

Abstract
Ketamine is a dissociative anaesthetic that is formulated as Ketalar, which contains the preservative benzethonium chloride (BCl). We have studied the effects of pure racemic ketamine, the preservative BCl and the Ketalar mixture on human neuronal nicotinic acetylcholine receptors (nAChRs) composed of the α7 subunit or α4 and β2 subunits expressed in Xenopus laevis oocytes.
Ketamine inhibited responses to 1 mM acetylcholine (ACh) in both the human α7 and α4β2 nAChRs, with IC50 values of 20 and 50 μM respectively. Inhibition of the α7 nAChRs occurred within a clinically relevant concentration range, while inhibition of the α4β2 nAChR was observed only at higher concentrations. The Ketalar formulation inhibited nAChR function more effectively than was expected given its ketamine concentration. The surprising increased inhibitory potency of Ketalar compared with pure ketamine appeared to be due to the activity of BCl, which inhibited both α7 (IC50 value of 122 nM) and α4β2 (IC50 value of 49 nM) nAChRs at concentrations present in the clinical formulation of Ketalar.
Ketamine is a noncompetitive inhibitor at both the α7 and α4β2 nAChR. In contrast, BCl causes a parallel shift in the ACh dose-response curve at the α7 nAChR suggesting competitive inhibition. Ketamine causes both voltage-dependent and use-dependent inhibition, only in the α4β2 nAChR.
Since α7 nAChRs are likely to be inhibited during clinical use of Ketalar, the actions of ketamine and BCl on this receptor subtype may play a role in the profound analgesia, amnesia, immobility and/or autonomic modulation produced by this anaesthetic.
Keywords: Neuronal nicotinic acetylcholine receptors, benzethonium chloride, ketamine, anaesthetic mechanism
Introduction
Ketamine is a dissociative anaesthetic, that is an effective analgesic, amnestic and hypnotic, but causes hallucinations that limit its usefulness as a clinical anaesthetic agent. Due to its potency as an analgesic and its sympathomimetic properties, ketamine finds extensive use in the treatment of burn patients in intensive care settings. In the early 1980's Lodge described ketamine as a selective inhibitor of NMDA receptor activity and neuronal nicotinic receptors (nAChRs) on Renshaw cells of the cat spinal cord (Anis et al., 1983). Ketamine is also an antagonist of NMDA receptors in rat cortex (Harrison & Simmonds, 1985) and blocks these receptors at sub-clinical concentrations, by acting both as a use-dependent open channel blocker (Honey et al., 1985) and through allosteric inhibition (Orser et al., 1997). Ketamine inhibits the response to acetylcholine or nicotine in both muscle and neuronal type nAChRs (Sumikawa et al., 1983; Wachtel & Wegrzynowicz, 1992; Scheller et al., 1996; Durieux, 1995 Furuya et al., 1999; Flood & Krasowski, 2000; Friederich et al., 2000; Sasaki et al., 2000; Yamakura et al., 2000). In the case of nAChRs containing the α4 subunit, inhibition occurs at concentrations similar to those effective in inhibiting NMDA responses (Flood & Krasowski, 2000; Friederich et al., 2000; Sasaki et al., 2000).
Ketamine is marketed in the United States as Ketalar, a racemic mixture of ketamine containing benzethonium chloride (BCl). BCl is included in Ketalar as a preservative, at a concentration of 0.1 mg with 10 mg of ketamine. BCl is also used as a topical antiseptic alone. BCl is an inhibitor of acetylcholine esterase (AChE) and choline esterase (Zaman et al., 1997) as well as muscarinic receptors for acetylcholine (ACh) (Durieux & Nietgen, 1997). The secondary structure of BCl consists of a quaternary ammonium atom as does acetylcholine, though it also contains 2 benzyl groups. (Figure 1).
Figure 1.
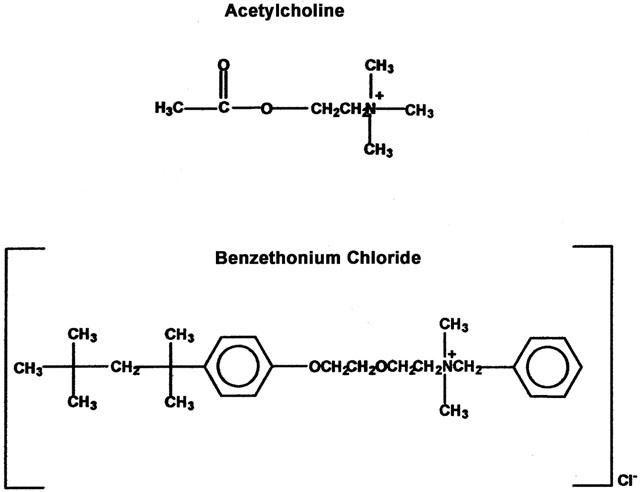
Chemical structures of acetylcholine, the native nicotinic agonist and benzethonium chloride, a nicotinic and muscarinic antagonist.
In the present study we investigated the mechanism of action of ketamine and BCl at both human heteromeric α4β2 and homomeric α7 type nAChRs that are found in the CNS. We compared the activity of each drug alone with the activity of the clinical formulation, Ketalar, for evidence of potentially clinically significant interaction.
Methods
Molecular biology
The human α4 (accession number L35901) and β2 (accession number 53971) type nAChR cDNAs were in a pSP64 expression vector and the α7 (accession number X70297) type nAChR was in a pMXT expression vector. The XbaI restriction enzyme was used to linearize the α7 containing vector, AseI was used to linearize the α4 vector and PvuII was used to linearize the β2 containing vector. Using the SP6 RNA polymerase, cRNA was made from the linearized vectors using a standard protocol.
Oocyte extraction and injection
Xenopus laevis oocytes were extracted from anesthetized females and placed in ND-96 medium (mM: NaCl 96, KCl 2, MgCl2 1, CaCl2·H2O 1.8, HEPES 5, Na-pyruvate 2.5, theophylline 0.5, and 10 mg l−1 of gentamicin, adjusted to pH 7.5). The oocyte clusters were incubated in 0.2% collagenase (type IA, Sigma-Aldrich) in ND-96 medium for defolliculation. Oocytes were agitated at 18.5°C for 4 h and afterwards were rinsed with Barth's medium (mM: NaCl 88, KCl 1, NaHCO3 2.4, HEPES 15, pH 7.6). The oocytes were left to recover for 24 h in L-15 oocyte medium (Specialty Media) before injection of cRNA. Approximately 10 ng of α7 cRNA or 10 ng of a 1 : 1 ratio of α4 to β2 cRNA were injected into individual oocytes in volumes of ∼100 nl using an automatic injector (Nanoject; Drummond Scientific, Broomall, PA, U.S.A.). The oocytes were incubated at 17°C for 2 – 5 days in ND-96 medium prior to electrophysiological recording.
Electrophysiology
Current recordings were made from whole oocytes at room temperature (19 – 23°C) using a Gene-Clamp 500 two-microelectrode voltage-clamp amplifier with an active ground circuit (Axon Instruments, Inc., Foster City, CA, U.S.A.). The recording electrodes were pulled from glass capillary tubing (Drummond Scientific, Broomall, PA, U.S.A.) to obtain a resistance between 1 and 5 MΩ and then filled with 3M KCl. The Ringer's solution (mM: NaCl 115, KCl 2.5, BaCl2 1.8, HEPES 10, and 1 μM atropine, pH 7.4) used for recordings, contained atropine to prevent muscarinic receptor stimulation and barium in place of calcium to avoid current amplification by calcium activated chloride currents. Oocytes were clamped at a holding potential of −60 mV unless otherwise indicated and held in a 125 μl cylindrical channel. ACh and other drugs were applied at a flow rate of 4 ml min−1 with a two second application. A saturating concentration of 1 mM ACh was used in all experiments unless otherwise noted. The oocytes were pre-equilibrated with potential inhibitors for 2 min prior to co-application with ACh. To minimize the contribution of nAChR desensitization, 5 min passed between ACh applications.
Analysis
A baseline control response to 1 mM ACh was measured before each agonist-antagonist co-application. The response to agonist was measured after each antagonist application. Responses that did not return to within 80% of baseline values were rejected for analysis. Concentration-response curves for ACh were fitted to a modified Hill equation, y=ymax/(1+(x/EC50)n), where EC50 is the concentration of ACh which elicited 50% of the maximal response, ymax is the maximal current elicited by ACh, and n is the Hill coefficient. Concentration-response relationships for the inhibition were constructed by calculating the current recorded in the presence of antagonist as a percentage of that elicited by ACh alone. ACh dose-response curves were normalized to the average response to a saturating concentration of ACh in the absence of ketamine (1 mM α4β2, 3 mM α7). The datapoints obtained at each antagonist concentration were averaged and the calculated mean and standard error were fit to a modified Hill equation, where IC50 is the concentration of antagonist at which 50% of the response is inhibited. Clampex 7 (Axon Instruments, Inc., Foster City, CA, U.S.A.) was used for data acquisition and Microcal Origin 5.0 (Microcal, Northampton, MA, U.S.A.) was used for graphics and statistical calculation. One-way analysis of variance (ANOVA) was used to compare the ketamine and Ketalar concentration response curves. P<0.05 was considered significant and data were represented as mean±s.e.
The interaction between ketamine and BCl was investigated further using an isobolographic analysis (Tallarida et al., 1989; Durieux & Nietgen, 1997). Briefly, concentration response curves were constructed for the activity of each compound alone. The concentrations of each compound that were calculated to produce an IC50 effect separately were combined and the effect measured. In this case Ketalar was used to look at the effect of the combined compounds. If a drug combination had an additive interaction, the effect of the combined compounds would be the sum of effects of its component parts. An antagonistic or subadditive interaction would produce less than the sum of the component parts and a superadditive interaction would be greater.
Materials
Molecular biology reagents used were obtained from Promega (Madison, WI, U.S.A.). Ketalar was from Parke-Davis (Morris Plains, NJ, U.S.A.). Ketamine, BCl, ACh and other chemicals used were obtained from Sigma-Aldrich (St. Louis, MO, U.S.A.). L-15 oocyte media was obtained from Specialty Media (Phillipsburg, NJ, U.S.A.). Ketamine, BCl and Ketalar were made as stock solutions and serially diluted to the appropriate concentrations on the day of the experiment.
Results
Actions of ketamine and Ketalar formulation at α4β2 and α7 nAChRs
Pure racemic ketamine inhibited the activation of human α4β2 and α7 nAChRs (Figure 2A,B). Ketamine inhibition was more potent at the α7 nAChR, with an IC50 value of 20±2 μM, than at the α4β2 nAChR, with an IC50 value of 50±4 μM. A parallel series of experiments were carried out using the clinical formulation of Ketalar. To our surprise, we found that a solution of Ketalar that contained a known concentration of ketamine produced significantly greater inhibition of ACh currents than would be predicted at both receptor subtypes. Careful examination of the concentration-response curves for ketamine and for the Ketalar formulation showed that Ketalar dilutions routinely had greater activity than expected with an IC50 value for α4β2 inhibition of 17±3 μM ketamine and 11±0.6 μM for α7 (Figure 2C,D) (ANOVA, P<0.001 for α4β2 and P<0.01 for α7). For example, a Ketalar solution that contained 20 μM ketamine had the same inhibitory activity as 50 μM of the pure racemic ketamine on the α4β2. One possible explanation for this discrepancy was that the preservative BCl might be responsible for the unexpected activity, and this hypothesis was therefore tested directly. In fact, the preservative BCl is a potent inhibitor of both subtypes of nAChRs (Figure 3A, B). Inhibition of receptor activation by BCl was concentration responsive with an IC50 of 49±11 nM at α4β2 and an IC50 of 122±19 nM at α7 nAChRs (Figure 3C,D).
Figure 2.
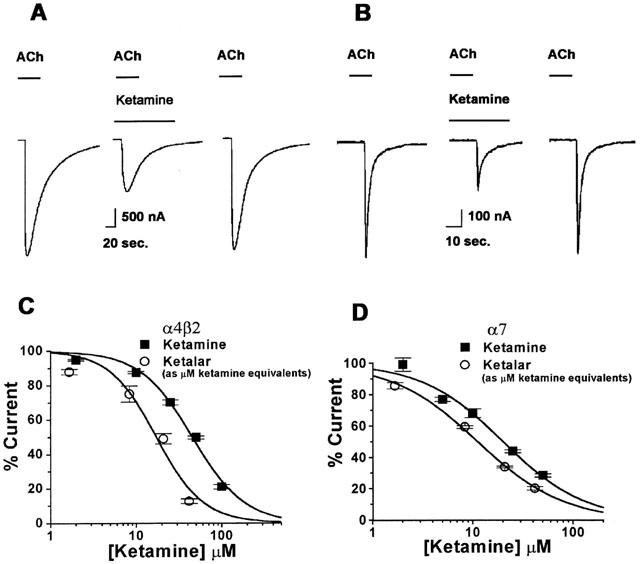
Ketalar, the clinically used formulation of ketamine, produced more potent inhibition of the activation of both the α4β2 and α7 nAChRs than ketamine. (A) A control current activated by 1 mM ACh, from α4β2 before (left), during a 2 s ACh application with 50 μM ketamine (middle), and after ketamine washout (right). (B) A control current activated by 1 mM ACh, from α7 before (left), during a 2 s application of 20 μM ketamine (middle), and after ketamine washout (right). (C) Concentration-response relationship for ketamine and Ketalar inhibition of the activation of the α4β2 nAChR, the IC50 for Ketalar was 17±3 μM (Hill coefficient is 1.59±0.48) compared to an IC50 of 50±4 μM (Hill coefficient is 1.55±0.22) for ketamine. (D) For the α7 nAChR, the IC50 for Ketalar was 11±0.6 μM (Hill coefficient is 1.02±0.06) compared to an IC50 of 20±2 μM (Hill coefficient is 1.07±0.14) for ketamine (number of oocytes for each data point (n)=5 – 7). Values are mean±s.e.
Figure 3.
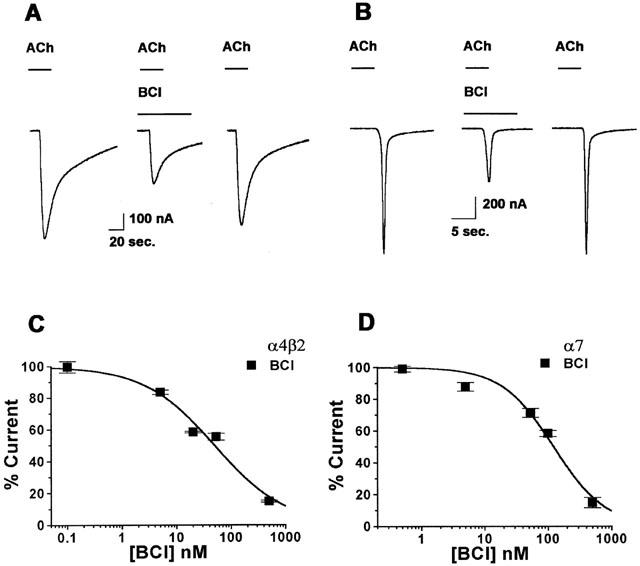
Benzethonium chloride (BCl), the preservative in Ketalar, caused inhibition of the activation of both the α7 and α4β2 nAChRs at concentrations consistent with those found in the clinically used Ketalar formulation. (A) A control current activated by 1 mM ACh, from α4β2 before (left), during a 2 s ACh application with 55 nM BCl (middle), and after BCl washout (right). (B) A control current activated by 1 mM ACh, from α7 before (left), during a 2 s ACh application with 110 nM BCl (middle), and after BCl washout (right). (C) A concentration-response curve for BCl inhibition of the activation of the α4β2 nAChR. For the α4β2 nAChR, the IC50 for BCl inhibition with 1 mM ACh was 49±11 nM (Hill coefficient is 0.67±0.11). (D) For the α7 nAChR, the IC50 for BCl inhibition with 1 mM ACh was 122±19 nM (Hill coefficient is 1.07±0.20) (n=5 – 7). Values are mean±s.e.
Mechanism of ketamine inhibition of α4β2 and α7 nAChRs
ACh concentration-response curves were constructed in the presence and absence of the calculated IC50 ketamine concentration for both the α4β2 (60 μM) and the α7 (25 μM) with activation by 1 mM ACh, these concentrations were used for further experiments as noted. In each case, the slopes of the ACh dose-response curves were noticeably shallower in the presence of ketamine and the maximal current amplitudes were reduced (Figure 4A,B). To determine whether the inhibition of the α4β2 and α7 nAChRs by ketamine was voltage-dependent, the percent inhibition by ketamine (60 μM for α4β2, 25 μM for α7) when activated by 1 mM ACh, was determined at a range of holding potentials from −100 to −30 mV. Inhibition of the α4β2 nAChR increased markedly with membrane hyperpolarization (Figure 4C) (P<0.0001), whereas there was a much smaller and statistically insignificant effect of membrane potential on ketamine inhibition of the α7 nAChR (Figure 4D). To determine whether inhibition by ketamine was dependent on channel activation, we measured peak current responses to repeated applications of ACh, at 5 min intervals, during a continuous application of ketamine. Inhibition of the peak current from the α4β2 nAChR increased significantly with the second and third application of agonist (ANOVA, P<0.05) (Figure 4E). In contrast, there was no increment in the degree of inhibition of the α7 nAChR by ketamine, with repeated agonist application (Figure 4F).
Figure 4.
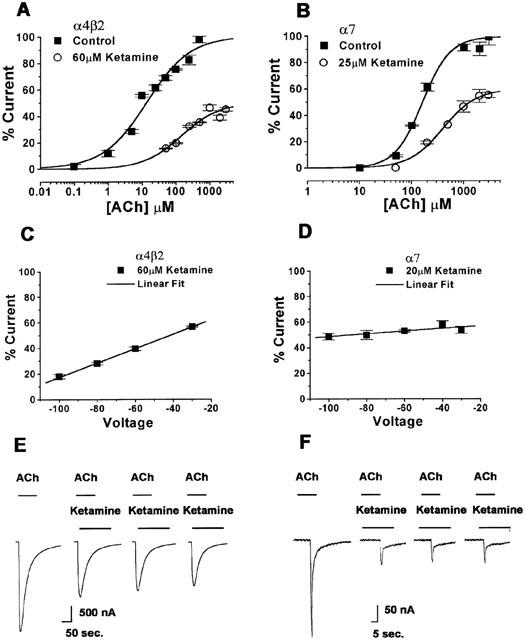
Ketamine inhibits ACh responses of the α4β2 and α7 nAChRs in a noncompetitive manner and this inhibition is voltage- and use-dependent in the α4β2 subtype. ACh dose-response curves were normalized to the average response to a saturating concentration of ACh in the absence of ketamine. (A) A concentration-response curve for the activation of α4β2 by varying concentrations of ACh in the absence and presence of 60 μM ketamine. Membrane potential was held at −60 mV (n=5 – 6). (B) A concentration-response curve for the activation of α7 by varying ACh concentrations with and without 25 μM ketamine. Membrane potential was held at −60 mV (n=5 – 6). (C) Voltage response relationship for the α4β2 nAChR inhibition by 60 μM ketamine when activated by 1 mM ACh. At more negative membrane potentials, inhibition is more pronounced (P<0.0001) (n=4 – 7). (D) Voltage response relationship for the α7 nAChR inhibition by 20 μM ketamine when activated by 1 mM ACh. There is no significant voltage-dependence as the slope is not significantly different from zero (P>0.05) (n=4 – 7). (E) Current traces of α4β2 in response to 1 mM ACh before the ketamine application (first), during the first 50 μM ketamine application (second), during the second and third applications of continuous ketamine exposure (third and fourth figures). (F) Current traces of the α7 subtype in response to 1 mM ACh before the ketamine application (first), during the first 50 μM ketamine application (second), during the second and third applications of continuous ketamine exposure (third and fourth figures). Values are mean±s.e.
Mechanism of BCl inhibition of α4β2 and α7 nAChRs
Concentration-response curves for ACh were constructed in the absence and presence of BCl for both nAChR subtypes (Figure 5A,B). In the case of α4β2, the maximal response to ACh in the presence of BCl was only about 50% of the maximal response of the curve without BCl. To determine if BCl inhibition was dependent on voltage, the inhibition by 55 nM of BCl when activated by 1 mM ACh, was determined at a range of holding potentials from −100 to −30 mV. There was only a small, statistically insignificant effect of membrane potential on either the α4β2 or the α7 nAChRs (Figure 5C,D). To determine whether inhibition by BCl was dependent on channel activation, peak current responses were measured with repeat applications of ACh in the continuous presence of BCl. Similarly, there was no statistically significant difference in the degree of inhibition by BCl with repeated agonist applications in either subtype (Figure 5E,F).
Figure 5.
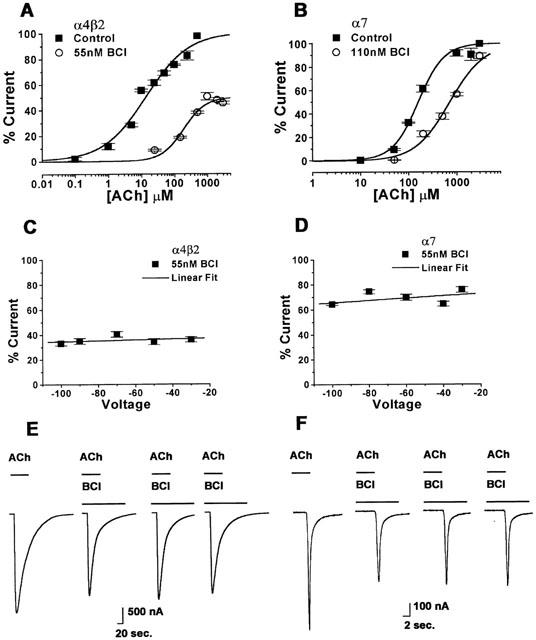
Benzethonium chloride inhibits acetylcholine responses in the α7 nAChRs in a mixed competitive and non-competitive manner but there is no voltage- or use-dependence of the response in either subtype. ACh dose-response curves were normalized to the average response to a saturating concentration of ACh in the absence of ketamine. (A) A concentration-response curve for the activation of the α4β2 nAChR with varying concentrations of ACh in the absence and presence of 55 nM BCl. Membrane potential was held at −60 mV (n=5 – 7). (B) A concentration-response curve for the activation of the α7 nAChR by varying ACh concentrations with and without 110 nM BCl. Membrane potential was held at −60 mV (n=5 – 7). (C) Voltage response relationship for the α4β2 nAChR inhibition by 55 nM BCl when activated by 1 mM ACh. There is no significant voltage-dependence (P>0.05) (n=5 – 7). (D) Voltage response relationship for the α7 nAChR inhibition by 55 nM BCl when activated by 1 mM ACh. There is no significant voltage-dependence (P>0.05) (n=5 – 7). (E) Current traces from the α4β2 nAChR in response to 1 mM ACh before BCl application (first), during the first 5 nM BCl application (second), during the second and third applications of continuous BCl exposure (third and fourth figures). (F) Currents traces from the α7 nAChR in response to 1 mM ACh before the BCl application (first), during the first 100 nM BCl application (second), during the second and third applications of continuous BCl exposure (third and fourth figures). Values are mean±s.e.
Interaction between ketamine and BCl
When ketamine and BCl were combined, the inhibitory effect on the α4β2 nAChR was less than the sum of the inhibition by each compound alone (Figure 6A). In contrast, the combination had the same inhibitory effect as the sum of the two component effects on the α7 nAChR (Figure 6B).
Figure 6.
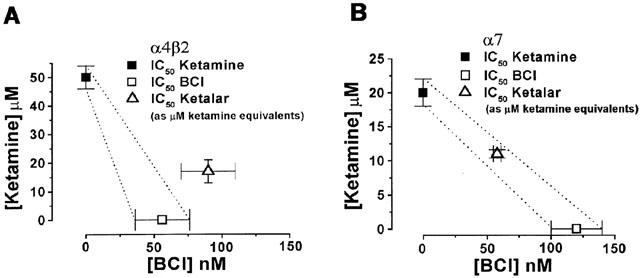
A graphical analysis of ketamine and benzethonium chloride's interactions as seen in the α4β2 and α7 nAChRs. (A) Using 1 mM ACh as the agonist, the IC50 concentrations of ketamine alone, BCl alone and the combined drugs for inhibition of the α4β2 nAChR are plotted. The 95% confidence intervals for additivity are displayed as dotted lines. A subadditive effect between ketamine and BCl for the α4β2 subtype is shown. (B) Using 1 mM ACh as the agonist, the IC50 concentrations of ketamine alone, BCl alone and the combined drugs for inhibition of the α7 nAChR are plotted. The 95% confidence intervals for additivity are displayed as dotted lines. The IC50 concentrations for the combined drugs fall within the 95% confidence intervals, suggesting additivity. Error bars represent standard error.
Discussion
The α4β2 and α7 nAChRs were chosen for our study because of their wide distribution in the CNS, including the hippocampus, thalamus and hypothalamus. In addition, these two nAChRs represent heteromeric and homomeric subtypes, which were shown to be differentially sensitive to volatile anaesthetics (Flood et al., 1997; Violet et al., 1997). As nAChR activation has been implicated in attention, arousal and analgesia, there is a potential role for these receptors in mediating the behavioural effects of ketamine such as hypnosis, amnesia, analgesia and autonomic regulation.
Ketamine produces an anaesthetic state at concentrations from 2.7 – 4.7 μM (Little et al., 1972; Idvall et al., 1979). Whereas, analgesia occurs at lower ketamine concentrations (Grant & Lovinger, 1995). Ketamine has previously been shown to inhibit the chick and human heteromeric nAChRs as well as those expressed in PC12 cells (Flood & Krasowski, 2000; Sasaki et al., 2000; Yamakura et al., 2000). In these studies we have demonstrated that unlike the volatile anaesthetics, ketamine inhibits the homomeric human α7 nAChR. In similar studies, Yamakura et al. (2000) reported less inhibition by ketamine of human α4β2 nAChRs, with an IC50 concentration of 72 μM. Whereas, their studies used an EC50 ACh concentration, the present study used a 1 mM saturating ACh concentration. If ketamine were acting as a pure open channel blocker, more potent inhibition would be expected at the higher agonist concentrations. In our experiments, we found more effective inhibition at lower agonist concentrations, suggesting a mixed mechanism for inhibition by ketamine. Additionally in our experiments on the α4β2 nAChR, voltage- and use-dependence were seen in the ACh response in the presence of ketamine (Figure 4C,E). These results differed from the results of Yamakura et al. (2000) which did not demonstrate use-dependence. This variation may be due to subtle differences in channel regulation. Conceivably, differences in subunit expression levels could result in altered stoichiometry of a heteromeric receptor. In the α7 nAChR, inhibition by ketamine of the ACh response was neither voltage- nor use-dependent (Figure 4D,F). Previous studies have demonstrated that ketamine acts via the open and closed states of the channel in the muscle type nAChRs (Scheller et al., 1996). On the NMDA receptor, ketamine acts both as an open channel blocker (Honey et al., 1985; MacDonald et al., 1991) and as an allosteric inhibitor (Orser et al., 1997). We can deduce from both previous studies and from the evidence of voltage- and use-dependence in our own studies, that ketamine is acting predominantly as an open channel blocker in the α4β2. As a result of the weak voltage-dependence and the lack of significant use-dependence in the α7 nAChR, ketamine may be acting in a more superficial and exposed portion of the channel lumen, accessible when the channel is in the closed state.
Although only about 50 nM of the preservative BCl is found in a 10 μM formulation of Ketalar, we found this concentration to be quite potent alone. Additionally, when Ketalar was tested the concentration-response curve was shifted showing greater inhibition by ketamine with BCl than without. In the α7 nAChR, the presence of BCl caused the log concentration-response curve for the action of ACh to shift to the right, as would be expected if BCl was acting as a competitive antagonist. This would be in keeping with the structural similarity between BCl and ACh. In contrast, BCl reduced the maximum response of α4β2 nAChR, suggesting a noncompetitive mechanism possibly involving allosteric inhibition.
Our isobolographic analysis of the interaction between ketamine and BCl suggests that the two compounds inhibit the α7 nAChR in an additive manner, while the interaction at the α4β2 nAChR suggests subadditivity. This subadditivity of the interaction at the α4β2 nAChR may be secondary to a partially overlapping site of action. This could be important since BCl is known to cause toxicity (Budavari, 1996; Elder, 1985) but nevertheless is used as a preservative in Ketalar in the United States. Although BCl is a permanently charged molecule, the fact that high concentrations cause coma and convulsions suggests that it has at least some access to the brain (Budavari, 1996; Elder, 1985). Since BCl on its own causes inhibition, it may increase the effect of ketamine when the two are used in combination.
Unlike volatile anaesthetics, ketamine inhibits the α7 type nAChR at clinically relevant concentrations. The α7 type nAChRs, like the NMDA receptors, are highly calcium permeable and thought to be involved in neuronal facilitation. Their inhibition by ketamine and BCl may cause some of the effects and side effects found with the clinical administration of Ketalar.
Acknowledgments
Supported by NIH GM00695 (P. Flood). The authors wish to thank Dr Jon Lindstrom for the human α7 and α4β2 nACh receptor clones, Dr Steve Siegelbaum for the availability of oocytes, Dr Andrew Jenkins and Dr Neil Harrison for their critical review of this manuscript.
Abbreviations
- BCl
benzethonium chloride
- nAChRs
neuronal nicotinic acetylcholine receptors
References
- ANIS N.A., BERRY S.C., BURTON N.R., LODGE D. The dissociative anaesthetics, ketamine and phencyclidine, selectively reduce excitation of central mammalian neurones by N-methylaspartate. Br. J. Pharmacol. 1983;79:565–575. doi: 10.1111/j.1476-5381.1983.tb11031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUDAVARI S.Benzethonium chloride Merck Index 12th edition 1996Whitehouse Station, NJ: Merck; ed. Budavari, S. pp1103 [Google Scholar]
- DURIEUX M.E. Inhibition by ketamine of muscarinic acetylcholine receptor function. Anesth. Analg. 1995;81:57–62. doi: 10.1097/00000539-199507000-00012. [DOI] [PubMed] [Google Scholar]
- DURIEUX M.E., NIETGEN G.W. Synergistic inhibition of muscarinic signaling by ketamine stereoisomers and the preservative benzethonium chloride. Anesthesiology. 1997;86:1326–1333. doi: 10.1097/00000542-199706000-00014. [DOI] [PubMed] [Google Scholar]
- ELDER R.L. Safety assessment of cosmetic ingredients, final report on the safety assessment of benzethonium chloride and methylbenzethonium chloride. J. Am. Coll. Toxicol. 1985;4:65–106. [Google Scholar]
- FLOOD P., KRASOWSKI M. Intravenous anesthetics differentially modulate ligand-gated ion channels. Anesthesiology. 2000;92:1418–1425. doi: 10.1097/00000542-200005000-00033. [DOI] [PubMed] [Google Scholar]
- FLOOD P., RAMIREZ-LATORRE J., ROLE L. Alpha 4 beta 2 neuronal nicotinic acetylcholine receptors in the central nervous system are inhibited by isoflurane and propofol, but alpha 7-type nicotinic acetylcholine receptors are unaffected. Anesthesiology. 1997;86:859–865. doi: 10.1097/00000542-199704000-00016. [DOI] [PubMed] [Google Scholar]
- FRIEDERICH P., DYBEK A., URBAN B.W. Stereospecific interaction of ketamine with nicotinic acetylcholine receptors in human sympathetic ganglion-like SH-SY5Y cells. Anesthesiology. 2000;93:818–824. doi: 10.1097/00000542-200009000-00032. [DOI] [PubMed] [Google Scholar]
- FURUYA R., OKA K., WATANABE I., KAMIYA Y., ITOH H., ANDOH T. The effects of ketamine and propofol on neuronal nicotinic acetylcholine receptors and P2x purinoceptors in PC12 cells. Anesth. Analg. 1999;88:174–180. doi: 10.1097/00000539-199901000-00033. [DOI] [PubMed] [Google Scholar]
- GRANT K.A., LOVINGER D.M. Cellular and behavioral neurobiology of alcohol: receptor-mediated neuronal processes. (Review) Clin. Neurosci. 1995;3:155–164. [PubMed] [Google Scholar]
- HARRISON N.L., SIMMONDS M.A. Quantitative studies on some antagonists of N-methyl D-aspartate in slices of rat cerebral cortex. Br. J. Pharmacol. 1985;84:381–391. doi: 10.1111/j.1476-5381.1985.tb12922.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HONEY C.R., MILJKOVIC Z., MACDONALD J.F. Ketamine and phencyclidine cause a voltage-dependent block of responses to L-aspartic acid. Neurosci. Lett. 1985;61:135–139. doi: 10.1016/0304-3940(85)90414-8. [DOI] [PubMed] [Google Scholar]
- IDVALL J., AHLGREN I., ARONSEN K.R., STENBERG P. Ketamine infusions: pharmacokinetics and clinical effects. Br. J. Anaesth. 1979;51:1167–1173. doi: 10.1093/bja/51.12.1167. [DOI] [PubMed] [Google Scholar]
- LITTLE B., CHANG T., CHUCOT L., DILL W.A., ENRILE L.L., GLAZKO A.J., JASSANI M., KRETCHMER H., SWEET A.Y. Study of ketamine as an obstetric anesthetic agent. Am. J. Obstet. Gynecol. 1972;113:247–260. doi: 10.1016/0002-9378(72)90774-0. [DOI] [PubMed] [Google Scholar]
- MACDONALD J.F., BARTLETT M.C., MODY I., PAHAPILL P., REYNOLDS J.N., SALTER M.W., SCHNEIDERMAN J.H., PENNEFATHER P.S. Actions ketamine, phencylcidine and MK-801 on NMDA receptor currents in cultured mouse hippocampal neurones. J. Physiol. 1991;432:483–508. doi: 10.1113/jphysiol.1991.sp018396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ORSER B.A., PENNEFATHER P.S., MACDONALD J.F. Multiple mechanisms of ketamine blockade of N-methyl-D-aspartate receptors. Anesthesiology. 1997;86:903–917. doi: 10.1097/00000542-199704000-00021. [DOI] [PubMed] [Google Scholar]
- SASAKI T., ANDOH T., WATANABE I., KAMIYA Y., ITOH H., HIGASHI T., MATSUURA T. Nonstereoselective inhibition of neuronal nicotinic acetylcholine receptors by ketamine isomers. Anesth. Analg. 2000;91:741–748. doi: 10.1097/00000539-200009000-00046. [DOI] [PubMed] [Google Scholar]
- SCHELLER M., BUFLER J., HERTLE I., SCHNECK H.J., FRANKE C., KOCHS E. Ketamine blocks currents through mammalian nicotinic acetylcholine receptor channels by interaction with both the open and the closed state. Anesth. Analg. 1996;83:830–836. doi: 10.1097/00000539-199610000-00031. [DOI] [PubMed] [Google Scholar]
- SUMIKAWA K., MATSUMOTO T., AMENOMORI Y., HIRANO H., AMAKATA Y. Selective actions of intravenous anesthetics on nicotinic- and muscarinic-receptor-mediated responses of the dog adrenal medulla. Anesthesiology. 1983;59:412–416. doi: 10.1097/00000542-198311000-00008. [DOI] [PubMed] [Google Scholar]
- TALLARIDA R.J., PORRECA F., COWAN A. Statistical analysis of drug-drug and site-site interactions with isobolograms. Life Sci. 1989;45:947–961. doi: 10.1016/0024-3205(89)90148-3. [DOI] [PubMed] [Google Scholar]
- VIOLET J.M., DOWNIE D.L., NAKISA R.C., LIEB W.R., FRANKS N.P. Differential sensitivities of mammalian neuronal and muscle nicotinic acetylcholine receptors to general anesthetics. Anesthesiology. 1997;86:866–874. doi: 10.1097/00000542-199704000-00017. [DOI] [PubMed] [Google Scholar]
- WACHTEL R.E., WEGRZYNOWICZ E.S. Kinetics of nicotinic acetylcholine ion channels in the presence of intravenous anaesthetics and induction agents. Br. J. Pharmacol. 1992;106:623–627. doi: 10.1111/j.1476-5381.1992.tb14385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMAKURA T., CHAVEZ-NORIEGA L.E., HARRIS R.A. Subunit-dependent inhibition of human neuronal nicotinic acetylcholine receptors and other ligand-gated ion channels by dissociative anesthetics ketamine and dizocilpine. Anesthesiology. 2000;92:1144–1153. doi: 10.1097/00000542-200004000-00033. [DOI] [PubMed] [Google Scholar]
- ZAMAN Z., SPEELEVELD E., SNEYERS L., DESMET K. Inhibition of acetylcholine esterase and choline esterase by benzethonium chloride and avoidance of the benzethonium chloride carry-over inhibitory effect. Eur. J. Clin. Chem. Clin. Biochem. 1997;35:603–607. doi: 10.1515/cclm.1997.35.8.603. [DOI] [PubMed] [Google Scholar]