

Abstract
In starved mice, the anorectic activity of methylamine (MET) and benzylamine (BZ), both substrates of semicarbazide-sensitive benzylamine oxidases (Bz-SSAO), was compared with that of the potassium channel blocking agents charybdotoxin (ChTX), tetraethylammonium (TEA), gliquidone (GLI), ammonium chloride (NH4+) and of the anoressants amphetamine (AMPH) and nicotine (NIC). After i.c.v. administration, an approximate ranking order of potency was: ChTX⩾AMPH>NIC=TEA⩾GLI⩾MET>BZ>NH4+.
Clorgyline (2.5 mg kg−1 i.p.) or deprenyl (10 mg kg−1 i.p.) potentiated the anorectic effect of i.c.v.-administered BZ, NIC and AMPH. The effect of TEA was increased only by deprenyl, while MET, NH4+, ChTX and GLI were not affected by either of the inhibitors.
The Bz-SSAO inhibitors α-aminoguanidine (50 mg kg−1 i.p.), B24 (100 mg kg−1 i.p.) and MDL 72274 (2.5 mg kg−1 i.p.) potentiated the effect of i.p., but not of i.c.v.-administered MET.
Antisense oligodeoxyribonucleotides (aODN) to Kv1.1 potassium channels abolished the effect of BZ and TEA, but was ineffective in reducing the activity of MET and other compounds.
These results suggest that MET is endowed with peculiar hypophagic effects at dosage levels that are not able to affect gross behaviour in mice. The effect of MET, differently from BZ, seems unrelated to an increase in the central release of monoaminergic mediators, as well as to a Kv1.1 blocking activity. Through a reduction of the endogenous breakdown of MET, Bz-SSAO inhibitors enhance the central pharmacological activity of this amine.
Keywords: Food consumption, methylamine, semicarbazide-sensitive benzylamine oxidases, K+ channels, Kv1.1, antisense oligodeoxyribonucleotide
Introduction
Methylamine (MET) is a short aliphatic amine that is normally present in several mammals, including humans. This compound, which can be detected in blood and urine, may derive endogenously from the metabolism of adrenaline, sarcosine, creatinine and lecithin. In addition, it has been reported that MET may also derive from exogenous compounds, such as nicotine, or be directly ingested from food and drink (Zeisel & Dacosta, 1986). Nevertheless, although MET has been known from many years, its physiopathological role has still not been completely clarified. It is known that the urinary excretion of MET is enhanced in particular physiological conditions that are characterized by an increase in creatine elimination, including pregnancy, parturition and muscular exertion (Dar et al., 1985; Precious et al., 1988). It has also been reported that the amine increases in typhoid fever, diabetes, hepatic or renal insufficiency where it is suspected of generating a variety of central or peripheral effects.
In the body, MET is deaminated to hydrogen peroxide and formaldehyde by semicarbazide-sensitive benzylamine oxidases (Bz-SSAO) (EC 1.4.3.6), a group of amine oxidases found predominantly in the vascular tissues, fat and connective tissue, but virtually absent in the central nervous system. Its physiological role, moreover is not completely understood (Lewinson et al., 1978; Buffoni, 1995; Castillo et al., 1998). Tissutal Bz-SSAO, which share the xenobiotic compound benzylamine (BZ) with MAO B as their best substrate (Buffoni, 1995), is insensitive to the MAO (EC 1.4.3.4) inhibitors clorgyline and deprenyl but is inhibited by semicarbazide or α-aminoguanidine (AG) and by the highly selective inhibitors 3,5-ethoxy-4-aminomethylpyridine (B24) (Buffoni et al., 1998) or some allylamine derivatives including (E)-2-phenyl-3-chloroallylamine (MDL 72274) (Ekblom, 1998).
MET, together with Bz-SSAO, is suspected of having possible relevance in diabetic cardiovascular degeneration (Yu & Zuo, 1993). This view seems to be confirmed by the observation that Bz-SSAO inhibitors have a protective role in this pathology (Yu & Zuo, 1997), probably by reducing the formation of the angiotoxic compound formaldehyde. In general, however, the inhibition of Bz-SSAO should also be considered as a condition capable of improving the potential biological activity of its own substrates. This should be of particular interest in every condition in which the formation of biologically active, endogenous substrates of Bz-SSAO, such as MET, is increased (Lyles & McDougall, 1989; Yu & Zuo, 1997). Recently, this view has received some indirect experimental support from the observation that, when administered i.p., BZ displays a novel central hypophagic activity in mice only after pretreatment with the Bz-SSAO inhibitors B24 or MDL 72274. We have also observed that BZ induces its effects probably by acting like the potassium channel blockers TEA or 4-aminopyridine (Pirisino et al., 1993; Banchelli et al., 2000; 2001). In other investigations, it has been found (Ghelardini et al., 1997) that potassium channel blockers reduce food intake in mice by acting as depolarizing compounds in the brain. The central role of potassium channels has been studied using aODN against the gene that encodes the expression of delayed rectifier K+ channels (Ik) in the cells (Hopkins & Tempel, 1992). It has been found that repeated i.c.v. administration of aODN, targeting the translation start region of the mKv1.1 channel, induced hyperphagic behaviour together with a transient specific reduction of the expression of this channel subtype in the brain.
MET has recently been reported as blocking outward potassium conductance (Ik) in intestinal epithelial cells and, similarly to the anorectic compound amphetamine (AMPH), as promoting Ca2+ dependent exocytosis in chromaffin cells isolated from adrenal medulla (Mundorf et al., 1999). Taken together, these results suggest to us that MET, like BZ and other well-known potassium channel blockers, may be endowed with hypophagic activity as well. We reasoned that, if our supposition is correct, this property could have a role, for example, in diabetes, where high levels of circulating MET are found and a reduction of dietary caloric intake would be beneficial. In this hypothesis, new selective inhibitors of Bz-SSAO would gain more attention as pharmaceutical compounds of potential interest as enhancers of the central effects of MET.
On the basis of these considerations, we decided to extend to MET the investigations previously made for assessing the hypophagic activity of BZ in mice.
In the present work we have compared the anorectic activity of MET with that of NH4+, TEA, GLI or ChTX, all compounds known to block different types of potassium channels (Aronson, 1992; Hrnjez et al., 1999). In addition, the effect of NIC or AMPH, anorexic compounds that are both generally regarded as having a mechanism of action different from that of potassium channel blockers, was evaluated. The experiments were conducted in the absence or presence of selective AO inhibitors, in order to clarify the potential role of Bz-SSAO inhibition in enhancing the activity of these compounds.
In an attempt to elucidate a potassium channel blocker activity potentially involved in the hypophagic effect of MET, BZ and the reference compounds, we have also planned to study the antagonism induced in mice by pretreatment with aODN against the Kv1.1 channel subtype.
Methods
Animals
Male Swiss albino mice (25 – 30 g) from the Morini (San Polo d'Enza, Italy) breeding farm were used. Fifteen mice were housed per cage. The cages were placed in the room of the experiment 24 h before the test for adaptation. The animals were kept at 23±1°C with a 12 h light – dark cycle, light on at 0700, and were fed a standard laboratory diet with water ad libitum. All experiments were carried out in accordance with the European Communities Council Directive of 24 November 1986 (86/609/EEC) for experimental animal care.
Intracerebroventricular injection technique
Intracerebroventricular (i.c.v.) administration was performed under ether anaesthesia, according to the method described by Haley & McCormick (1957). Briefly, under anaesthesia, the mice were grasped firmly by the loose skin behind the head. A 0.4 mm external diameter, hypodermic needle attached to a 10 micro syringe was inserted perpendicularly through the skull, no more than 2 mm into the brain of the mouse, where 5 μl were then administered. The injection site was 1 mm to the right or left from the midpoint, on a line drawn through to the anterior base of the ears. Injections were performed randomly into the right or left ventricle. To make sure that the drugs had been administered exactly into the cerebral ventricle, some mice were injected with 5 μl of 1 : 10 diluted India ink and their brains were examined macroscopically after sectioning. When i.c.v. administration was used, it was previously assessed that the pH values of the nM compound solutions, ranging from 7.2 to 6.7, did not vary significantly from that of the saline (pH=6.8±0.4).
Evaluation of food consumption
The mice did not have access to food for 12 h, but water was available ad libitum. A weighed amount of food (standard laboratory pellets) was given, and the weight consumed (evaluated as the difference between the original amount and the food left in the cage, including spillage) was measured 15, 30, 45 and 60 min after i.c.v. or i.p. administration of saline or drug solutions, with an accuracy of 0.1 g. An arbitrary cut-off time of 60 min was used. To obtain selective inhibition of monoamine oxidase A and B activities in mouse brain, we administered 2.5 mg kg−1 i.p. clorgyline or 10 mg kg−1 deprenyl (Finnegan et al., 1995). With regard to the dosage regimen of MDL 72274, B24 or AG as inhibitors of semicarbazide-sensitive benzylamine oxidases, the same dosage (2.5 mg kg−1 i.p.; 100 mg kg−1; 50 mg kg−1 respectively) previously found to be effective for these enzymes (Yu & Zuo, 1997; Banchelli et al., 2001) was given i.p. to the mice. Each inhibitor was administered i.p. to the mice 3 h before the injection (i.c.v. or i.p.) of either saline or test compounds.
Antisense oligonucleotides
24mer phosphodiester oligonucleotides (ODNs) were capped by a terminal phosphorothioate double substitution and purified by high-performance liquid chromatography (HPLC; Genosys, The Woodlands, TX, U.S.A.). The antisense oligonucleotide (5′-CGA CAT CAC CGT CAT GAT GAA AGG-3′) was designed by targeting the 5′ portion of the murine Kv1.1 mRNA, residues 575 – 598 of the published cDNA sequence (Chandy et al., 1990). To evaluate the specific, antisense effects of the oligodeoxynucleotides, a fully degenerated phosphorodiester phosphorothioate-capped oligonucleotide was used as negative control. The fully degenerated 24mer is a collection of about 3×1014 different molecular species. Therefore, for the nanomolar-micromolar range concentrations obtained in antisense experiments, every oligonucleotide of a defined sequence was present at the site of action in a sub-attomolar concentration, which is totally insufficient for any antisense effect. For more details on the aODN and dODN synthesis and the RT – PCR analysis of mKv1.1 mRNA in the mouse brain tissue the reader may refer to our previous papers (Ghelardini et al., 1997; Galeotti et al., 1997).
Administration of antisense oligonucleotide
In experiments in vivo, to favour cell penetration and to minimize the degradation of the oligonucleotides, phosphorothioate-capped phosphorodiester oligonucleotides associated with an artificial cationic lipid (DOTAP) to enhance cellular uptake, has been proved to be suitable (Quattrone et al., 1994). Mice were randomly assigned to an antisense oligonucleotide (aODN), degenerated oligonucleotide (dODN), vector (DOTAP; N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium methyl sulphate), saline or naïve group. A suitable amount of oligonucleotide was preincubated at 37°C for 30 min with 13 μM DOTAP. Each group received a single intracerebroventricular (i.c.v.) injection (5 μl; 3 nmol of ODN) on days 1, 4 and 7.
Rota rod test
The apparatus consisted of a base platform and a rotating rod with a diameter of 3 cm and non-skid surface. The rod was placed at a height of 15 cm from the base. The rod, 30 cm in length, was divided into five equal sections by six disks. Up to five mice were then tested simultaneously on the apparatus, with a rod rotation speed of 16 r.p.m. The integrity of motor coordination was assessed on the basis of the number of falls from the rod in 30 s. Performance time was measured before and 15, 30, 45 and 60 min after the i.c.v. administration of saline or drug solutions.
Reagents and drugs
Oligonucleotides used for the antisense strategy were from Genosys (The Woodlands, U.S.A.). DOTAP was from Boheringer-Mannheim (Mannheim, Germany). Deprenyl hydrochloride, clorgyline hydrochloride, benzylamine hydrochloride, methylamine hydrochloride, amphetamine sulphate, tetraethylammonium HCl, charybdotoxin, nicotine and gliquidone were purchased from the Sigma Chemical Company (St. Louis, MO, U.S.A.). MDL 72274 was a gift of the Marion Merrell Dow Research Institute (Strasbourg Cedex, France). B24 was synthesized by Prof V. Bertini (Department of Organic Chemistry of the University of Genoa).
The drugs were dissolved in isotonic (NaCl 0.9%) saline solution; drug concentrations were prepared in such a way that the necessary dose could be administered in a volume of 5 μl/mouse by i.c.v. injection and 10 ml kg−1 by i.p. injection. Antisense and degenerated oligonucleotides were dissolved in the vector (DOTAP) at least 30 min before injection.
Statistical analysis
All experimental results are expressed as the mean±s.e.mean. Analysis of variance (ANOVA), followed by Fisher's Protected Least Significant Difference (PLSD) procedure for post hoc comparison, were used to verify significance between two means. Data were analysed using the StatView software for Machintosh (1992). The fitting of the sigmoid dose-response curves and the ED50 values with their confidence limits (C.L.), were obtained from a non-linear regression analysis (Prism program, Graph Pad Software Inc., San Diego, CA, U.S.A.).
Results
Food intake behaviour
In the mice starved for 12 h, 15 μg MET given i.c.v. significantly reduced food consumption, as compared to the controls in a 60 min test. At this dosage (Table 1), MET was more active, as a hypophagic compound, than BZ (30 μg), NH4+ (12 μg), TEA (5 μg), ChTX (1 μg), GLI (6 μg) or NIC (5 μg) were. From the dose-response relationship (Figure 1) an ED50 value was calculated of 146.3 nmol/mouse (CL=36.2 – 591.1) and 63.2 nmol/mouse (CL=13.7 – 262.9), for BZ and MET, respectively. The i.p. pretreatment of mice with clorgyline (2.5 mg kg−1) or deprenyl (10 mg kg−1) to selectively inhibit MAO A or MAO B activities (Banchelli et al., 2001), did not affect the basal food consumption of the controls, but differently modified the anorectic effect of some i.c.v.-administered compounds. Namely, the anorectic effect of BZ, AMPH and NIC was potentiated by clorgyline (40, 67 and 18% respectively) and deprenyl (64, 88 and 27% respectively), the effect of TEA only by deprenyl (64%), while the activity of MET, ChTX, GLI remained completely unmodified after selective MAOs inhibition (Table 1). After the i.p. pretreatment with MDL 72274, the anorectic effect of MET given i.c.v. was unmodified; on the contrary, this inhibitor significantly potentiated the hypophagic effect of MET when this compound was administered i.p. (Figure 2). The EC50 values for MET were reduced from 334.6 mg kg−1 (CL=280.8 – 398.8) to 43.05 mg kg−1 (CL=38.51 – 48.13) in controls and MDL 72274 pretreated mice, respectively. Similar results (Figure 2) were also obtained when the Bz-SSAO inhibitors B24 (100 mg kg−1) or AG (50 mg kg−1) was given i.p. to mice; again, the EC50 values for MET were reduced approximately to 45.72 mg kg−1 and 37.68 mg kg−1 respectively.
Table 1.
Anorectic effect of MET, BZ and other treatments in mice food-deprived for 12-h
Figure 1.
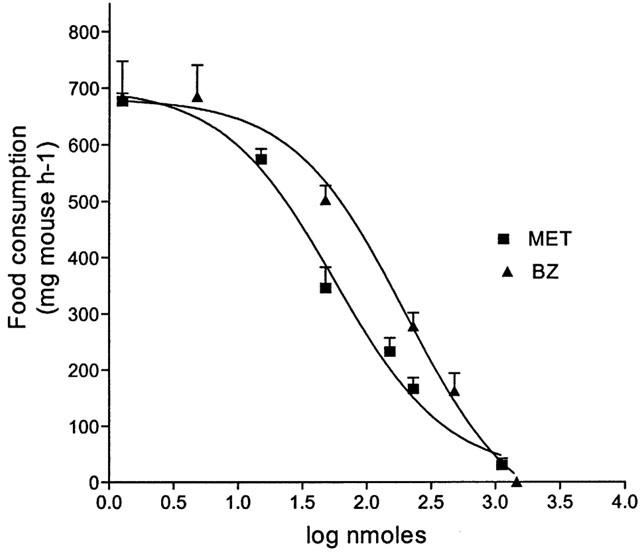
Dose-food consumption curves of i.c.v. injected MET, in mice food-deprived for 12-h, as compared to the anorectic effect of BZ. Each point represents the mean±s.e.mean for 10 – 20 mice.
Figure 2.
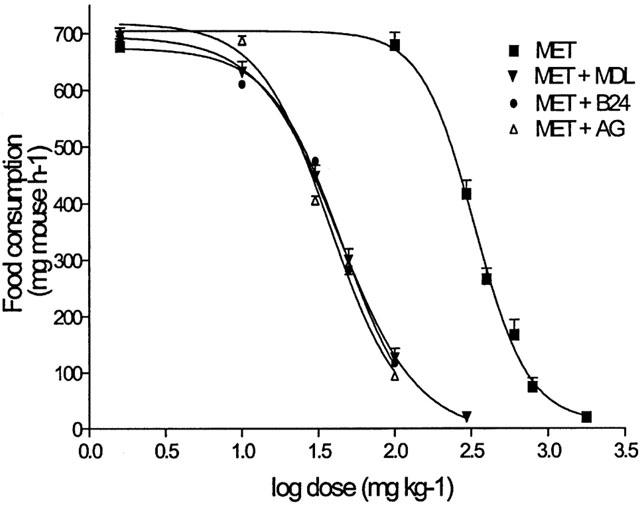
Shift to the left of the dose-food consumption curves of i.p.-injected MET, in mice food-deprived for 12-h by the inhibition of semicarbazide-sensitive benzylamine oxidases (B24 100 mg kg−1; MDL 72274 2.5 mg kg−1; AG 50 mg kg−1). Mice were injected i.p. with saline or MET solution 15 min before the test. Amine oxidase inhibitors were administered 60 min before treatment with MET. Each point represents the mean±s.e.mean for 10 – 20 mice.
Effect of aODN to mKv1.1 pretreatments
The effect induced by repeated administration of aODN against mKv1.1 on the anorectic activity of MET compared to those of BZ and other reference compounds was investigated in food-deprived mice. The experiments were performed 48 h after the last aODN administration, because at this time a substantial reduction (>70%) in Kv1.1 mRNA levels was previously obtained in brain homogenates, which returned to control levels only after 7 days (Ghelardini et al., 1997)
In our experiments, the i.c.v. injection of 3 nmol of aODN as well as of dODN, as negative controls, did not modify food intake in comparison with the vehicle-treated mice. On the contrary, the aODN decreased the hypophagic effect induced by BZ and TEA or AMPH, while the same treatment did not affect the anorectic activity of MET, NH4+, NIC, ChTX or GLI (Table 2). The hypothesis that the aODN pretreatment effectively decreased the hypophagic effect of some compounds through a reduction in the Kv1.1 channel expression was further confirmed by the recovery of the anorectic effect of 5 μg TEA administered 7 days after the last of aODN pretreatment (food consumption=674±25 mg per mouse after 48 h, in comparison with 365±18 mg per mouse after 7 days of aODN pretreatment; P<0.01, n=10). It has been previously seen, in fact, that, after 7 days, the inhibitory effect of aODN on Kv1.1 mRNA levels completely disappeared in the brain of the treated mice (Ghelardini et al., 1997).
Table 2.
Effect of the aODN pretreatment on the anorexia induced by MET, BZ and other treatments
Effect of treatments on mouse behaviour
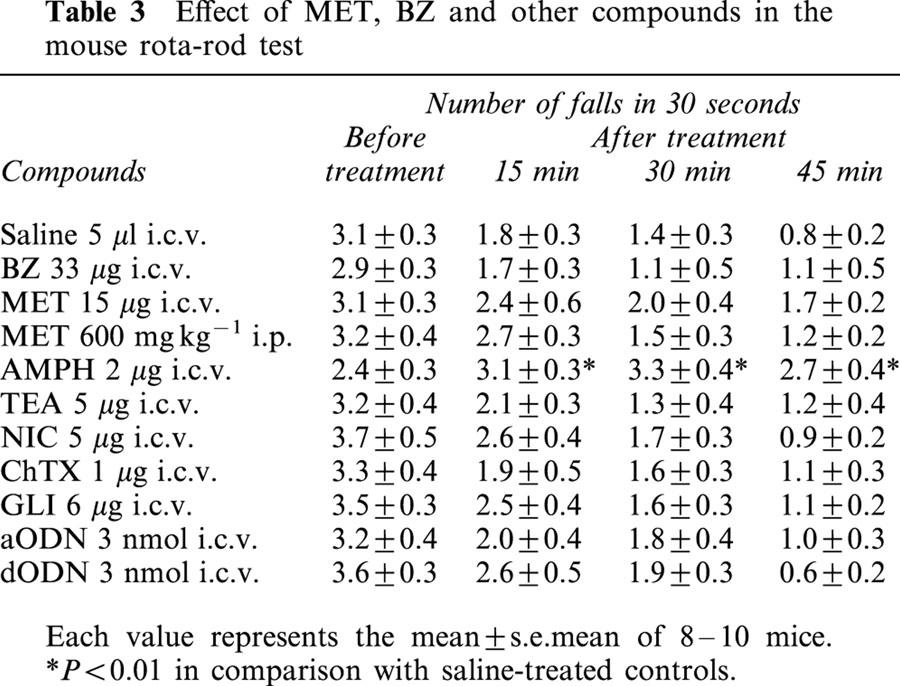
The motor coordination in mice after different pharmacological treatments was evaluated by the rota-rod test starting 15 min after the i.c.v. injection of the test compounds or 48 h after the last ODN injection. As shown in Table 3, with the exclusion of AMPH, the motor coordination of the mice treated with the highest dosage of compounds used in food intake experiments was not modified. In comparison with the control mice, the treated animals reduced the number of falls during observation to a similar extent indicating that they learned how to balance on the rotating rod in a similar manner.
Table 3.
Effect of MET, BZ and other compounds in the mouse rota-rod test
Discussion
We compared the central hypophagic effect of MET, an endogenous substrate of Bz-SSAO, to that of BZ, a synthetic amine deaminated by both Bz-SSAO and MAO B enzymes. It was found that MET induced anorectic effects in mice at lower dosages than those of BZ.
One of the main purposes of this work was to verify the influence of the selective inhibition of MAO A, MAO B and Bz-SSAO, respectively on the pharmacological activity of MET compared to that of BZ and other reference compounds, after clorgyline, deprenyl and MDL 72274 mice pretreatments. To this aim we used the same dosage schedule of the inhibitors previously reported to induce a selective and statistically significant (more than 85%) inhibition of the appropriate AO subtype in the tissues of the mice (Banchelli et al., 2001). Depending on the absence of Bz-SSAO in the brain, the effect of MET, injected directly into cerebral ventricles, was unaffected by pretreatments with MDL 72274, a selective and irreversible inhibitor of these enzymes. Clorgyline or deprenyl were also without effect in modifying the hypophagic activity of MET, while they significantly potentiated the anorexia induced by BZ, TEA or AMPH.
As reported previously in more detail (Banchelli et al., 2001), the observation that the hypophagic effect of BZ is potentiated by both MAO A and MAO B inhibitors has been explained by the inhibition of the deamination of 5HT (MAO A substrate) or DA (MAO B substrate) probably released by BZ in the brain. Furthermore the selective inhibition of MAO B may effectively increase the levels of unchanged BZ active in the brain.
It is known that NIC, TEA or AMPH can reduce food intake in animals by stimulating, with different mechanisms, the neuronal release of either 5HT or DA (Parada et al., 1988; Dawson & Routledge, 1995; Miyata et al., 1999). The observation that the hypophagic effect of these compounds, which are not MAOs substrates, was potentiated by the inhibition of these enzymes can only be explained as a reduction in the oxidative deamination of monoaminergic mediators released by these compounds.
A number of novel mediators have been identified in the central regulation of feeding including, for example, peptidergic compounds like NPY, CCK, CRF, etc (Inui, 2000). Because both clorgyline and deprenyl were without effect in potentiating the hypophagic activity of MET, NH4+, ChTX or GLI, it is possible that, at the dosages employed and differently from AMPH, BZ or TEA, these compounds might preferentially influence the release of some anorectic, non monoaminergic mediators, the nature of which warrants further investigations.
In contrast to the results obtained in i.c.v. experiments, the selective inhibition of Bz-SSAO potentiated the hypophagic effect of MET given i.p. to mice. Once more, this feature seems very similar to the one previously observed with BZ (Banchelli et al., 2001) and confirms that Bz-SSAO could be regarded as scavenger enzymes capable of protecting the central nervous system from the permeation of some centrally active amines.
It is known that circulating MET increases in different types of diabetes, together with the activity of vascular Bz-SSAO (Boor et al., 1992; Meszaros et al., 1999) which oxidize them into formaldehyde and H2O2. There has been considerable debate about the physiopathological significance of these observations. Because formaldehyde is an angiotoxic compound, the high levels of both MET and Bz-SSAO may have a role in the cardiovascular degeneration observed in diabetes, and may contribute to explaining the favourable effects of Bz-SSAO inhibitors in this pathology (Lyles & McDougall, 1989; Hammes et al., 1994; Yu & Zuo, 1997; Ekblom, 1998). Moreover, in considering the hypophagic effect of MET, it can also be hypothesized that the increased levels of this aliphatic amine in diabetes represent a part of the physiological mechanisms involved in the control of hyperglycaemia. Thus, notwithstanding the fact that a hypophagic effect after chronic Bz-SSAO inhibition has not yet been reported, our results could suggest that, through an increase in circulating MET, Bz-SSAO inhibitors, could be useful in diabetes by reducing food consumption and caloric intake. This could be particularly true in insulin-independent diabetes mellitus, a dismetabolic condition that is often associated with obesity, in which increases of both MET levels and Bz-SSAO activity have been observed (Ekblom, 1998).
More than sixty potassium channel subunits have been cloned to date. According to currently accepted nomenclature, the acronym KvX.Y defines a population of voltage-activated channels which are structurally different, for example, to ligand gated (KirX.Y) or Ca2+-operated (mlso) ones (Snyders, 1999).
It is known that TEA blocks different types of potassium channels in neurons, including voltage- and calcium-activated ones (Halliwell, 1990). On the other hand, ChTX has been reported to block currents through the calcium activated potassium channels, while sulphonylureas, such as gliquidone, block KATP channels in different cells, including neurons (Cook, 1988; Edwards & Weston, 1993).
We reported that i.c.v. injections of K+ channel blockers induced a decrease in food consumption in mice, whereas the openers did the opposite. The observation that aODN, which inhibited the expression in the C.N.S. of Shaker-like Kv1.1 potassium channels, also showed orexic properties in starved mice, further indicate a role of these voltage-activated channels in the physiological control of food intake (Ghelardini et al., 1997).
We investigated the possible potassium channel blocking effect of BZ, MET and other hypophagic compounds in mice, using an antisense strategy. The aODN specifically bind to targeted mRNA, preventing translation and/or mediating mRNA cleavage by Rnase and thus down-regulating the synthesis of the encoded protein. The aODN was used assuming that a reduction in the expression of Kv1.1 potassium channels would decrease the pharmacological activity of compounds believed to interact with this molecular target. This strategy has proved to be successful, for example, in studies on the role of potassium channels in the central activity of different analgesic drugs (Chen et al., 1995; Galeotti et al., 1997).
The administration schedule of aODN (a single i.c.v. injection on days 1, 4 and 7 before the administration of compounds) was chosen on the basis of previous results with quantitative RT – PCR analysis that showed that this treatment significantly reduced the expression of Kv1.1 channels in mouse brain (Galeotti et al., 1997). In this regard, it must be mentioned that this aODN pretreatment was unable to further increase food consumption in the animals, which was maximally stimulated after 12 h of fasting.
In our experiments, the anorectic effect of TEA was completely antagonized by aODN. The antagonism was also present for BZ, supporting our previous observation on its action mechanism and further indicating that, similarly TEA and at the dosage used, the effect of these compounds was probably due to a block of the Kv1.1 channel subtype.
It was shown that NH4+, as well as MET, could modify the exocitotic process by blocking potassium channels of different types (Hrnjez et al., 1999) and because of their elevated circulating levels, may be contributory neurodepressant compounds in cirrhotic and uremic patients (Simenoff, 1975; Butterworth, 1992).
Differently from TEA or BZ, the hypophagic effect of NH4+ and MET was not antagonized by the pretreatment with the aODN to mKv1.1 subtype. Therefore, it is possible to hypothesize that both NH4+ and MET induce their hypophagic effect probably by blocking voltage dependent channels different to Kv1.1.
A generally accepted action mechanism for AMPH and for several related psychostimulants proposes that these amines increase intersynaptic catecholamine levels indirectly, acting on vesicular transporters. It has recently been found that AMPH release neuromediators also by increasing the intragranular pH in neurons, thus disrupting the association of catecholamines with Ca2+, ATP and vesicular proteins (Mundorf et al., 1999). We now observed that the aODN pretreatment almost completely prevented the hypophagic effect of AMPH; considering that no previous observations on a possible potassium channel blocking activity of AMPH have yet been reported, these results were unexpected. However, the observation that dexfenfluramine, an anorectic compound structurally related to AMPH and known to be a serotoninergic releasing agent, may act by blocking delayed rectifier K+ channels (Hu et al., 1998) might indirectly support a similar activity of AMPH.
In our opinion, these results are interesting in the view of an alternative mechanism of action of AMPH and structurally related drugs. Moreover, our results suggest that aODN against Kv1.1 potassium channels could be useful tools for investigating the relationship between channel modulation and other behavioural properties of AMPH, such as, for example, central excitation, motor stimulation or memory improvement.
To conclude, in the present work we have demonstrated that MET is endowed with peculiar hypophagic effects at dosage levels that are unable to affect motility and gross behaviour in mice. Indirect evidence indicates that, differently from BZ, the anorectic effect of MET is probably related neither to an increase in the central release of monoaminergic mediators or to a Kv1.1 channel blocking activity. This effect is potently increased by selective Bz-SSAO inhibitors, indicating that, through a reduction in the endogenous breakdown of MET, these agents may improve the central physiopharmacological activity of this amine.
Acknowledgments
The authors wish to thank Professor Franca Buffoni for her critical revision of the manuscript and the Marrion Merrel Dow Research Institute (Strasbourg Cedex, France) for the generous gift of MDL 72274. This work was partially supported by a grant from the MURST (Italy), COFIN 1998.
Abbreviations
- AG
α-aminoguanidine
- NH4+
ammonium chloride
- AMPH
amphetamine
- aODN
antisense oligodeoxyribonucleotide
- BZ
benzylamine
- ChTX
charybdotoxin
- GLI
gliquidone
- MET
methylamine
- Bz-SSAO
semicarbazide-sensitive benzylamine oxidases
- TEA
tetraethylammonium
References
- ARONSON J.K. Potassium channels in nervous tissue. Biochem. Pharmacol. 1992;43:11–14. doi: 10.1016/0006-2952(92)90653-z. [DOI] [PubMed] [Google Scholar]
- BANCHELLI G., RAIMONDI L., GHELARDINI C., PIRISINO R., BERTINI V., DE MUNNO A., LUCCHESINI F. Benzylamine-related compounds stimulate rat vas deferens neurotransmission and potentiate memory in the mouse acting as potassium channel blockers. Pharmacol. Res. 2000;41:151–162. doi: 10.1006/phrs.1999.0571. [DOI] [PubMed] [Google Scholar]
- BANCHELLI G., GHELARDINI C., RAIMONDI L., GALEOTTI N., PIRISINO R. Selective inhibition of amine oxidases differently potentiate the hypophagic effect of benzylamine in mice. Eur. J. Pharmacol. 2001;413:91–99. doi: 10.1016/s0014-2999(01)00739-7. [DOI] [PubMed] [Google Scholar]
- BOOR P.J., TRENT M.B., LYLES G.A., TAO M., ANSARI G.A. Methylamine metabolism to formaldehyde by vascular semicarbazide-sensitive amine oxidase. Toxicology. 1992;73:251–258. doi: 10.1016/0300-483x(92)90067-o. [DOI] [PubMed] [Google Scholar]
- BUFFONI F. Semicarbazide-sensitive amine oxidases: some biochemical properties and general considerations. Prog. Brain Res. 1995;106:323–331. doi: 10.1016/s0079-6123(08)61228-5. [DOI] [PubMed] [Google Scholar]
- BUFFONI F., BERTINI V., DINI G. The mechanism of inhibition of benzylamine oxidase by 3,5-diethoxy-4-aminomethylpyridine (B24) J. Enzyme Inhibition. 1998;13:253–266. doi: 10.3109/14756369809021474. [DOI] [PubMed] [Google Scholar]
- BUTTERWORTH R.F. Pathogenesis, and treatment of portal-systemic encephalopathy: an update. Dig. Dis. Sci. 1992;37:321–327. doi: 10.1007/BF01307722. [DOI] [PubMed] [Google Scholar]
- CASTILLO V., LIZCANO J.M., VISA J., UNZETA M. Semicarbazide-sensitive amine oxidase (SSAO) from human and bovine cerebrovascular tissues: biochemical and immunohistological characterization. Neurochem. Int. 1998;33:415–423. doi: 10.1016/s0197-0186(98)00045-x. [DOI] [PubMed] [Google Scholar]
- CHANDY K.G., WILLIAMS C.B., SPENCER R.H., AGUILAR B.A., GHANSHANI S., TEMPEL B.L., GUTMAN G.A. A family of three mouse potassium channel genes with intronless coding regions. Science. 1990;247:973–975. doi: 10.1126/science.2305265. [DOI] [PubMed] [Google Scholar]
- CHEN X.H., ADAMS J.U., GELLER E.B., DERIEL J.K., ADLER M.W., LIU-CHEN L.Y. An antisense oligodeoxynucleotide to μ-opioid inhibits μ-opioid receptor agonist-induced analgesia in rats. Eur. J. Pharmacol. 1995;275:105–108. doi: 10.1016/0014-2999(95)00012-a. [DOI] [PubMed] [Google Scholar]
- COOK N.S. The pharmacology of potassium channels and their therapeutic potential. Trends Pharmacol. Sci. 1988;9:21–28. doi: 10.1016/0165-6147(88)90238-6. [DOI] [PubMed] [Google Scholar]
- DAR M.S., MORSELLI P.L., BOWMAN E.R. The enzymatic systems involved in the mammalian metabolism of methylamine. Gen. Pharmacol. 1985;16:557–560. doi: 10.1016/0306-3623(85)90142-9. [DOI] [PubMed] [Google Scholar]
- DAWSON L.A., ROUTLEDGE C. Differential effects of potassium channel blockers on extracellular concentrations of dopamine and 5-HT in the striatum of conscious rats. Br. J. Pharmacol. 1995;116:3260–3264. doi: 10.1111/j.1476-5381.1995.tb15133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., WESTON A.H. The pharmacology of ATP-sensitive potassium channels. Annu. Rev. Pharmacol. Toxicol. 1993;33:597–637. doi: 10.1146/annurev.pa.33.040193.003121. [DOI] [PubMed] [Google Scholar]
- EKBLOM J. Potential therapeutic value of drugs inhibiting semicarbazide-sensitive amine oxidase: vascular cytoprotection in diabetes mellitus. Pharmacol. Res. 1998;37:87–92. doi: 10.1006/phrs.1997.0272. [DOI] [PubMed] [Google Scholar]
- FINNEGAN T.K., IRWIN I., DELANNEY L.E., LANGSTON W. Age-dependent effects of the 2′-methyl analog of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine: prevention by inhibitors of monoamine oxidase B. J. Pharmacol. Exp. Ther. 1995;237:716–720. [PubMed] [Google Scholar]
- GALEOTTI N., GHELARDINI C., CAPACCIOLI S., QUATTRONE A., NICOLIN A., BARTOLINI A. Blockade of clomipramine and amitriptyline analgesia by an antisense oligonucleotide to mKv1.1, a mouse Shaker-like K+channel. Eur. J. Pharmacol. 1997;330:15–25. doi: 10.1016/s0014-2999(97)10134-0. [DOI] [PubMed] [Google Scholar]
- GHELARDINI C., GALEOTTI N., PECORI V.A., CAPACCIOLI S., QUATTRONE A., BARTOLINI A. Effect of K+channel modulation on mouse feeding behaviour. Eur. J. Pharmacol. 1997;329:1–8. doi: 10.1016/s0014-2999(97)10102-9. [DOI] [PubMed] [Google Scholar]
- HALEY T.J., MCCORMICK G.L. Pharmacological effects produced by intracerebral injections of drugs in the conscious mouse. Br. J. Pharmacol. Chemother. 1957;12:12–15. doi: 10.1111/j.1476-5381.1957.tb01354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HALLIWELL J.V.K+ channels in the central nervous system Potassium channels: Structure, classification, function and therapeutic potential 1990Chichester: Ellis Horwood Limited; 348–381.ed. Cook, N. S. pp [Google Scholar]
- HAMMES H.P., BROWNLEE M., EDELSTEIN D., SALECK M., MARTIN S., FEDERLIN K. Aminoguanidine inhibits the development of accelerated diabetic retinopathy in the spontaneous hypertensive rat. Diabetologia. 1994;37:32–35. doi: 10.1007/BF00428774. [DOI] [PubMed] [Google Scholar]
- HOPKINS W.F., TEMPEL B.L. Members of a mouse subfamily of genes encoding voltage-gated potassium channel subunits from heteromultimers when co expressed in Venous oocytes. Soc. Neurosci. Abstr. 1992;18:1093. [Google Scholar]
- HRNJEZ B.J., SONG J.C., PRASAD M., MAYOL J.M., MATTHEWS J.B. Ammonia blockade of intestinal epithelial K+ conductance. Am. J. Physiol. 1999;277:G521–G532. doi: 10.1152/ajpgi.1999.277.3.G521. [DOI] [PubMed] [Google Scholar]
- HU S.H., WANG S., GIBSON J., GILBERTSON T.A. Inhibition of delayed rectifier K+channels by dexfenfluramine (Redux) J. Pharmacol. Exp. Ther. 1998;287:480–486. [PubMed] [Google Scholar]
- INUI A. Transgenic approach to the study of body weight regulation. Pharmacol. Rev. 2000;52:35–61. [PubMed] [Google Scholar]
- LEWINSON R., BOHM H.K., GLOVER V., SANDLER M. A benzylamine oxidase distinct from monoamine oxidase B. Widespread distribution in man and rat. Biochem. Pharmacol. 1978;27:1857–1863. doi: 10.1016/0006-2952(78)90033-3. [DOI] [PubMed] [Google Scholar]
- LYLES G.A., MCDOUGALL S.A. The enhanced daily excretion of urinary methylamine in rats treated with semicarbazide or hydralazin may be related to the inhibition of semicarbazide-sensitive amine oxidase activities. J. Pharm. Pharmacol. 1989;41:97–100. doi: 10.1111/j.2042-7158.1989.tb06401.x. [DOI] [PubMed] [Google Scholar]
- MESZAROS Z., SZOMBATHY T., RAIMONDI L., KARADI I., ROMICS L., MAGYAR K. Elevated serum semicarbazide-sensitive amine oxidase activity in non-insulin-dependent diabetes mellitus: correlation with body mass index and serum triglyceride. Metabolism. 1999;48:113–117. doi: 10.1016/s0026-0495(99)90019-7. [DOI] [PubMed] [Google Scholar]
- MIYATA G., MEGUID M.M., FETISSOV S.O., TORELLI G.F., KIM H.J. Nicotine's effect on hypothalamic neurotransmitters and appetite regulation. Surgery. 1999;126:255–263. [PubMed] [Google Scholar]
- MUNDORF M.L., HOCHSTETLER S.E., WIGHTMAN R.M. Amine weak bases disrupt vesicular storage and promote exocytosis in chromaffin cells. J. Neurochem. 1999;73:2397–2405. doi: 10.1046/j.1471-4159.1999.0732397.x. [DOI] [PubMed] [Google Scholar]
- PARADA M.A., HERNANDEZ L., SHWARTZ D., HOEBEL B.G. Hypothalamic infusions of amphetamine increase serotonin, dopamin and norepinephrine. Physiol. Behav. 1988;44:607–610. doi: 10.1016/0031-9384(88)90325-3. [DOI] [PubMed] [Google Scholar]
- PIRISINO R., BANCHELLI G., IGNESTI G., MANTELLI L., MATUCCI R., RAIMONDI L., BUFFONI F. Calcium modulatory properties of 2,6-dibutylbenzylamine (B25) in rat isolated vas deferens, cardiac and smooth muscle preparations. Br. J. Pharmacol. 1993;109:1038–1045. doi: 10.1111/j.1476-5381.1993.tb13726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRECIOUS E., GUNN C.E., LYLES G.A. Deamination of methylamine by semicarbazide-sensitive amine oxidase in human umbilical artery and rat aorta. Biochem. Pharmacol. 1988;37:707–713. doi: 10.1016/0006-2952(88)90145-1. [DOI] [PubMed] [Google Scholar]
- QUATTRONE A., PAPUCCI L., SCHIAVONE N., MINI E., CAPACCIOLI S. Intracellular enhancement of intact antisense oligonucleotide steady-state levels by cationic lipids. Anticancer Drug Des. 1994;9:549–553. [PubMed] [Google Scholar]
- SIMENOFF M.L. Metabolism and toxicity of aliphatic amines. Kidney Int. 1975;7:S314–S317. [PubMed] [Google Scholar]
- SNYDERS D.J. Structure and function of cardiac potassium channels. Cardiovasc. Res. 1999;42:377–390. doi: 10.1016/s0008-6363(99)00071-1. [DOI] [PubMed] [Google Scholar]
- YU P.H., ZUO D.M. Oxidative deamination of methylamine by semicarbazide-sensitive amine oxidase leads to cytotoxic damage in endothelial cells. Possible consequences for diabetes. Diabetes. 1993;42:594–603. doi: 10.2337/diab.42.4.594. [DOI] [PubMed] [Google Scholar]
- YU P.H., ZUO D.M. Aminoguanidine inhibits semicarbazide-sensitive amine oxidase activity: implications for advanced glycation and diabetic complications. Diabetologia. 1997;40:1243–1250. doi: 10.1007/s001250050816. [DOI] [PubMed] [Google Scholar]
- ZEISEL S.H., DACOSTA K.A. Increase in human exposure to methylamine precursors of N-nitrosamines after eating fish. Cancer Res. 1986;46:6136–6138. [PubMed] [Google Scholar]