described elsewhere for the B5 anti-PAR2 polyclonal antibody (Kong et al., 1997)) to a peptide that corresponded to sequences (bold type) representing the potentially antigenic epitopes in the pre-cleavage domain of wt-rPAR2 (SLAWLLGGPNSKGR-GGYGGC). These epitopes would be lost from the cell surface upon cleavage/activation of the receptor by proteases. The rabbit polyclonal B5 antiserum targets the cleavage/activation site of wt-rPAR2 (30GPNSKGRSLIGRLDT45P) and can recognize both the cleaved/activated receptors as well as the uncleaved receptor. During attempts to use either of these antisera for Western blot detection of PAR2, we found that both the SLAW and B5 antibodies recognized multiple bands both in non-transfected (or vector-alone transfected) KNRK cells and PAR2-expressing KNRK cells. Thus, because multiple bands were detected by the Western blot procedure, the antisera did not prove of use for a ‘gel-shift analysis' to compare fully glycosylated versus glycosylation-deficient PAR2.
Immunocytochemistry
The SLAW antiserum was employed to demonstrate a loss of the N-terminal pre-cleavage epitope upon proteolytic activation of PAR2. Further, in order to show a relationship between the calcium signal generated by enzyme activation and the loss of immunoreactivity for SLAW, we harvested both responder and non-responder cells from the calcium signalling cell suspension assay following treatment with trypsin (10 nM) or tryptase (300 nM). Cytospins of nontreated and treated cells were prepared using a Shandon cytopsin (Pittsburgh, PA, U.S.A.) and a 3,3′-diamino benzidine (DAB) substrate immunocytochemistry protocol was utilized as described in detail elsewhere (Saifeddine et al., 2001). Using this protocol, both the B5 and SLAW antibodies were able to detect PAR2 in transfected receptor expressing cells, but not in empty vector transfected cells. Further, in PAR2 expressing cells, for both antibodies, immunoreactivity was abolished by preincubation with the corresponding immunizing peptides (see below).
Calcium signalling in suspension
Calcium signalling was measured as described previously (Compton et al., 2000). Harvested cells grown to near confluence in 75 cm2 culture flasks were incubated in 1 ml of DMEM, 10% FCS and 0.25 mM sulphinpyrazone, 22 μM Fluo-3 acetoxymethyl ester (Molecular Probes Inc., Eugene, OR, U.S.A.) for 25 min at room temperature with gentle shaking. Cells were then washed and resuspended in calcium assay buffer (mM: NaCl 150, KCl 3, CaCl2 1.5, glucose 10, HEPES 20, sulphinpyrazone 0.25, pH 7.4). Fluorescence measurements were performed on a Perkin-Elmer fluorescence spectrometer 650-10S, with an excitation wavelength of 480 nm and emission recorded at 530 nm. Cell suspensions (1 ml) in 4 ml cuvettes were stirred with a magnetic flea bar and maintained at 24°C. The signal produced (E530) by the addition of a test agonist was measured as a percentage of the fluorescence peak height yielded by the addition of 2 μM calcium ionophore (A23187).
Cross-desensitization studies
As antagonists for PAR2 are currently unavailable, we employed a receptor cross-desensitization approach. This assay makes use of the principle that repeated exposure of a tissue or cell to a receptor-selective agonist leads to diminution/desensitization of the receptor's response to that agonist (Kawabata et al., 1999).
Calcium signalling in monolayers
The wt-hPAR2 cells (as described elsewhere Compton et al., 2000) were seeded on 12 mm coverslips in fresh medium until 60 – 80% confluent. Cells were washed with PBS before and after incubation with DMEM containing 10% FCS, 0.25 mM sulphinpyrazone, 0.01% Pluronic F127 and Fura2/AM at room temperature for 40 min. Coverslips were mounted in a perfusion chamber (Warner Instruments) on a Zeiss Axiovert 135 microscope. Cells were stimulated with test agents by replacement of the 500 μl calcium assay buffer with prediluted agonists in buffer. Fura-2 fluorescence was measured through a Fluar x20 objective and a Chroma filter set using ImageMaster System software and DeltaRAM rapid wavelength switching illuminator (Photon Technology International). Fura-2 fluorescence was expressed as the ratio of emission at 510 nm with excitation at 340 and 380 nm. Twenty cells were analysed for each coverslip of cells treated.
Neuraminidase treatment of rPAR2 and HEK
Cells in 75 cm2 culture flasks were rinsed once with PBS (without calcium and magnesium), lifted with non-enzymatic cell dissociation fluid and washed with PBS. The cell pellet was resuspended in calcium assay buffer (as described in calcium signalling in suspension assay) and a final concentration of 10 U ml−1 of Vibrio cholerae neuraminidase was incubated with the cells for 30 min at room temperature with mild shaking. Treated cells were subsequently washed with PBS before being prepared for calcium signalling using the suspension assay (as described above).
Tunicamycin treatment of HEK
HEK cells were resuspended in PBS and seeded into 75 cm2 culture flasks and incubated overnight. The following day, tunicamycin (1 μg ml−1) was applied to the cells and incubated for a further 48 h, as described previously (Servant et al., 1996). Cells were lifted with PBS, centrifuged and then washed with PBS before being prepared for calcium signalling using the suspension assay (as described above).
Statistics
Results were analysed for statistical significance by the paired Student's t-test, taking P<0.05 as statistically significant.
Results
Tryptase purification
Considerable care was taken to ensure that the tryptase used in the study was of the highest purity. Tryptase was purified to apparent homogeneity as assessed by SDS – PAGE and Western blot analysis using the tryptase specific monoclonal antibody AA5 (Walls et al., 1990). Further, a Western blot of the purified enzyme stained with Coomassie Blue reagent was sequenced for the first 10 amino acids and found to be IVGGQEAPRS, which is identical to the first 10 amino acids of human mast cell tryptase (Miller et al., 1990). The specific activities of tryptase preparations were routinely from 2 – 2.5 U μg−1, as assessed in the absence of heparin.
Tryptase efficiently cleaves a peptide corresponding to the cleavage/activation site of wt-rPAR2
Our preliminary calcium signalling experiments with wt-rPAR2 cells failed to demonstrate any appreciable activation of PAR2 with tryptase (see below). We therefore investigated the ability of tryptase to cleave a 20 mer peptide (P20) that corresponded to the cleavage/activation site of wt-rPAR2. As verified by HPLC analysis and sequencing of the cleavage products, tryptase catalyzed the release of the receptor-activating peptide SLIGRL . . . from P20 at both concentrations tested, although 10 nM tryptase cleaved considerably more of the starting material, compared to 1 nM tryptase (see Table 1). Prior incubation of tryptase with STI had a negligible effect on its ability to cleave P20, but completely inhibited trypsin cleavage of the P20 peptide. However, leupeptin completely abolished tryptase release of SLIGRL . . . from the P20 peptide. Purified human lung tryptase purchased from Calbiochem generated identical results to those obtained with the inhouse purified tryptase (data not shown). The data showed that tryptase was able to hydrolyze the PAR2 sequence at its cleavage/activation site (36R/37S) as efficiently as did trypsin.
Table 1.
Tryptase and trypsin cleavage of a 20mer peptide (P20) corresponding to the cleavage/activation sequence of wt-rPAR2

Tryptase at concentrations sufficient to hydrolyze P20 peptide fail to activate/disarm wt-rPAR2 expressed on KNRK cells
Having shown efficient cleavage of P20, we next sought to demonstrate, using the calcium signalling assay, (1) whether tryptase (100 nM) might activate or disarm rPAR2 on wt-rPAR2 cells and (2) whether tryptase present in the cell suspension was capable of cleaving the P20 peptide to release the receptor-activating sequence (SLIGRL...). Tryptase at concentrations well above those sufficient to cleave the P20 peptide failed to activate rPAR2 in the wt-rPAR2 transfected cell line (Figure 1, upper (i)). Trypsin, added following the addition of tryptase provoked a calcium signal equivalent to that of the trypsin control. Addition of the P20 peptide on its own to the wt-rPAR2 cell suspension had no observable effect in the calcium assay (Figure 1, upper (ii)). However, a robust calcium signal was generated when tryptase was added to the cells following the addition of P20 peptide. Further, following stimulation of the cells by the combination of P20 peptide and tryptase, the response to trypsin was markedly reduced compared to its respective control (Figure 1, upper (ii)). Prior desensitization of rPAR2 with two consecutive additions of SLIGRL-NH2 desensitized the subsequent response obtained with the combined addition of P20 followed by tryptase (Figure 1, upper (iii)). Addition of P20 peptide following the prior addition of tryptase stimulated a robust calcium signal. Addition of SLIGRL-NH2 after this response to the combined P20/tryptase treatment resulted in a calcium signal that was reduced when compared to its respective control (Figure 1, upper (iv)). Further, by measuring tryptase enzymatic activity in cell suspensions, we assessed the possibility that sequestration of tryptase by cell surface proteoglycans might be responsible for the lack of PAR2 activation. We found no detectable loss of tryptase enzymatic activity using BAPNA as a substrate in supernatants that previously contained wt-rPAR2 cells compared to tryptase in buffer alone (data not shown).
Figure 1.

Inability of tryptase to activate either wt-rPAR2 (upper) or wt-hPAR2 (lower) in contrast to its ability to release the PAR2-AP, SLIGRL... from the synthetic cleavage/activation peptide, P20. Upper, (i – iv) Wt-rPAR2: activation by tryptase hydrolyzed P20, and by trypsin, but not by tryptase alone. Arrows indicate when test agents were added to the cells. Controls are shown in the right panel of each individual figure, except for (iii). Cells were lifted with non-enzymatic cell dissociation fluid and loaded with Fluo-3 (22 μM) prior to incubation for 25 min at room temperature. Cells were challenged with different agents and responses were monitored by fluorescence spectrophotometry (excitation 480 nm, emission 530 nm). Results are representative of three separate experiments using separately grown crops of cells. Lower, (i – ii) Wt-hPAR2: activation by tryptase hydrolyzed P20 and by trypsin, but not by tryptase alone. Arrows indicate when test agents were added to the cells. Cell monolayers were rinsed and loaded with fura-2AM at room temperature for 40 min. Fluorescence was measured in individual cells using an ICCD video camera and video microscopy acquisition program. The fluorescence ratio (340/380 nm) was measured. Twenty cells were analysed for each coverslip of cells treated. TPZ, tryptase; TPN, trypsin; SL-NH2, SLIGRL-NH2.
We also investigated whether cells in a monolayer as opposed to in suspension might be susceptible to tryptase activation of PAR2. In addition, for these experiments we used the human PAR2 transfected cell line (wt-hPAR2) instead of wt-rPAR2 to determine if the previous results might have been dependent on receptor species. Results are shown in Figure 1, lower (i) and (ii). Tryptase (100 nM) added to the cell monolayer did not produce a calcium signal. However, addition of P20 peptide following tryptase addition stimulated a robust calcium signal, as was the case for the cell suspension assay described above (Figure 1, lower (i)). Trypsin, used as the positive control, stimulated a robust calcium signal in this monolayer assay (Figure 1, lower (ii)). The data with both the suspension and monolayer assay therefore indicated that whilst neither tryptase (100 nM) nor P20 peptide on their own can activate PAR2, tryptase was capable of cleaving P20 peptide into an active fragment that was then able to activate PAR2 expressed on the wt-PAR2 cell lines.
Tryptase activates PAR2 in KNRK cells transfected with wt-rPAR2 and HEK cells pretreated with neuraminidase
Given that a glycosylation sequon is in close proximity to the cleavage/activation site of PAR2 (13 amino acids N-terminal in rPAR2 and only six amino acids in hPAR2), and that sialic acid on cell surface glycoproteins is known to regulate receptor function (Cuatrecasas & Illiano, 1971; Hayes & Lockwood, 1986), we investigated whether preincubation of wt-rPAR2 or the endogenously hPAR2 expressing HEK cells with the sialidase, neuraminidase, would facilitate tryptase activation of PAR2. Results are shown in Figure 2. Although tryptase stimulated no observable response in wt-rPAR2 cells, a prominent calcium signal was generated by tryptase in the neuraminidase treated wt-rPAR2 cells (Figure 2i). In HEK cells, tryptase stimulated a small but viable response (Figure 2ii). Strikingly, the HEK cell response to tryptase was increased by over 7 fold following incubation with neuraminidase. The response provoked by trypsin in wt-rPAR2 and HEK cells was not significantly altered following incubation of the cells with neuraminidase when compared to non-treated controls (Figure 2i and ii). Tryptase (50 nM) caused no observable calcium signal in human wt-rPAR1 cells or empty vector transfected cells (pcDNA3) either before or after incubation of the cells with neuraminidase (data not shown). Further, preincubation of tryptase (100 nM) with neuraminidase (50 mU ml−1) had no observable effect on the ability of tryptase to induce a calcium signal in either untreated or neuraminidase treated wt-rPAR2 cells (data not shown).
Figure 2.
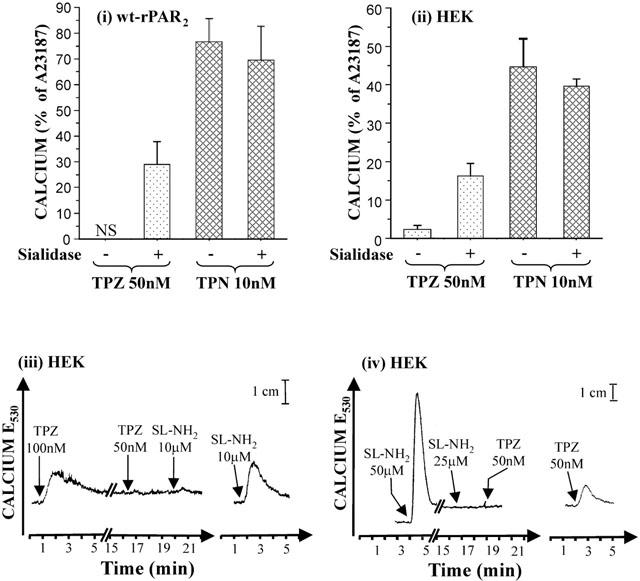
The sialidase, neuraminidase, unmasks PAR2 to activation by tryptase. (i & ii), enhancement of tryptase, (but not trypsin) activation of PAR2 on wt-rPAR2 and HEK cells by treatment with neuraminidase. Wt-rPAR2 or HEK cells were lifted with non-enzymatic cell dissociation fluid and incubated either with or without neuraminidase (10 mU ml−1) for 30 min at room temperature. Cells were washed then loaded with Fluo-3 (22 μM) prior to incubation for 25 min at room temperature. Cells were challenged with test agonists and responses were monitored by fluorescence spectrophotometry (excitation 480 nm, emission 530 nm). Responses were normalized to the peak height obtained with 2 μM calcium ionophore. Each treatment represents the mean±s.e.mean (bars) from 3 – 6 independent experiments from separately grown crops of cells. NS, no signal detected. (iii & iv) Cross-desensitization of tryptase induced calcium signalling in neuraminidase treated HEK cells. Arrows indicate when test agents were added to the cells. Control traces are shown in the right panel of each individual figure. Results are representative of two separate experiments using separately grown crops of cells. TPZ, tryptase; TPN, trypsin; SL-NH2, SLIGRL-NH2.
Cross-desensitization studies were conducted on neuraminidase treated wt-rPAR2 and hPAR2 expressing HEK cells to confirm that the increased tryptase responsiveness was due directly to an increase in PAR2 activation. In HEK cells pretreated with neuraminidase an initial application of tryptase desensitized the response to further additions of tryptase at the same concentration (Figure 2iii). Further, the signal normally induced by SLIGRL-NH2 following two applications of tryptase was desensitized. Moreover, repeated addition of high concentrations of SLIGRL-NH2, prior to the addition of tryptase also resulted in a loss of the tryptase induced calcium response (Figure 2iv). Repeated applications of trypsin (10 nM) also desensitized the tryptase (50 nM) response and vice versa (data not shown). Identical PAR2 cross-desensitization results were also observed for wt-rPAR2 cells (data not shown). Thus, neuramindase treatment was able to unmask PAR2 activation by tryptase both in cells that naturally express the hPAR2 receptor (HEK) and in cells transfected with wt-rPAR2 (KNRK).
Inhibition of the glycosylation process by tunicamycin enhances the ability of tryptase to activate hPAR2
To provide further evidence that glycosylation may play a role in regulating tryptase activation of PAR2, we incubated HEK cells with tunicamycin (which inhibits the N-linked glycosylation of proteins (Tkacz & Lampen, 1975)) for 48 h before investigating the ability of tryptase to activate PAR2 in the calcium signalling assay. Results are shown in Figure 3. Tryptase (300 nM) had failed to generate a calcium signal in HEK cells, but in tunicamycin-treated cells, tryptase generated a calcium signal that was of the same magnitude as trypsin (Figure 3i). The calcium signal generated by trypsin in the tunicamycin treated cells was considerably reduced compared to control cells, as might be expected if tunicamycin treatment reduced overall cell surface receptor density, as has been found with PAR1 (Tordai et al., 1995). Cross-desensitization studies were performed to confirm that tryptase was indeed activating PAR2 and are shown in Figure 3ii and iii. Two additions of the selective PAR2 peptide SLIGRL-NH2, desensitized completely the tryptase response in tunicamycin treated cells (Figure 3ii). In addition, two additions of tryptase desensitized the SLIGRL-NH2 response (Figure 3iii). In further experiments tryptase desensitized a subsequent application of trypsin and vice versa (data not shown). It would appear that by inhibiting HEK cells from glycosylating cell surface proteins including PAR2, tryptase was then able to gain access to the cleavage/activation site of the receptor.
Figure 3.

Inhibition of protein glycosylation in HEK cells by tunicamycin results in the expression of tryptase responsive PAR2. (i) tryptase activation of PAR2 following pretreatment of HEK cells with tunicamycin for 48 h. Semi-confluent HEK cells in 75 cm2 culture flasks were propagated with or without tunicamycin (1 μg ml−1) for 48 h. Cells were lifted with non-enzymatic cell dissociation fluid and washed, then loaded with Fluo-3 (22 μM) prior to incubation for 25 min at room temperature. Cells were challenged with test agonists and responses were monitored by fluorescence spectrophotometry (excitation 480 nm, emission 530 nm). Responses were normalized to the peak height obtained with 2 μM calcium ionophore. Each treatment represents the mean±s.e.mean (bars) from three independent experiments from separately grown crop of cells. NS, no signal detected. (ii & iii) Cross-desensitization of tryptase induced calcium signalling in tunicamycin treated HEK cells. Arrows indicate when test agents were added to the cells. Control traces are shown in the right panel of each individual figure. Results are representative of two separate experiments using separately grown crops of cells. TPZ, tryptase; TPN, trypsin; SL-NH2, SLIGRL-NH2; Tunica, tunicamycin.
We performed a number of Western blots in order to provide direct evidence of receptor glycosylation. However, shifts in the molecular weight of PAR2 in transfected cells or HEK cells following either neuraminidase or tunicamycin treatment could not be observed as the PAR2 antisera (B5 and SLAW) consistently failed to recognize PAR2 on Western blots (data not shown). Therefore we employed an alternative approach of deleting the PAR2 N-terminus glycosylation sequon and examined the efficiency of tryptase to activate the mutated receptor that could not be N-terminally glycosylated.
Tryptase mediated activation of PAR2 is dramatically enhanced by deletion of the receptor N-terminus glycosylation sequon
We performed concentration-effect curves for tryptase on wt-rPAR2 and on a mutated rPAR2 receptor (rPAR2T25−) where threonine25 was deleted to nullify the N-terminus glycosylation sequon N23R24T25. Results are shown in Figure 4. In wt-rPAR2 tryptase (100 – 300 nM) stimulated a very small but concentration-dependent activation (Figure 4, upper, i). In rPAR2T25−, there was a dramatic increase in receptor activation by tryptase. Tryptase was at least 30 times more potent for rPAR2T25− than for wt-rPAR2, stimulating activation of the receptor from 10 – 300 nM. In contrast, trypsin displayed no appreciable alteration in its ability to activate either wt-rPAR2 or rPAR2T25−, stimulating activation from 1 – 20 nM (Figure 4, upper, ii). Tryptase (100 nM) also activated mutated human and rat PAR2 receptors where the asparagine at the N-terminal glycosylation sequon was substituted with alanine (hPAR2N30A and rPAR2N23A): 33.9±5.4 and 25.0±7.0% of the calcium signal caused by 2 μM calcium ionophore respectively; and 63.0±16.5 and 60±3.0% of the calcium signal caused by 10 nM trypsin, respectively (results are the mean±1/2 range from two separate experiments). Finally, tryptase (but not trypsin) failed to induce a calcium signal in a mutated rPAR2 receptor where the only other glycosylation sequon on extracellular loop 2 was deleted (threonine224 substituted with alanine224 : rPAR2T224A; data not shown).
Figure 4.

Tryptase activation of rPAR2 is dramatically enhanced by deletion of the N-linked glycosylation motif on the receptor N-terminus. Upper, (i and ii) Calcium signalling in the wt-rPAR2 and PAR2T25− cells in response to tryptase and trypsin. Concentration effect curves are shown for tryptase (i) and trypsin (ii) in wt-rPAR2 and PAR2T25−. Cells were lifted with non-enzymatic cell dissociation fluid and loaded with Fluo-3 (22 μM) prior to incubation for 25 min at room temperature. Cells were challenged with different agents and responses were monitored by fluorescence spectrophotometry (excitation 480 nm, emission 530 nm). Each data point for PAR2T25− and wt-rPAR2 represents the mean±s.e.mean (bars) of three separate experiments respectively from separately grown crops of cells, except for tryptase (300 nM) on wt-rPAR2, n=11. Lower, (i – iv) Cross-desensitization of tryptase induced calcium signalling in PAR2T25−. Arrows indicate when test agents were added to the cells. Control traces are shown in the right panel of each individual figure. Results are representative of two separate experiments using separately grown crops of cells. TPZ, tryptase; TPN, trypsin; SL-NH2, SLIGRL-NH2.
Cross-desensitization studies were also conducted to confirm that tryptase was indeed activating PAR2T25− (Figure 4, lower). The calcium signal in response to tryptase (100 nM) was abolished by two prior consecutive additions of the PAR2 selective activating peptide SLIGRL-NH2 (Figure 4, lower (i)). Two applications of trypsin (10 nM) 10 min apart were found to desensitize the ability of tryptase (100 nM) to induce a calcium signal (Figure 4, lower (ii)). An initial application of tryptase (500 nM) stimulated a robust calcium signal in rPAR2T25−, that desensitized PAR2 to a further addition of tryptase (300 nM) 10 min later (Figure 4, lower (iii)). Addition, of trypsin (10 nM) following the two previous applications of tryptase resulted in a negligible calcium response compared to the trypsin control. Two prior applications of tryptase markedly reduced the response to SLIGRL-NH2 compared to its respective control (Figure 4, lower (iv)). Preincubation of tryptase (100 nM) with the protease inhibitor, leupeptin (50 μg ml−1), completely blocked its ability to cause a calcium signal in the rPAR2T25− cells (data not shown).
Tryptase removes the pre-cleavage/activation SLAW epitope from rPAR2T25− but not from wt-rPAR2
To confirm that tryptase was activating rPAR2T25− by revealing the tethered ligand, we employed an antiserum (SLAW) that selectively recognizes the N-terminal wt-rPAR2 sequence that is lost upon cleavage/activation by protease treatment. As outlined in Methods, initial studies confirmed the specificity of the SLAW antibody for PAR2. No staining was evident on KNRK cells transfected with the empty vector (data not shown) and preincubation of the antiserum with the immunizing peptide abolished staining of PAR2 transfected cells (Figure 5A,B insets). Further, trypsin pretreatment of PAR2 transfected cells resulted in a complete loss of staining by the SLAW antibody but not by the B5 antiserum (see Figure 5). Thus, we could specifically detect intact PAR2 receptors and monitor activation by the loss of the SLAW-targeted epitope. The amount of immunostaining was confirmed by quantitative morphometric analysis of stained cell cytospins (Figure 5G,H). Non treated wt-rPAR2 and PAR2T25− cells display prominent ring-like staining with SLAW antiserum (Figure 5A,B respectively). Identical results were obtained with the B5 antiserum (data not shown). Pretreatment of wt-rPAR2 or rPAR2T25− cells with trypsin resulted in essentially a complete loss of this ring like immunostaining with the SLAW antiserum compared to the untreated control (Figure 5A and E,B and F,G). However, tryptase treatment had no effect on the immunostaining of SLAW antiserum, compared to untreated controls in the wt-rPAR2 cells (Figure 5A and C,G). Nevertheless, tryptase treatment substantially reduced the immunostaining with SLAW antiserum in rPAR2T25− cells (Figure 5B and D,G). Immunostaining of the cells was also performed with the B5 antiserum that recognizes an epitope (SLIGRLDTP) that remains after PAR2 cleavage-activation. On both wt-rPAR2 and rPAR2T25− cells B5 reactivity showed no appreciable change in staining amongst the various treatments, confirming that PAR2 was still present on the cell surface (Figure 5H). Preincubation of SLAW and B5 antisera with the immunizing peptides (Figure 5A,B insets and data not shown for B5) abolished staining in both wt-rPAR2 and rPAR2T25− cells.
Figure 5.

Tryptase removes the precleavage/activation domain on PAR2T25− but not on wt-rPAR2 cells. Immunostaining with the SLAW antiserum on wt-rPAR2 and PAR2T25− cells following: (A,B) no treatment, (C,D) tryptase 300 nM, and (E,F) trypsin 10 nM at room temperature for 10 min. (G,H): Histograms showing quantitative morphometric counting analysis of stained wt-rPAR2 and PAR2T25− cells (e.g. arrows, A,B) demonstrating the degree of immunostaining for SLAW (G) and B5 (H) antiserum following various treatments. Arrows in A, B and C, show cell surface staining, whilst D, E and F show a loss of cell surface SLAW immunoreactivity. The insets in A and B show the elimination of immunostaining by peptides preabsorbed with the SLAW antiserum. Calcium responses of cells to test agonists were confirmed in the calcium assay (as described in the Methods) before cytospins were prepared. Immunostaining with the SLAW antiserum was conducted as described in the Methods. Results are expressed as the mean counts±s.e.mean from 10 different fields (>100 cells/field) for each treatment. Bar=25 μm.
Heparin concentration dependently inhibits tryptase activation of PAR2
In our initial experiments we found that when heparin was added to the tryptase preparation, activation of rPAR2T25− by tryptase was abolished. Therefore, concentration effect curves were performed to establish the tryptase : heparin molar ratios at which heparin blocked tryptase triggered rPAR2T25− activation. Both porcine intestinal mucosal (PIM) heparin (MW, 16.6 kDa) and the clinically used low molecular weight heparin (fraxiparine®, MW, 4.3 kDa) were used to determine the influence of the molecular weight of heparin on its ability to inhibit tryptase activation of rPAR2T25−. Results are shown in Figure 6. Both forms of heparin concentration-dependently inhibited tryptase (50 nM) activation of rPAR2T25−. Inhibition of tryptase (50 nM) activation of rPAR2T25− by PIM heparin was observed at heparin concentrations as low as 3 nM and reached complete inhibition at 100 nM heparin at which concentrations the molar ratio of heparin : tryptase was 2 : 1. Fraxiparine® was also found to inhibit tryptase activation of rPAR2T25−, but with a lower potency, inhibiting tryptase (50 nM) from 100 nM and reached complete inhibition at 2000 nM. Heparin (100 nM) had a no effect on trypsin (10 nM) activation of wt-rPAR2 (data not shown).
Figure 6.

Inhibition of tryptase induced calcium signalling in PAR2T25− by PIM heparin and Fraxiparine®. Concentration-effect curves are shown for PIM heparin (MW, 16.5 kDa) and Fraxiparine® (MW, 4.3 kDa). Cells were lifted with non-enzymatic cell dissociation fluid and loaded with Fluo-3 (22 μM) prior to incubation for 25 min at room temperature. Cells were challenged with tryptase (50 nM) containing increasing concentrations of heparin and increases in intracellular calcium were monitored by fluorescence spectrophotometry (excitation 480 nm, emission 530 nm). Responses were normalized to the peak height obtained with 2 μM calcium ionophore. For PIM heparin each data point represents the mean±s.e.mean (bars) of four separate experiments, and for Fraxiparine® each data point represents the mean±s.e.mean. TPZ, tryptase; PIM heparin, porcine intestinal mucosa heparin.
Discussion
The principle observation that triggered our study was the finding that human lung tryptase had little efficacy in activating PAR2 in a KNRK cell background, although tryptase was as efficient as trypsin in releasing the active peptide, SLIGRL, from a peptide (P20), representing the cleavage/activation sequence of wt-rPAR2. We demonstrated that by treating transfected KNRK wt-rPAR2 cells, or endogenously expressing hPAR2 HEK cells with either neuraminidase or tunicamycin, we were able to enhance tryptase activation of PAR2. Further, by generating several mutant PAR2 receptors whereupon the N-terminus glycosylation sequon was eliminated, we showed that the ability of tryptase to activate PAR2 was selectively and substantially enhanced. In contrast, elimination of the glycosylation sequon in extracellular loop 2 of PAR2 had no such potentiating effect on tryptase activation. Finally, heparin inhibited tryptase activation of this receptor. Our results suggest that glycosylation of PAR2 and heparin-inhibition of PAR2 activation by tryptase could provide novel mechanisms for regulating receptor activation by tryptase and possibly other proteases.
Our initial experiments studying the ability of tryptase and trypsin to recognize the cleavage/activation site of P20 and release the expected ‘tethered ligand' sequence, SLIGRL..., were in accord with a previous study by Molino et al. (1997). Our results indicated that tryptase was as efficient or more so than trypsin in hydrolyzing the P20 peptide to release SLIGRL, again in agreement with Molino et al. (1997). However, in a wt-rPAR2 cell based calcium signalling assay, tryptase failed to stimulate a calcium response. Tryptase also appeared not to disarm the receptor, since the trypsin response following the addition of tryptase was unaltered compared to the trypsin control. In addition, evidence that tryptase could recognize and cleave the wt-rPAR2 activation/cleavage sequence, but not that on the wt-rPAR2 cells, came from the fact that addition of the P20 peptide either prior to or following the addition of tryptase to the cell suspension generated a robust PAR2-dependent calcium signal. Further, the possibility that the lack of PAR2 activation by tryptase was a result of the enzyme being sequestered by cell surface proteoglycans was excluded as no significant loss in enzymatic activity in supernatants from tryptase challenged cells could be detected. Further, the data also suggest that the cells were not secreting an inhibitor of tryptase, but rather that tryptase access to the cleavage/activation site of wt-rPAR2 on these cells was restricted.
Given that both human and rat PAR2 possess two N-linked glycosylation sequons, of which one is in close proximity to the cleavage/activation site, we hypothesized that N-linked glycosylation may be responsible for restricting tryptase access to the receptor. Therefore, we tested if pretreatment of wt-rPAR2 cells or endogenously human PAR2 expressing HEK cells with neuraminidase to remove sialic acid residues from oligosaccharides, might facilitate tryptase access to the cleavage/activation site of PAR2. Tryptase induced a prominent calcium signal in HEK and wt-rPAR2 cells following but not prior to incubation with neuraminidase. In contrast, neuraminidase had no effect on the lack of ability of tryptase to activate PAR1 in the wt-hPAR1 transfected cells or to cause a calcium signal in empty vector transfected cells. Cross-desensitization studies provided further evidence that the tryptase response unmasked in the wt-rPAR2 cells by neuraminidase was a result of PAR2 activation and was not due to some other receptor. In addition, preincubation of tryptase with neuraminidase had no effect on the ability of tryptase to activate PAR2, indicating that glycans on tryptase probably do not play a part in its ability to activate PAR2. We conclude that cell surface sialic acid plays a role in regulating the ability of tryptase to activate PAR2.
We next decided to take a pharmacological approach by treating HEK cells with tunicamycin, an agent that blocks protein glycosylation (Tkacz & Lampen, 1975). Fortunately, in the presence of tunicamycin, although reduced, sufficient PAR2 was expressed at the cell surface, as indicated by the calcium signal response to trypsin and the PAR2-AP, SLIGRL-NH2. In the tunicamycin treated HEK cells, tryptase activated PAR2 to the same magnitude, as did trypsin, suggesting that tryptase could gain access to all of the trypsin sensitive receptors. Cross-desensitization studies with the selective PAR2-AP SLIGRL-NH2 or trypsin confirmed that tryptase was indeed activating PAR2. It would thus appear that like for PAR1 (Tordai et al., 1995), some functional PAR2 expression does occur in the presence of tunicamycin, and that tryptase is able to activate those receptors that lack N-linked glycosylation.
We attempted to provide direct evidence that PAR2 was indeed a glycosylated receptor. However, both with receptor-expressing and control vector-transfected KNRK cells, we observed multiple bands on Western blots when employing either the SLAW or B5 antibodies. Therefore, we could not, with confidence, positively identify PAR2 for a gel-shift analysis of the mutant glycosylation-deficient receptors. Interestingly Kong et al. (1997) detected two major bands on Western blots using the B5 antibody, whilst Napoli et al. (2000) showed only one band. It appears, at least from our experiments, that the SLAW and B5 antibodies recognize a number of cellular proteins in addition to the receptor, when employed for Western blotting and that these bands might be due to the unmasking of non-receptor protein epitopes by the Western blotting procedure, resulting in a ‘non-specific' cross-reactivity with the antibodies. Nevertheless, a recent survey of 300 multi-span proteins shows that 92% (211/229) of N-linked glycosylation sequons within the N-terminus of seven transmembrane receptors are glycosylated (Landolt-Marticorena & Reithmeier, 1994). Therefore, we tested whether a rat mutant receptor (rPAR2T25−) devoid of the N-terminus glycosylation sequon (N23R24T25) would display enhanced activity toward tryptase. Indeed, there was at least a 30 fold increase in rPAR2T25− sensitivity toward tryptase that was selective, as trypsin was found to activate both wt-rPAR2 and rPAR2T25− with equal potency. Cross-desensitization studies confirmed that tryptase was indeed activating rPAR2T25−. We also generated a mutant rPAR2 where the only other glycosylation sequon situated on extracellular loop 2 was eliminated (rPAR2T224A). The lack of response to tryptase, (but not trypsin) in rPAR2T224A indicated that the N-terminal glycosylation sequon was of primary importance in regulating tryptase activation of PAR2. Therefore, the site directed mutagenesis studies strongly imply, but do not prove, that PAR2 may well be glycosylated on the N-terminus and that removal of this sequon allows tryptase access to the cleavage/activation sequence of the receptor.
In order to provide physical evidence that tryptase activation of rPAR2T25− resulted in the removal of the precleavage domain of the receptor, we employed immunocytochemistry, using a polyclonal antibody (SLAW) that only recognizes the precleavage domain of wt-rPAR2. The inability of tryptase, but not trypsin, to remove the SLAW epitope from the wt-rPAR2 cells was entirely in accord with the calcium assay results showing that tryptase was inefficient at activating wt-rPAR2. However, the loss of the SLAW epitope following tryptase treatment of the rPAR2T25− cells clearly demonstrated that the precleavage N-terminal domain of rPAR2T25− had been removed. This observation was also in agreement with the calcium signalling assay, which demonstrated activation of rPAR2T25− in response to tryptase. This loss in the SLAW epitope was not simply due to a generalized loss of cell surface receptors, since the B5 antiserum (that recognizes both activated and non activated receptors) showed equivalent staining in both wt-rPAR2 and rPAR2T25− cell lines following tryptase or trypsin treatment when compared to untreated control cells. Thus the immunocytochemistry confirmed that tryptase removed the precleavage domain from rPAR2T25− but not for wt-rPAR2.
As tryptase is thought to be released from mast cells in association with heparin and since heparin has been reported both to inhibit (Schechter et al., 1998) and to be required (Steinhoff et al., 2000) for tryptase activation of PAR2, we explored the ability of different heparin preparations to affect rPAR2T25− activation by tryptase. Interestingly, both PIM heparin (16.5 kDa) and the Fraxiparine® (4.3 kDa) heparin were found, concentration-dependently, to inhibit tryptase activation of rPAR2T25−. This result is in agreement with a recent report (Schechter et al., 1998) but not another (Steinhoff et al., 2000). The reason(s) for these apparent differences may be dependent on the cell types, experimental conditions, or the preparations of heparin employed. Nevertheless, at least in HEK cells and keratinocytes (Schechter et al., 1998), tryptase inactivation by heparin provides a further mechanism whereby tryptase activation of PAR2 may be tightly regulated.
Although tryptase at comparatively high concentrations stimulated a small calcium signalling response in wt-rPAR2 and untreated HEK cells, we found this result to be extremely variable. In only three out of the 11 experiments that were performed with wt-rPAR2 cells did we observe a calcium response for tryptase (300 nM). For the HEK cells, we observed a calcium response to tryptase (50 nM) in only three out of the six experiments. Given our data on glycosylation it is tempting to speculate that the small tryptase responses observed in the wt-rPAR2, HEK cells and those from previous studies (Molino et al., 1997; Schechter et al., 1998), may represent the activation of differentially glycosylated PAR2. Differential glycosylation would, in effect, restrict the number of receptors available for tryptase activation. Indeed it has been reported previously that tryptase only appears to activate a subpopulation of PAR2 in keratinocytes and other cell types (Schechter et al., 1998). Not only did Molino et al. (1997) demonstrate that tryptase was unable to stimulate activation of PAR2 in transfected COS cells to the same extent as trypsin, but also that the tryptase-induced calcium signalling in HUVEC differed considerably between various cultures. Therefore, our present finding that glycosylation of PAR2 can regulate receptor activation by tryptase provides a plausible rationale for the ability of tryptase to activate PAR2 with an efficacy comparable to that of trypsin in some cell types (Corvera et al., 1997; Steinhoff et al., 2000) but not others (Alm et al., 2000; Corvera et al., 1999; Molino et al., 1997; Schechter et al., 1998).
In summary, the present results show that glycosylation of PAR2 may provide a novel mechanism whereby cells or tissues may specifically regulate receptor activation by tryptase and possibly other proteases. The potential differences in PAR2 glycosylation that may occur, either in the settings of neoplasia or in the context of infection by neuraminidase-producing viruses may thus be of pathophysiological importance in terms of PAR2 function.
Acknowledgments
This study was supported by a grant from the Canadian Institutes of Health Research (formerly MRC of Canada), an Alberta Heritage Foundation for Medical Research Post-Doctoral Fellowship (S.J. Compton), and ancillary support provided by grants from the Heart and Stroke Foundation of Alberta and the Kidney Foundation of Canada. We are grateful to Laurie Robertson for help with the FACS analysis and to Dr Shalini Mokashi for the production of the SLAW antisera. We also wish to thank Dr Francis Green for help in obtaining the lung tissue; Dr Andrew Walls for supplying the tryptase monoclonal antibody; Dr Graham Pineo for supplying the Fraxiaprine®, and Dr Shaun Coughlin for supplying the human PAR1 cDNA. The insightful discussions provided by Dr Nigel Bunnett are also gratefully acknowledged.
Abbreviations
- AP
agonist peptide
- BAPNA
Nα-Benzoyl-DL-Arginine 4-Nitro-anilide
- HEK
human embryonic kidney cells
- KNRK
Kirsten virus sarcoma transformed rat kidney epithelial cells
- PAR
proteinase-activated receptor
- PIM
porcine intestinal mucosa
- STI
soya trypsin inhibitor
References
- AL ANI B., SAIFEDDINE M., KAWABATA A., RENAUX B., MOKASHI S., HOLLENBERG M.D. Proteinase-activated receptor 2 (PAR(2)): development of a ligand- binding assay correlating with activation of PAR(2) by PAR(1)- and PAR(2)-derived peptide ligands. J. Pharmacol. Exp. Ther. 1999;290:753–760. [PubMed] [Google Scholar]
- ALM A.K., GAGNEMO-PERSSON R., SORSA T., SUNDELIN J. Extrapancreatic trypsin-2 cleaves proteinase-activated receptor-2. Biochem. Biophys. Res. Commun. 2000;275:77–83. doi: 10.1006/bbrc.2000.3267. [DOI] [PubMed] [Google Scholar]
- BOHM S.K., KONG W., BROMME D., SMEEKENS S.P., ANDERSON D.C., CONNOLLY A., KAHN M., NELKEN N.A., COUGHLIN S.R., PAYAN D.G., BUNNETT N.W. Molecular cloning, expression and potential functions of the human proteinase-activated receptor-2. Biochem. J. 1996;314:1009–1016. doi: 10.1042/bj3141009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BROWN J.K., TYLER C.L., JONES C.A., RUOSS S.J., HARTMANN T., CAUGHEY G.H. Tryptase, the dominant secretory granular protein in human mast cells, is a potent mitogen for cultured dog tracheal smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 1995;13:227–236. doi: 10.1165/ajrcmb.13.2.7626290. [DOI] [PubMed] [Google Scholar]
- CAIRNS J.A., WALLS A.F. Mast cell tryptase is a mitogen for epithelial cells. Stimulation of IL- 8 production and intercellular adhesion molecule-1 expression. J. Immunol. 1996;156:275–283. [PubMed] [Google Scholar]
- CAIRNS J.A., WALLS A.F. Mast cell tryptase stimulates the synthesis of type I collagen in human lung fibroblasts. J. Clin. Invest. 1997;99:1313–1321. doi: 10.1172/JCI119290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COMPTON S.J., WALLS A.F.Mast cell proteases as new targets for therapeutic intervention in asthma ‘Anti-inflammatory drugs in asthma.' 1999Birkhauser Verlag: Basal; 229–249.ed. Sampson A.P. & Church M.K. pp [Google Scholar]
- COMPTON S.J., CAIRNS J.A., HOLGATE S.T., WALLS A.F. The role of mast cell tryptase in regulating endothelial cell proliferation, cytokine release, and adhesion molecule expression: tryptase induces expression of mRNA for IL-1 beta and IL-8 and stimulates the selective release of IL-8 from human umbilical vein endothelial cells. J. Immunol. 1998;161:1939–1946. [PubMed] [Google Scholar]
- COMPTON S.J., CAIRNS J.A., PALMER K.J., AL ANI B., HOLLENBERG M.D., WALLS A.F. A polymorphic protease-activated receptor 2 (PAR2) displaying reduced sensitivity to trypsin and differential responses to PAR agonists. J. Biol. Chem. 2000;275:39207–39212. doi: 10.1074/jbc.M007215200. [DOI] [PubMed] [Google Scholar]
- CORVERA C.U., DERY O., MCCONALOGUE K., BOHM S.K., KHITIN L.M., CAUGHEY G.H., PAYAN D.G., BUNNETT N.W. Mast cell tryptase regulates rat colonic myocytes through proteinase- activated receptor 2. J. Clin. Invest. 1997;100:1383–1393. doi: 10.1172/JCI119658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CORVERA C.U., DERY O., MCCONALOGUE K., GAMP P., THOMA M., AL ANI B., CAUGHEY G.H., HOLLENBERG M.D., BUNNETT N.W. Thrombin and mast cell tryptase regulate guinea-pig myenteric neurons through proteinase-activated receptors-1 and -2. J. Physiol (Lond) 1999;517:741–756. doi: 10.1111/j.1469-7793.1999.0741s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUATRECASAS P., ILLIANO G. Membrane sialic acid and the mechanism of insulin action in adipose tissue cells. Effects of digestion with neuraminidase. J. Biol. Chem. 1971;246:4938–4946. [PubMed] [Google Scholar]
- DERY O., CORVERA C.U., STEINHOFF M., BUNNETT N.W. Proteinase-activated receptors: novel mechanisms of signaling by serine proteases. Am. J. Physiol. 1998;274:C1429–C1452. doi: 10.1152/ajpcell.1998.274.6.C1429. [DOI] [PubMed] [Google Scholar]
- HAYES G.R., LOCKWOOD D.H. The role of cell surface sialic acid in insulin receptor function and insulin action. J. Biol. Chem. 1986;261:2791–2798. [PubMed] [Google Scholar]
- HE S., PENG Q., WALLS A.F. Potent induction of a neutrophil and eosinophil-rich infiltrate in vivo by human mast cell tryptase: selective enhancement of eosinophil recruitment by histamine. J. Immunol. 1997;159:6216–6225. [PubMed] [Google Scholar]
- HE S., WALLS A.F. Human mast cell tryptase: a stimulus of microvascular leakage and mast cell activation. Eur. J. Pharmacol. 1997;328:89–97. doi: 10.1016/s0014-2999(97)83033-6. [DOI] [PubMed] [Google Scholar]
- HUANG C., DE SANCTIS G.T., O'BRIEN P.J., MIZERD J.P., FRIEND D.S., DRAZEN J.M., BRASS L.F., STEVENS R.L. Human Mast Cell Tryptase BetaI: evaluation of its substrate specificity and demonstration of its importance in bacterial infections of the lung. J. Biol. Chem. 2001;276:26276–26284. doi: 10.1074/jbc.M102356200. [DOI] [PubMed] [Google Scholar]
- ISHIHARA H, , CONNOLLY A.J., ZENG D., KAHN M.L., ZHENG Y.W., TIMMONS C., TRAM T., COUGHLIN S.R. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature. 1997;386:502–506. doi: 10.1038/386502a0. [DOI] [PubMed] [Google Scholar]
- ISHII K., HEIN L., KOBILKA B., COUGHLIN S.R. Kinetics of thrombin receptor cleavage on intact cells. Relation to signaling. J. Biol. Chem. 1993;268:9780–9786. [PubMed] [Google Scholar]
- KAHN M.L., ZHENG Y.W., HUANG W., BIGORNIA V., ZENG D., MOFF S., FARESE R.V., JR, TAM C., COUGHLIN S.R. A dual thrombin receptor system for platelet activation. Nature. 1998;394:690–694. doi: 10.1038/29325. [DOI] [PubMed] [Google Scholar]
- KAWABATA A., SAIFEDDINE M., AL ANI B., LEBLOND L., HOLLENBERG M.D. Evaluation of proteinase-activated receptor-1 (PAR1) agonists and antagonists using a cultured cell receptor desensitization assay: activation of PAR2 by PAR1 -targeted ligands. J. Pharmacol. Exp. Ther. 1999;288:358–370. [PubMed] [Google Scholar]
- KONG W., MCCONALOGUE K., KHITIN L.M., HOLLENBERG M.D., PAYAN D.G., BOHM S.K., BUNNETT N.W. Luminal trypsin may regulate enterocytes through proteinase-activated receptor 2. Proc. Natl. Acad. Sci. U.S.A. 1997;94:8884–8889. doi: 10.1073/pnas.94.16.8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANDOLT-MARTICORENA C., REITHMEIER R.A. Asparagine-linked oligosaccharides are localized to single extracytosolic segments in multi-span membrane glycoproteins. Biochem. J. 1994;302:253–260. doi: 10.1042/bj3020253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MILLER J.S., MOXLEY G., SCHWARTZ L.B. Cloning and characterization of a second complementary DNA for human tryptase. J. Clin. Invest. 1990;86:864–870. doi: 10.1172/JCI114786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIRZA H., SCHMIDT V.A., DERIAN C.K., JESTY J., BAHOU W.F. Mitogenic responses mediated through the proteinase-activated receptor- 2 are induced by expressed forms of mast cell alpha or beta-tryptases. Blood. 1997;90:3914–3922. [PubMed] [Google Scholar]
- MOLINO M., BARNATHAN E.S., NUMEROF R., CLARK J., DREYER M., CUMASHI A., HOXIE J.A., SCHECHTER N., WOOLKALIS M., BRASS L.F. Interactions of mast cell tryptase with thrombin receptors and PAR-2. J. Biol. Chem. 1997;272:4043–4049. doi: 10.1074/jbc.272.7.4043. [DOI] [PubMed] [Google Scholar]
- NAKANISHI-MATSUI M., ZHENG Y.W., SULCINER D.J., WEISS E.J., LUDEMAN M.J., COUGHLIN S.R. PAR3 is a cofactor for PAR4 activation by thrombin. Nature. 2000;404:609–613. doi: 10.1038/35007085. [DOI] [PubMed] [Google Scholar]
- NAPOLI C., CICALA C., WALLACE J.L., DE NIGRIS F., SANTAGADA V., CALIENDO G., FRANCONI F., IGNARRO L.J., CIRINO G. Protease-activated receptor-2 modulates myocardial ischemia-reperfusion injury in the rat heart. Proc. Natl. Acad. Sci. U.S.A. 2000;97:3678–3683. doi: 10.1073/pnas.97.7.3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NYSTEDT S., RAMAKRISHNAN V., SUNDELIN J. The proteinase-activated receptor 2 is induced by inflammatory mediators in human endothelial cells. Comparison with the thrombin receptor. J. Biol. Chem. 1996;271:14910–14915. doi: 10.1074/jbc.271.25.14910. [DOI] [PubMed] [Google Scholar]
- PEREIRA P.J., BERGNER A., MACEDO-RIBEIRO S., HUBER R., MATSCHINER G., FRITZ H., SOMMERHOFF C.P., BODE W. Human beta-tryptase is a ring-like tetramer with active sites facing a central pore. Nature. 1998;392:306–311. doi: 10.1038/32703. [DOI] [PubMed] [Google Scholar]
- RUOSS S.J., HARTMANN T., CAUGHEY G.H. Mast cell tryptase is a mitogen for cultured fibroblasts. J. Clin. Invest. 1991;88:493–499. doi: 10.1172/JCI115330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAIFEDDINE M., AL ANI B., SANDHU S., WIJESURIYA S.J., HOLLENBERG M.D. Contractile actions of proteinase-activated receptor-derived polypeptides in guineapig gastric and lung parenchymal strips: evidence for distinct receptor systems. Br. J. Pharmacol. 2001;132:556–566. doi: 10.1038/sj.bjp.0703839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHECHTER N.M., BRASS L.F., LAVKER R.M., JENSEN P.J. Reaction of mast cell proteases tryptase and chymase with protease activated receptors (PARs) on keratinocytes and fibroblasts. J. Cell Physiol. 1998;176:365–373. doi: 10.1002/(SICI)1097-4652(199808)176:2<365::AID-JCP15>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- SCHWARTZ L.B., BRADFORD T.R. Regulation of tryptase from human lung mast cells by heparin. Stabilization of the active tetramer. J. Biol. Chem. 1986;261:7372–7379. [PubMed] [Google Scholar]
- SCHWARTZ L.B., IRANI A.M., ROLLER K., CASTELLS M.C., SCHECHTER N.M. Quantitation of histamine, tryptase, and chymase in dispersed human T and TC mast cells. J. Immunol. 1987;138:2611–2615. [PubMed] [Google Scholar]
- SCHWARTZ L.B., LEWIS R.A., AUSTEN K.F. Tryptase from human pulmonary mast cells. Purification and characterization. J. Biol. Chem. 1981;256:11939–11943. [PubMed] [Google Scholar]
- SERVANT G., DUDLEY D.T., ESCHER E., GUILLEMETTE G. Analysis of the role of N-glycosylation in cell-surface expression and binding properties of angiotensin II type-2 receptor of rat pheochromocytoma cells. Biochem. J. 1996;313:297–304. doi: 10.1042/bj3130297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEINHOFF M., VERGNOLLE N., YOUNG S.H., TOGNETTO M., AMADESI S., ENNES H.S., TREVISANI M., HOLLENBERG M.D., WALLACE J.L., CAUGHEY G.H., MITCHELL S.E., WILLIAMS L.M., GEPPETTI P., MAYER E.A., BUNNETT N.W. Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat. Med. 2000;6:151–158. doi: 10.1038/72247. [DOI] [PubMed] [Google Scholar]
- TKACZ J.S., LAMPEN O. Tunicamycin inhibition of polyisoprenyl N-acetylglucosaminyl pyrophosphate formation in calf-liver microsomes. Biochem. Biophys. Res. Commun. 1975;65:248–257. doi: 10.1016/s0006-291x(75)80086-6. [DOI] [PubMed] [Google Scholar]
- TORDAI A., BRASS L.F., GELFAND E.W. Tunicamycin inhibits the expression of functional thrombin receptors on human T-lymphoblastoid cells. Biochem. Biophys. Res. Commun. 1995;206:857–862. doi: 10.1006/bbrc.1995.1122. [DOI] [PubMed] [Google Scholar]
- VERGNOLLE N. Proteinase-activated receptor-2-activating peptides induce leukocyte rolling, adhesion, and extravasation in vivo. J. Immunol. 1999;163:5064–5069. [PubMed] [Google Scholar]
- VERGNOLLE N., HOLLENBERG M.D., SHARKEY K.A., WALLACE J.L. Characterization of the inflammatory response to proteinase-activated receptor-2 (PAR2)-activating peptides in the rat paw. Br. J. Pharmacol. 1999;127:1083–1090. doi: 10.1038/sj.bjp.0702634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VU T.K., HUNG D.T., WHEATON V.I., COUGHLIN S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- WALLS A.F., BENNETT A.R., MCBRIDE H.M., GLENNIE M.J., HOLGATE S.T., CHURCH M.K. Production and characterization of monoclonal antibodies specific for human mast cell tryptase. Clin. Exp. Allergy. 1990;20:581–589. doi: 10.1111/j.1365-2222.1990.tb03153.x. [DOI] [PubMed] [Google Scholar]
- XU W.F., ANDERSEN H., WHITMORE T.E., PRESNELL S.R., YEE D.P., CHING A., GILBERT T., DAVIE E.W., FOSTER D.C. Cloning and characterization of human protease-activated receptor 4. Proc. Natl. Acad. Sci. U.S.A. 1998;95:6642–6646. doi: 10.1073/pnas.95.12.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]