

Abstract
This study demonstrated aloe-emodin- and emodin-induced apoptosis in lung carcinoma cell lines CH27 (human lung squamous carcinoma cell) and H460 (human lung non-small cell carcinoma cell). Aloe-emodin- and emodin-induced apoptosis was characterized by nuclear morphological changes and DNA fragmentation.
During apoptosis, an increase in cytochrome c of cytosolic fraction and activation of caspase-3, identified by the cleavage of its proform, were observed.
To elucidate whether the expression of protein kinase C (PKC) isozymes are involved in aloe-emodin- and emodin-induced apoptosis, this study examined the changes of PKC isozymes by Western blotting techniques during aloe-emodin- and emodin-induced apoptosis.
The expression of PKC isozymes involved in aloe-emodin- and emodin-induced apoptosis of CH27 and H460 cells. In this study, aloe-emodin and emodin induced the changes of each of PKC isozymes in CH27 and H460 cells.
The decrease in the expression of PKCδ and ε may play a critical role in aloe-emodin- and emodin-induced apoptosis in CH27 and H460 cells.
The present study also demonstrated that PKC stimulation occurs at a site downstream of caspase-3 in the emodin-mediated apoptotic pathway.
Keywords: Aloe-emodin (1,8-dihydroxy-3-(hydroxymethyl)-anthraquinone); emodin (1,3,8-trihydroxy-6-methylanthraquinone); human lung squamous carcinoma cell line CH27; human lung non-small cell carcinoma cell line H460; apoptosis; cytochrome c; caspase-3; protein kinase C
Introduction
Aloe-emodin (1,8-dihydroxy-3-(hydroxymethyl)-anthraquinone) and emodin (1,3,8-trihydroxy-6-methylanthraquinone) are the active components contained in the root and rhizome of Rheum palmatum L. (Polygonaceae) (Tsai & Chen, 1992; Liang et al., 1993; Yang et al., 1999). Pecere et al. (2000) have reported that aloe-emodin has a specific anti-neuroectodermal tumor activity. Emodin has also been reported to sensitize HER-2/neu-overexpressing lung cancer cells to chemotherapeutic drugs (Zhang & Hung, 1996) and repress transformation and metastasis-associated properties of HER-2/neu overexpression breast cancer cells (Zhang et al., 1995; 1998). However, the reasons why the molecular mechanisms of aloe-emodin and emodin produced their biological effects remained unknown. The present study served to determine whether aloe-emodin and emodin induced cytotoxicity on lung carcinoma cell lines CH27 and H460. Furthemore, this study investigated the mechanisms of the aloe-emodin- and emodin-induced cytotoxicity on lung carcinoma cell lines CH27 and H460. The present study demonstrates the cytotoxicity of lung carcinoma cells by aloe-emodin and emodin, and the anti-tumor activity is based on apoptotic cell death.
Caspases, a family of cysteine proteases, play a critical role in the apoptosis and are responsible for many of the biochemical and morphological changes associated with apoptosis (Cohen, 1997; Cryns & Yuan, 1998). Two major pathways of apoptotic signalling have been identified. The first involves ligation of death receptors (e.g. Fas) by their ligands, leading to recruitment of adaptor proteins and activation of the initiator, caspase-8 (Ashkenazi & Dixit, 1998). In the second pathway, mitochondrial cytochrome c is released into the cytosol and binds Apaf-1, which in turn associates and activates the initiator, caspase-9 (Li et al., 1997). However, caspases have been proposed that ‘initiator' caspases, such as caspase-8 and caspase-9, either directly or indirectly activate ‘effector' caspases, such as caspase-3 (Fraser & Evan, 1996; Sun et al., 1999). Therefore, the activation of caspase-3 is requisite for apoptosis. This study investigated whether the activation of caspase-3 is involved in aloe-emodin- and emodin-induced the CH27 and H460 cell death.
Protein kinase C represents a family of 11 isozymes that have been categorized into three groups: calcium-dependent or ‘classical' (PKCα, βI, βII and γ), calcium-independent or ‘novel' (PKCδ, ε, ζ and θ) and ‘atypical' (PKCζ and ι/λ) (Stabel & Parker, 1991; Basu, 1993). The protein kinase C family has been implicated in the regulation of apoptosis (Bialik et al., 1999; Koriyama et al., 1999; Gomez-Angelats et al., 2000). However, the contribution of individual PKC isozymes to this process is not well understood. This study investigated the role of PKC isozymes in apoptotic signalling induced by aloe-emodin and emodin. The relationship between the activation of the caspase and the activation of PKC was investigated in many reports. It is generally believed that PKCδ lie downstream of caspase-3 and proteolytic activation of PKCδ is responsible for apoptotic execution (Tsujio et al., 2000; Tanaka et al., 2000). However, some investigators have found that caspase-3 inhibitors did not prevent down-regulation of PKCδ (Jun et al., 1999; Basu & Akkaraju, 1999). It seems to suggest that PKC activation act upstream of caspase-3. This study examined the specificity of the PKC-caspase-3 relationship on aloe-emodin- and emodin-induced apoptosis.
Methods
Materials
N-Acetyl-Asp-Glu-Val-Asp-al (Ac-DEVD-CHO), Aloe-emodin (1,8-dihydroxy-3-(hydroxymethyl)-anthraquinone), emodin (1,3,8-trihydroxy-6-methylanthraquinone), antipain, aprotinin, dithiothreitol, 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI), ethylenediaminetetraacetic acid (EDTA), ethyleneglycol-bis-(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), leupeptin, pepstatin, phenylmethylsulphonyl fluoride, propidium iodide and tris (hydroxymethyl) aminomethane (Tris) were purchased from Sigma Chemical Company (St. Louis, MO, U.S.A.); anti-goat, anti-mouse and anti-rabbit IgG peroxidase-conjugated secondary antibody were purchased from Amersham (Buckinghamshire, U.K.). Antibodies to various proteins were obtained from the following sources: caspase-3, PKCα, β, δ, ε, θ, ι and μ were obtained from Transduction Laboratory (Lexington, KY, U.S.A.); PKCζ and ζ were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, U.S.A.); cytochrome c and poly(ADP-ribose)polymerase (PARP) were purchased from PharMingen (San Diego, CA, U.S.A.). Pierce Colorimetric PKC Assay Kit was obtained from PIERCE (Rockford, IL, U.S.A.). Enhanced chemiluminescent (Renaissance) detection reagents was obtained from NEN Life Science Products (Boston, MA, U.S.A.).
Cell culture
The human lung squamous carcinoma cell line CH27 and human lung non-small carcinoma cell line H460 were kindly provided by S.L. Hsu. CH27 and H460 cells were grown in monolayer culture in Dulbecco's modified Eagle's medium containing 5% foetal bovine serum, antibiotics (100 u ml−1 penicillin and 100 μg ml−1 stretomycin) and 2 mM glutamine at 37°C in a humidified atmosphere comprised of 95% air and 5% CO2. When CH27 and H460 cells were treated with aloe-emodin or emodin, the culture medium containing 1% foetal bovine serum was used. All data presented in this report are from at least three independent experiments showing the same pattern of expression.
Cell viability assay
Cells were seeded at a density of 1×105 cells per well onto 12-well plate 24 h before drugs treated. Drugs were added to medium, at various indicated times and concentrations. The control cultures were treated with 0.1% DMSO (dimethylsulphoxide). After incubation, cells were washed with PBS (phosphate-buffered saline). The number of viable cells was determined by staining cell population with Trypan blue. One part of 0.2% Trypan blue dissolved in PBS was added to one part of the cell suspension, and the number of unstained (viable) cells was counted.
4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI) staining
DAPI staining was performed by a modification of the method of Hsu et al. (1999). Cells were seeded at a density of 1×105 cells per well onto 12-well plate 24 h before drugs were treated. Cells were cultured with vehicle alone (0.1% DMSO), 40 μM aloe-emodin or 50 μM emodin for 16 h in 1% serum medium. After treatment, cells were fixed with 3.7% formaldehyde for 15 min, permeabilized with 0.1% Triton X-100 and stained with 1 μg ml−1 DAPI for 5 min at 37°C. The cells were then washed with PBS and examined by fluorescence microscopy (Olympus IX 70).
DNA fragmentation assay
DNA fragmentation was assayed as previously described (Hsu et al., 1999). Adherent and floating cells were collected and lysed in 400 μl of ice-cold lysis buffer (containing 50 mM Tris-HCl, pH 7.5, 10 mM EDTA, 0.3% Triton X-100), incubated on ice for 30 min and then centrifuged. RNase A (100 μg ml−1) was added to the supernatant, which was then incubated at 50°C for 30 min, followed by the addition of 200 μg ml−1 proteinase K and further incubation at 37°C for 1 h. Fragmented DNA was extracted with phenol/chloroform and precipitated at −20°C with ethanol/sodium acetate. The DNA fragments were electrophoresed on a 1.5% agarose gel containing 0.1 μg ml−1 ethidium bromide.
Flow cytometry analysis
The percentage of hypodiploid cells was determined as described previously (Hsu et al., 1999). Briefly, 2×106 cells were trypsinized, washed twice with PBS and fixed in 80% ethanol. Fixed cells were washed with PBS, incubated with 100 μg ml−1 RNase for 30 min at 37°C, stained with propidium iodide (50 μg ml−1) and analysed on a FACScan flow cytometer (Becton Dickinson Instruments). The percentage of cells that had undergone apoptosis was assessed to be the ratio of the fluorescent area smaller than the G0 – G1 peak to the total area of fluorescence. The average of the results from at least three samples of cells for each experimental condition is presented.
Protein preparation
Preparation of total protein
Protein was extracted by a modification of the method of Hsu et al. (1999). Adherent and floating cells were collected at the indicated times and washed twice in ice-cold PBS. Cell pellets were resuspended in modified RIPA buffer (mM: Tris-HCl 50, pH 7.5, NaCl 150, EGTA 1, dithiothreitol 1, phenylmethylsulphonyl fluoride 1, sodium orthovanadate 1, sodium fluoride 1, aprotinin 5 μg ml−1, leupeptin 5 μg ml−1, antipain 5 μg ml−1, Nonidet P-40 1%, sodium deoxycholate 0.25%) for 30 min at 4°C. Lysates were clarified by centrifugation at 100,000×g for 30 min at 4°C and the resulting supernatant was collected, aliquoted (50 μg per tube) and stored at −80°C until assay. The protein concentrations were estimated with the Bradford method (Bradford, 1976).
Preparation of cytosolic fractions
Cell fractionation was performed as described previously (Jun et al., 1999) with some modifications. Briefly, adherent and floating cells were collected at the indicated times and washed twice in ice-cold PBS. Cell pellets were frozen at −80°C, thawed at 4°C and resuspended in cytosol extraction buffer (mM: Tris-HCl 50, pH 7.5, EDTA 5, EGTA 10, phenylmethylsulphonyl fluoride 0.2, sodium fluoride 1, sodium orthovanadate 1, aprotinin 1 μg ml−1, leupeptin 5 μg ml−1, antipain 5 μg ml−1, β-mercaptoethanol 0.3%) for 20 min at 4°C until >95% of the cells were Trypan blue positive. Lysates were clarified by centrifugation at 100,000×g for 30 min at 4°C and the resulting supernatant was collected as the ‘cytosolic' fraction, aliquoted (10 μg per tube for cytochrome c) and stored at −80°C until assay.
Western blot analysis
Samples were separated by various appropriate concentrations (8, 10, 12 and 15%) of sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS – PAGE). The SDS-separated proteins were equilibrated in transfer buffer (50 mM Tris, pH 9.0 – 9.4, 40 mM glycine, 0.375% SDS, 20% methanol) and electro-transferred to Immobilon-P Transfer Membranes. The blot was blocked with a solution containing 5% non-fat dry milk in Tris-buffered saline (10 mM Tris, 150 mM NaCl) with 0.05% Tween 20 (TBST) for 1 h, washed and incubated with antibodies to PARP (1 : 2000), PKCα (1 : 5000), PKCβ (1 : 2500), PKCδ (1 : 500), PKCε (1 : 500), PKCζ (1 : 500), PKCζ (1 : 500), PKCθ (1 : 250), PKCι (1 : 250), PKCμ (1 : 1000) and cytochrome c (1 : 500). Secondary antibody consisted of a 1 : 20,000 dilution of horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (for PKCζ and cytochrome c) or HRP-conjugated goat anti-mouse IgG (for caspase-3, PARP, PKCα, β, δ, ε, θ, ι and μ) or HRP-conjugated anti-goat IgG (for PKCζ). The enhanced chemiluminescent (Renaissance) detection system was used for immunoblot protein detection.
Measurement of protein kinase C activity
Protein kinase C activity was determined as described previously (Lee & Wu, 2000) with some modification. After treatment, cells were washed twice with PBS and scraped, on ice, into ice-cold lysis buffer containing 20 mM Tris-HCl, pH 8.0, 0.5 mM EDTA, 0.5 mM EGTA, 2.5 mM phenylmethylsulphonyl fluoride, 5 μg ml−1 leupeptin and 5 μg ml−1 antipain. The cells were collected and sonicated for 10 pulses. The sonicated samples were centrifuged at 14,000×g for 30 min at 4°C and the resulting supernatant was collected, aliquoted (200 μg per tube) and measured PKC activity immediately. PKC activity in the supernatant was determined by Pierce Colorimetric PKC Assay Kit. The PKC-dependent phosphorylated peptide was quantified by 570 nm.
Results
Aloe-emodin- and emodin-induced lung carcinoma cell death in a dose- and time-dependent manner
Since aloe-emodin and emodin were found to have anti-tumor effects on neuroectodermal and breast cancer cells, respectively, the present study served to determine whether aloe-emodin and emodin-induced cytotoxicity on lung carcinoma cell lines CH27 and H460. This study determined the effect of aloe-emodin or emodin on CH27 and H460 cell viability by Trypan blue dye exclusion. The number of viable cells was counted by Trypan blue dye exclusion. As shown in Figure 1A, 72 h of continuous exposure to various concentrations of aloe-emodin (1, 10 and 40 μM) or emodin (1, 10 and 50 μM) on CH27 resulted in time- and dose-dependent decreases in cell number relative to control cultures. The similar results of the effect of various concentrations of aloe-emodin or emodin for various indicated times (2, 4, 8, 16, 24, 48 and 72 h) on H460 cell viability were obtained (Figure 1B). The concentration of aloe-emodin- and emodin-induced cell death was significant at 40 and 50 μM, respectively. Therefore, 40 μM aloe-emodin and 50 μM emodin were chosen for further experiments. These results suggested that aloe-emodin and emodin induced CH27 and H460 cell death.
Figure 1.
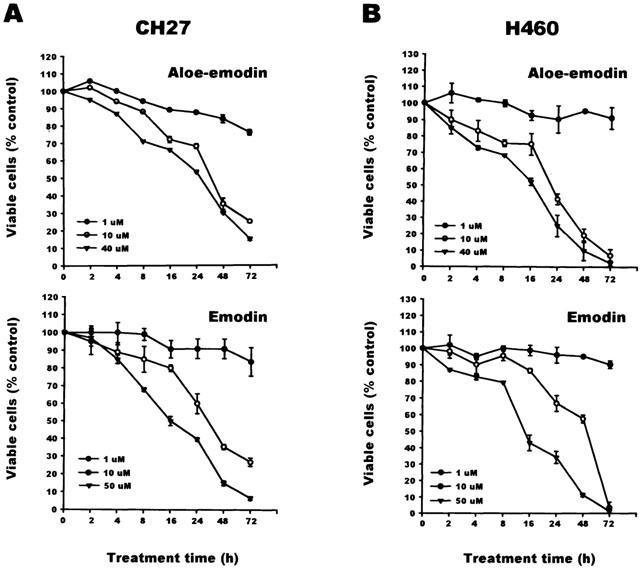
Effects of aloe-emodin and emodin on cell death in CH27 and H460 cells. Cells were cultured 24 h before drug treatment in 12-well plates. CH27 (A) and H460 (B) cells were treated with 0.1% DMSO, aloe-emodin or emodin in the presence of 1% serum at 37°C for different times (2, 4, 8, 16, 24, 48 and 72 h), and cells were washed and counted by Trypan blue exclusion with hemocytometer. All determinations are expressed as the mean percentage of control±s.d.mean of triplicate from three independent experiments.
Aloe-emodin- and emodin-induced apoptosis of CH27 and H460 cells
To further investigate whether the induction of cell death by aloe-emodin and emodin could be linked to apoptosis in lung carcinoma cells, both nuclear morphological changes and DNA fragmentation were performed. Treatment of CH27 with 40 μM aloe-emodin or 50 μM emodin for 16 h resulted in changes in nuclear morphology, evidenced by the DAPI staining, a DNA binding dye (Figure 2A). There was an increase in the number of irregular nuclear, fragmented nucleus, convoluted nucleus and giant nucleus after treatment with aloe-emodin (Figure 2A). Treatment with emodin also resulted in changes in nuclear morphology (Figure 2A). There was a gradual increase in the number of nuclear condensation after treatment with emodin in CH27 cells (Figure 2A). H460 cells also showed an increase in the number of irregular nuclear, fragmented nucleus, convoluted nucleus and giant nucleus after treatment with aloe-emodin and emodin (Figure 2B). Treatment with 40 μM aloe-emodin or 50 μM emodin for 24 h resulted in internucleosomal DNA fragmentation, evidenced by the formation of a DNA ladder on agarose gels (Figure 3A,B), a hallmark of cells undergoing apoptosis. No DNA ladders were detected in the sampled isolation from control cells. Apoptosis was also confirmed on the appearance of a sub-G1 peak of DNA content by flow cytometry, suggesting that the presence of cells with fragmented DNA. According to the DNA histogram shown in Figure 4A,B, a sub-G1 peak was detected following 24 h of 40 μM aloe-emodin or 50 μM emodin exposure. In this study, the aloe-emodin- and emodin-induced lung carcinoma cells nuclear morphological change, DNA fragmentation and cell death were observed. Based on the above results, aloe-emodin- and emodin-induced CH27 and H460 cell death were indicative of a typical apoptosis.
Figure 2.
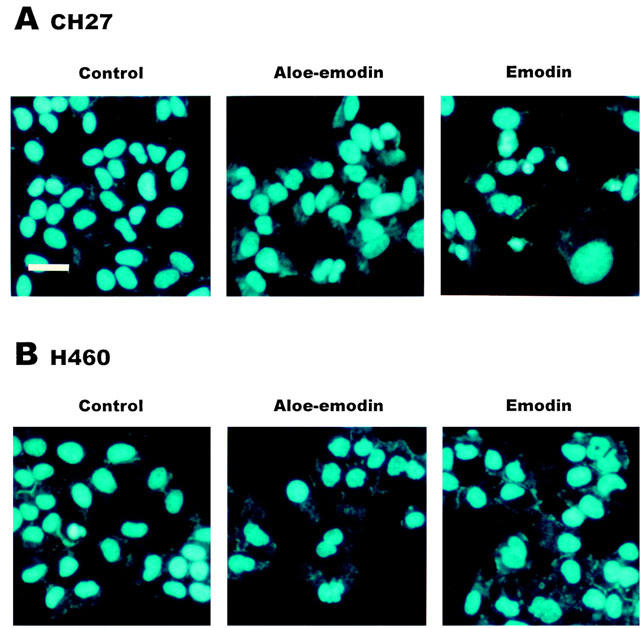
Aloe-emodin and emodin-induced phenotypic changes in CH27 and H460 cell nucleus. CH27 (A) and H460 (B) cells were cultured for 16 h in 1% serum medium with 0.1% DMSO, 40 μM aloe-emodin or 50 μM emodin. After treatment, cells were fixed with 3.7% formaldehyde for 15 min, permeabilized with 0.1% Triton X-100 and stained with 1 μg ml−1 DAPI for 5 min at 37°C. The cells were then washed with PBS and examined by fluorescence microscopy. Results are representative of three independent experiments. Bar=50 μm.
Figure 3.
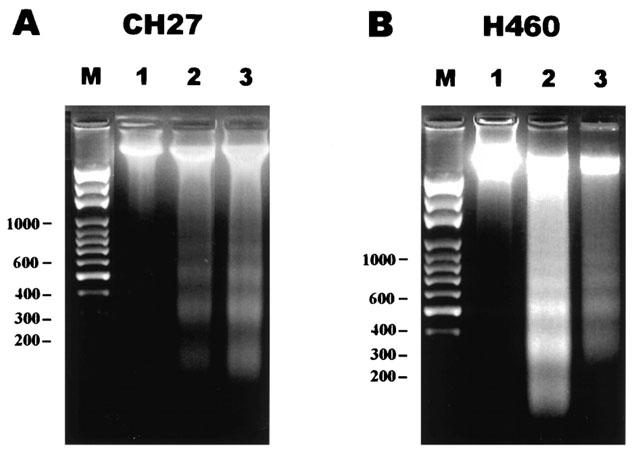
Aloe-emodin and emodin-induced internucleosomal DNA fragmentation in CH27 and H460 cell nucleus. CH27 (A) and H460 (B) cells were incubated with 0.1% DMSO (lane 1), 40 μM aloe-emodin (lane 2), or 50 μM emodin (lane 3) for 24 h in 1% serum medium. Total DNA was extracted from cells and separated by electrophoresis on 1.5% agarose gel. The molecular weight marker lane (lane M) represents DNA base pairs. Results are representative of three independent experiments.
Figure 4.
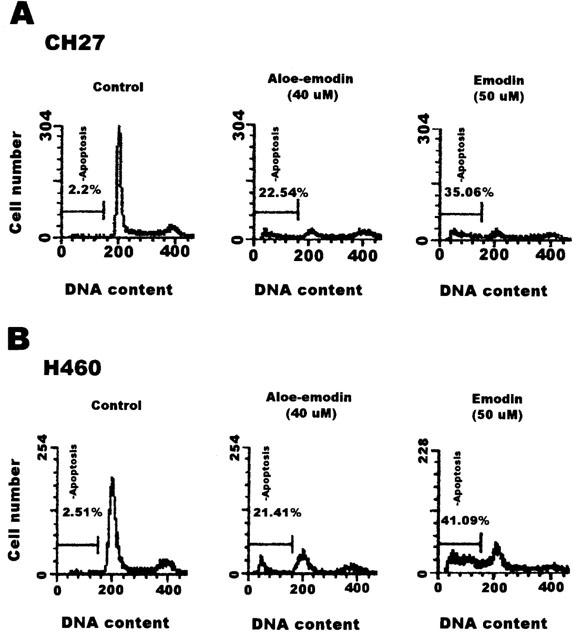
Aloe-emodin- and emodin-induced the appearance of a sub-G1 peak in CH27 and H460 cells by flow cytometry assay. CH27 (A) and H460 (B) cells were treated with vehicle alone (0.1% DMSO), 40 μM aloe-emodin, or 50 μM emodin in the presence of 1% serum for 24 h. After treatment, cells were harvested and subjected to cytometric analysis. Apoptosis was measured by cell cycle analysis with propidium iodide staining and the percentage of hypodiploid cells (apoptotic population of cells) was calculated. Results are representative of three independent experiments.
Effect of aloe-emodin and emodin on the release of cytochrome c and activation of caspase-3 in lung carcinoma cells
This study characterized the effect of aloe-emodin (40 μM) and emodin (50 μM) on the release of cytochrome c in CH27 and H460 cells. Western blotting analysis of the cytosolic fraction of aloe-emodin- and emodin-treated CH27 and H460 cells revealed increases in the relative abundance of cytochrome c for the indicated time intervals (Figure 5A,B). This study has also demonstrated that the activation of caspase-3 is involved in aloe-emodin- and emodin-induced the CH27 and H460 cell death. The proform of caspase-3 (32 kDa) was significantly decreased during aloe-emodin (40 μM) and emodin (50 μM) treated for 24 h by Western blotting analysis (Figure 5A,B). Caspase-3 was present in control cells primarily as 32 kDa protein. Treatment with 40 μM aloe-emodin or 50 μM emodin resulted in a time-dependent processing of caspase-3 accompanied by the formation of two major products, 22 and 17 kDa fragments (Figure 5A,B). It is worthy of note that the amount of these fragments of caspase-3 was significantly increased after treatment with aloe-emodin or emodin. In control cells, a low level of processing of caspase-3 (22 and 17 kDa) was observed; this may reflect basal caspase activity. Proteolysis of caspase-3 substrate provides a marker for apoptosis and caspase activity. To further determine whether caspase-3 was activated in aloe-emodin- or emodin-treated lung carcinoma cells, Western blot analysis of caspase-3 substrate PARP was performed. PARP (116 kDa) was processed to its predicted caspase cleavage product of 85 kDa during aloe-emodin or emodin treatment (Figure 5A,B). Furthermore, the cleavage product of 85 kDa appeared to be further processed in the aloe-emodin- and emodin-induced the cleavage of PARP in CH27 cells (Figure 5A). In emodin-induced caspase-3 activation and PARP cleavage, the caspase-3 had significantly processed at 2 and 4 h but the cleavage of PARP was not significantly increased (Figure 5A). When the time of immunoblot protein detection lengthened, the cleavage of PARP was observed at 2 and 4 h (data not shown). These above data (DNA fragmentation, cytochrome c release and the activation of caspase-3) suggested that the aloe-emodin and emodin induced apoptotic cell death in CH27 and H460 cells.
Figure 5.
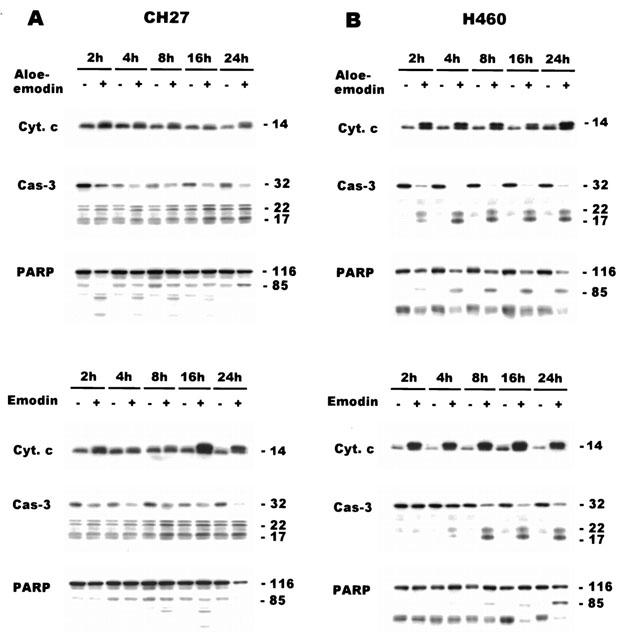
Effects of aloe-emodin and emodin on the release of cytochrome c and the expression of caspase-3 and PARP in CH27 and H460 cells. The effect of aloe-emodin and emodin on cytochrome c (Cyt. c), caspase-3 (Cas-3) and PARP was detected by Western blot analysis in CH27 (A) and H460 (B) cells. Cells were incubated with 40 μM aloe-emodin or 50 μM emodin in the presence of 1% serum for 2, 4, 8, 16 and 24 h. Cell lysates were analysed by 8% (PARP), 12% (caspase-3) and 15% (cytochrome c) SDS – PAGE, and then probed with primary antibody as described in Materials and methods. Results are representative of three independent experiments.
Effect of aloe-emodin and emodin on the protein kinase C isozymes in lung carcinoma cells
To investigate the role of PKC isozymes in apoptotic signalling induced by aloe-emodin and emodin, this study detected the expression of various PKC isozymes by Western blot analysis using isozyme-specific anti-PKC antibodies. In this study, PKCβ, γ and θ were not found in CH27 cell extracts even when various dilutions of primary and secondary antibodies were used. The very faint immunoreactive bands of PKCζ were observed in CH27 cells (data not shown). In H460 cells, PKCβ, γ, ζ and μ were not observed. Isozymes α, δ, ε, ζ, ζ, θ and ι had apparent molecular masses of 82, 78, 90, 72, 82, 79 and 74 kDa, respectively. The expression of PKCα showed a time-dependent decrease in aloe-emodin-treated CH27 cell extracts during 24 h (data not shown). In contrast to aloe-emodin-treated CH27, the expression of PKCα was significantly increased in aloe-emodin-treated H460, emodin-treated CH27 and emodin-treated H460 (data not shown). The changes of PKCζ and ι were not the same manner, i.e. some treatments were increased and some decreased, in four conditions (data not shown). It is worthy of note that the expression of PKCδ and ε was consistently decreased in aloe-emodin or emodin-treated CH27 and H460 cells (Figure 6). Proteolytic cleavage of PKCδ by caspase-3 at the V3 (hinge) domain of the enzyme releases a catalytically active fragment of approximately 40 kDa. However, this study could not detect the presence of PKCδ catalytic fragment after aloe-emodin and emodin treatment. These above data suggest that the changes of PKCδ and ε play a critical role during apoptosis but the PKCδ catalytic fragment may be rapidly degraded to smaller fragment, which cannot be detected in this study.
Figure 6.
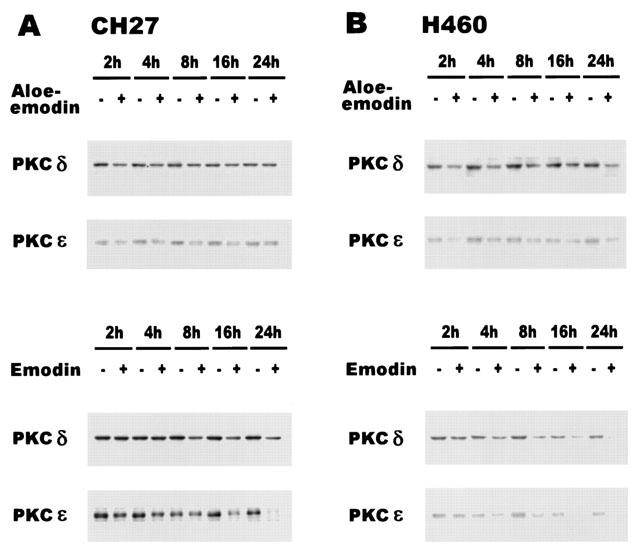
Effects of aloe-emodin and emodin on the PKCδ and ε in CH27 and H460 Cells. The effect of aloe-emodin and emodin on PKCδ and ε was detected by Western blot analysis in CH27 (A) and H460 (B) cells. Cells were incubated with 40 μM aloe-emodin or 50 μM emodin in the presence of 1% serum for 2, 4, 8, 16 and 24 h. Cell lysates were analysed by 10% SDS – PAGE, and then probed with antibodies against peptides specific for PKCδ and ε. Western blot analysis with a mAb to detect PKCδ and ε was performed as described in Materials and methods. Results are representative of three independent experiments.
Effects of aloe-emodin and emodin on protein kinase C activity in lung carcinoma cells
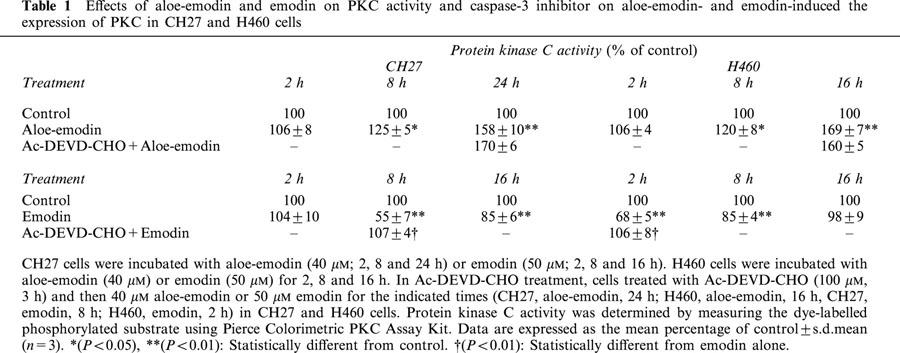
The effects of aloe-emodin and emodin on PKC activity were investigated in CH27 and H460 cells. As shown in Table 1, treatment of CH27 cells with 40 μM aloe-emodin for 2, 8 and 24 h resulted in increased of PKC activity. However, emodin (50 μM)-induced a decrease of PKC activity was observed at 2, 8 and 16 h (Table 1). In H460 cells, aloe-emodin also increased the PKC activity at 2, 8 and 16 h and emodin induced the decrease of PKC activity as well as emodin in CH27 cells (Table 1). These results indicated that treatment of CH27 and H460 cells with 40 μM aloe-emodin resulted in increase in PKC activity; however, the PKC activity was suppressed by treatment with 50 μM emodin.
Table 1.
Effects of aloe-emodin and emodin on PKC activity and caspase-3 inhibitor on aloe-emodin- and emodin-induced the expression of PKC in CH27 and H460 cells
Effects of caspase-3 inhibitor on aloe-emodin- and emodin-induced the expression of protein kinase C in lung carcinoma cells
To further investigate whether the changes of PKC activity by aloe-emodin or emodin could be linked to activation of the caspase-3, the caspase-3 inhibitor, Ac-DEVD-CHO, was used in this study. Cells treated with Ac-DEVD-CHO (100 μM, 3 h) and then 40 μM aloe-emodin or 50 μM emodin in CH27 and H460 cells for the indicated times (CH27, aloe-emodin, 24 h; H460, aloe-emodin, 16 h; CH27, emodin, 8 h; H460, emodin, 2 h). The response to pretreatment with Ac-DEVD-CHO and then emodin compared with the response to emodin alone showed that Ac-DEVD-CHO significantly reversed the emodin effect on PKC activity in CH27 and H460 cells (Table 1). The results indicated that caspase-3 inhibitor, Ac-DEVD-CHO, reversed the activity of PKC after being inhibited by emodin. It was also noted that aloe-emodin-induced increase in PKC activity was not significantly less in the presence of Ac-DEVD-CHO than that in the absence of Ac-DEVD-CHO in CH27 and H460 cells (Table 1). This result indicated that caspase-3 inhibitor, Ac-DEVD-CHO, had no effect on the aloe-emodin-induced increase in PKC activity in CH27 and H460 cells. This study also investigated the effect of caspase-3 inhibitor on aloe-emodin- or emodin-induced the decrease of PKCδ by Western blot analysis. As shown in Figure 7A, pretreatment with Ac-DEVD-CHO (100 μM, 3 h) and then aloe-emodin (40 μM, 2, 8 and 16 h) had no effect on the aloe-emodin (40 μM)-induced decrease in PKCδ in CH27 and H460 cells. However, Ac-DEVD-CHO (100 μM, 3 h) reversed the emodin (50 μM, 2, 8 and 16 h) -induced decrease in PKCδ in CH27 and H460 cells (Figure 7B).
Figure 7.
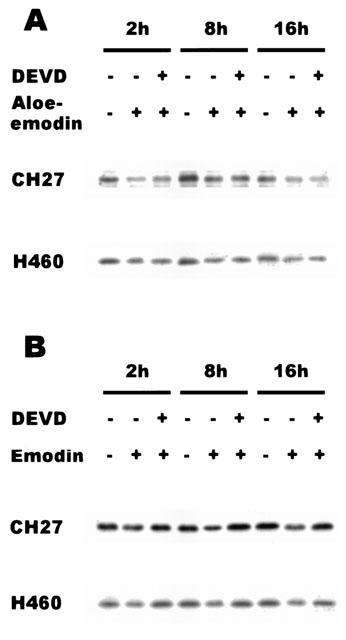
Effects of Ac-DEVD-CHO on aloe-emodin- and emodin-induced the expression of PKCδ in CH27 and H460 Cells. Cells were cultured for 3 h in medium that contained Ac-DEVD-CHO (100 μM, DEVD). Then, the cells were treated with 40 μM aloe-emodin (A) or 50 μM emodin (B) for 2, 8 and 16 h. Cell lysates were analysed by 10% SDS – PAGE, and then probed with antibody against PKCδ. Western blot analysis with a mAb to detect PKCδ was performed as described in Materials and methods. Results are representative of three independent experiments.
Discussions
Aloe-emodin and emodin are the active components contained in the root and rhizome of Rheum palmatum L. (Polygonaceae) (Tsai & Chen, 1992; Liang et al., 1993; Yang et al., 1999). Aloe-emodin and emodin were found to have anti-tumor effects on neuroectodermal and breast cancer cells, respectively (Zhang et al., 1995; 1998; Pecere et al., 2000). However, the reasons why the molecular mechanisms of aloe-emodin and emodin produced their biological effects remained unknown. The present study served to determine whether aloe-emodin and emodin induced cytotoxicity on lung carcinoma cell lines CH27 and H460. Furthermore, this study investigated the mechanisms of the aloe-emodin- and emodin-induced cytotoxicity on lung carcinoma cell lines CH27 and H460. The present study demonstrates the cytotoxicity of lung carcinoma cells by aloe-emodin and emodin, and the anti-tumor activity is based on apoptotic cell death.
Apoptosis is a major form of cell death and essential for normal development and for the maintenance of homeostasis. In addition, current anti-neoplastic therapies, chemotherapy and radiation-therapy, are likely to be affected by the apoptotic tendencies of cells; thus this process has obvious therapeutic implications (Green et al., 1994). During apoptosis, certain characteristic morphologic events, such as nuclear condensation, nuclear fragmentation and cell shrinkage, and biochemical events such as DNA fragmentation occur (Hsu et al., 1999; Shinoura et al., 1999). Aloe-emodin- and emodin-induced apoptosis was characterized by nuclear morphological changes and DNA fragmentation. Many investigators have suggested that the apoptotic effect of cells is mediated by a well-characterized transduction process of apoptotic signals, such as mitochondria cytochrome c efflux and the activation of caspase-3 in the cytosol (Kluck et al., 1997; Bossy-Wetzel & Green, 1999; Dong et al., 2000). Cytochrome c, which is usually present in the mitochondrial intermembrane space, is released into the cytosol following the induction of apoptosis by many different stimuli including Fas (CD95), tumor necrosis factor (TNF) and chemotherapeutic and DNA-damaging agents (Liu et al., 1996; Reed, 1997). In this study, Western blotting analysis of the cytosolic fraction of aloe-emodin- and emodin-treated CH27 and H460 cells revealed increases in the relative abundance of cytochrome c.
Caspases, a family of cysteine proteases, play a critical role in the apoptosis and are responsible for many of the biochemical and morphological changes associated with apoptosis (Cohen, 1997; Cryns & Yuan, 1998). Caspases have been proposed that ‘initiator' caspases, such as caspase-8 and caspase-9, either directly or indirectly activate ‘effector' caspases, such as caspase-3 (Fraser & Evan, 1996; Sun et al., 1999). During apoptosis, the cleavage and activation of caspase-3 is requisite. This study has demonstrated that the activation of caspase-3 is involved in aloe-emodin- and emodin-induced the CH27 and H460 cell death. The cleavage of caspase-3 substrate PARP, as an indicator of caspase-3 activation, was significantly observed after treatment with aloe-emodin and emodin. These above data (DNA fragmentation, cytochrome c release and the activation of caspase-3) suggested that the aloe-emodin and emodin induced apoptotic cell death in CH27 and H460 cells.
Protein kinase C is an attractive target for modulation of apoptosis as there is mounting evidence implicated PKC as a multifaceted regulator of cellular sensitivity to chemotherapeutic agents. Many other cellular models of apoptosis have been used to demonstrate that, during the transduction of cell death signals, there is selective inhibition/activation of PKC isoforms, depending on cell type and apoptotic stimuli considered (Jun et al., 1999; Koriyama et al., 1999; Pae et al., 2000; Jao et al., 2001). Pae et al. (2000) have demonstrated that TPA, a PKC activator, mediated protection from taxol-induced apoptosis of HL-60 cells. It has also reported that inactivation of PKCα may play an important role in modulating hepatic apoptosis (Jao et al., 2001). Overexpression of PKCβII, δ and ζ prevents NO-induced cell death in RAW 264.7 macrophage (Jun et al., 1999). In addition, recent report demonstrates proteolytic activation of PKCδ and ε in U937 cells during chemotherapeutic agent-induced apoptosis (Koriyama et al., 1999). Therefore, the contribution of individual PKC isozymes to this process is not well understood. The present study investigated the role of PKC isozymes in apoptotic signalling induced by aloe-emodin and emodin using Western blot analysis. Each of PKC isozymes has different expressions in CH27 and H460 after treatment with aloe-emodin or emodin in this study. These results suggest that PKC signalling pathways, in which the expression of the PKC isozymes is increased or decreased, play an important role in aloe-emodin- and emodin-induced CH27 and H460 apoptosis. However, it is worthy of note that the expression of PKCδ and ε was consistently decreased in aloe-emodin or emodin-treated CH27 and H460 cells. This result is consistent with previous observations in which the proteolysis of PKCδ and ε plays a critical role during apoptosis (Bialik et al., 1999; Koriyama et al., 1999; Gomez-Angelats et al., 2000). The present study also investigated aloe-emodin- and emodin-induced the change of PKC activity in CH27 and H460 by PKC activity assay kit. This study demonstrated that treatment of CH27 and H460 cells with 40 μM aloe-emodin resulted in increase in PKC activity; however, the PKC activity was suppressed by treatment with 50 μM emodin. These results are consistent with other observations that PKC-dependent signalling processes may depend on the diverse stimuli and specific cell types, such as the activation of PKC is sufficient for initiation of a apoptotic program (Fujii et al., 2000; Tanaka et al., 2000) and the inhibition of PKC activity may promote cells sensitive to drug-mediated apoptosis (Frutos et al., 1999; Pae et al., 2000).
The relationship between the activation of the caspase and the activation of PKC was investigated in many reports. It is generally believed that PKCδ lie downstream of caspase-3 and proteolytic activation of PKCδ is responsible for apoptotic execution (Tanaka et al., 2000; Tsujio et al., 2000). However, some investigators have found that caspase-3 inhibitors did not prevent down-regulation of PKCδ (Basu & Akkaraju, 1999; Jun et al., 1999). Fujii et al. (2000) have suggested that PKCδ-mediated apoptosis does not involve its proteolytic cleavage by caspase-3. It was also shown that PKCδ-mediated apoptosis in keratinocytes involves the alteration of mitochondria function (Li et al., 1999). It seems to suggest that PKC activation occurs at a site upstream of caspase-3 or involves different signalling pathway. Since caspase-3 has been implicated in the execution of cell death by aloe-emodin and emodin, this study examined the specificity of the PKC-caspase-3 relationship on aloe-emodin- and emodin-induced apoptosis. In this study, caspase-3 inhibitor Ac-DEVD-CHO reversed the activity of PKC after being inhibited by emodin. However, aloe-emodin-induced increase in PKC activity was not significantly effect by pretreatment of caspase-3 inhibitor. This study also demonstrated that caspase-3 inhibitor had no effect on the aloe-emodin-induced decrease in PKCδ, but could reverse emodin-induced decrease in PKCδ by Western blot analysis in CH27 and H460. Taken together, these findings are consistent with other observations that the specificity of the PKC-caspase relationship on apoptotic cell death may depend on the diverse stimuli and specific cell types (Jun et al., 1999; Koriyama et al., 1999; Pae et al., 2000; Jao et al., 2001). In this study, PKC lies downstream of caspase-3 in the emodin-induced apoptosis. However, the PKC-caspase-3 relationship can be proposed two different assumptions in the aloe-emodin-induced apoptosis. The first assumption may be involved the alteration of mitochondria function by PKCδ. Mitochondrial cytochrome c is released into the cytosol and binds Apaf-1, which in turn associates and activates the initiator caspase-9. This results in activation of caspase-9, which then processes caspase-3. In the second assumption, the activation of caspase-3 and PKC may proceed through two distinct mechanisms in the aloe-emodin-induced apoptosis. The PKCδ activity could be regulated by diacylglycerol, tyrosine phosphorylation, or tyrosine kinase (Denning et al., 1993; Li et al., 1994; Shanmugam et al., 1998; Song et al., 1998). However, the activation of caspase-3 is associated with two prototypical pathways for induction of apoptosis, such as Fas and Bax pathway (Fraser & Evan, 1996; Sun et al., 1999).
In summary, this study demonstrated aloe-emodin- and emodin-induced apoptosis in CH27 and H460. During apoptosis, an increase in cytochrome c of cytosolic fraction and activation of caspase-3, identified by the cleavage of its proform, were observed. The expression of PKC isozymes involved in aloe-emodin- and emodin-induced apoptosis of CH27 and H460 cells. In this study, aloe-emodin and emodin-induced the changes of each of PKC isozymes in CH27 and H460 cells. Particularly, the types of change of PKCδ and ε were decreased in the same manner in four conditions (aloe-emodin-treated CH27, aloe-emodin-treated H460, emodin-treated CH27 and emodin-treated H460). Therefore, the decrease in the expression of PKCδ and ε may play a critical role during apoptosis in CH27 and H460 cells. The present study also demonstrated that PKC stimulation occurs at a site downstream of caspase-3 in the emodin-mediated apoptotic pathway. However, the relationship between PKC and caspase-3 in the aloe-emodin-induced apoptosis would be investigated thoroughly in the future.
Acknowledgments
The author thanks S.L. Hsu for her generous support throughout this study. This work was supported by National Science Council Grant NSC 89-2745-P-039-001 and the China Medical College Grant CMC 89-P-12 of the Republic of China.
Abbreviations
- Aloe-emodin
1,8-dihydroxy-3-(hydroxymethyl)-anthraquinone
- CH27 cell
human lung squamous cell carcinoma cell line
- DAPI
4′,6-Diamidino-2-phenylindole dihydrochloride
- DMSO
dimethylsulphoxide
- Emodin
1,3,8-trihydroxy-6-methylanthraquinone
- H460 cell
human lung non-small carcinoma cell line
- PARP
poly(ADP-ribose)polymerase
- PBS
phosphate-buffered saline
- PKC
protein kinase C
- SDS – PAGE
sodium dodecyl sulphate-polyacrylamide gel electrophoresis
- TBST
Tris-buffered saline with Tween
- Tris
tris (hydroxymethyl) aminomethane
References
- ASHKENAZI A., DIXIT V.M. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- BASU A. The potential of protein kinase C as a target for anticancer treatment. Pharmacol. Ther. 1993;59:257–280. doi: 10.1016/0163-7258(93)90070-t. [DOI] [PubMed] [Google Scholar]
- BASU A., AKKARAJU G.R. Regulation of caspase activation and cis-diamminedichloroplatinum(II)-induced cell death by protein kinase C. Biochem. 1999;38:4245–4251. doi: 10.1021/bi982854q. [DOI] [PubMed] [Google Scholar]
- BIALIK S., CRYNS V.L., DRINCIC A., MIYATA S., WOLLOWICK A.L., SRINIVASAN A., KITSIS R.N. The mitochondrial apoptotic pathway is activated by serum and glucose deprivation in cardiac myocytes. Circ. Res. 1999;85:403–414. doi: 10.1161/01.res.85.5.403. [DOI] [PubMed] [Google Scholar]
- BOSSY-WETZEL E., GREEN D.R. Caspases induced cytochrome c release from mitochondria by activating cytosolic factors. J. Biol. Chem. 1999;274:17484–17490. doi: 10.1074/jbc.274.25.17484. [DOI] [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein using the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- COHEN G.M. Caspases: the executioners of apoptosis. Biochem. J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CRYNS V., YUAN J. Proteases to die for. Genes Dev. 1998;12:1551–1570. doi: 10.1101/gad.12.11.1551. [DOI] [PubMed] [Google Scholar]
- DENNING M.F., DLUGOSZ A.A., HOWETT M.K., YUSPA S.H. Expression of an oncogenic rasHa gene in murine keratinocytes induces tyrosine phosphorylation and reduced activity of protein kinase C delta. J. Biol. Chem. 1993;268:26079–26081. [PubMed] [Google Scholar]
- DONG Z., SAIKUMAR P., PATEL Y., WEINBERG J.M., VENKATACHALAM M.A. Serine protease inhibitors suppress cytochrome c-mediated caspase-9 activation and apoptosis during hypoxia-reoxygenation. Biochem. J. 2000;347:669–677. [PMC free article] [PubMed] [Google Scholar]
- FRASER A., EVAN G. A license to kill. Cell. 1996;85:781–784. doi: 10.1016/s0092-8674(00)81005-3. [DOI] [PubMed] [Google Scholar]
- FRUTOS S., MOSCAT J., DIAZ-MECO M.T. Cleavage of zetaPKC but not lambda/iotaPKC by caspase-3 during UV-induced apoptosis. J. Biol. Chem. 1999;274:10765–10770. doi: 10.1074/jbc.274.16.10765. [DOI] [PubMed] [Google Scholar]
- FUJII T., GARCIA-BERMEJO M.L., BERNABO J.L., CAAMANO J., OHBA M., KUROKI T., LI L., YUSPA S.H., KAZANIETZ M.G. Involvement of protein kinase C δ (PKCδ) in phorbol ester-induced apoptosis in LNCaP prostate cancer cells. J. Biol. Chem. 2000;275:7574–7582. doi: 10.1074/jbc.275.11.7574. [DOI] [PubMed] [Google Scholar]
- GOMEZ-ANGELATS M., BORTNER C.D., CIDLOWSKI J.A. Protein kinase C (PKC) inhibits fas receptor-induced apoptosis through modulation of the loss of K+ and cell shrinkage. A role for PKC upstream of caspases. J. Biol. Chem. 2000;275:19609–19619. doi: 10.1074/jbc.M909563199. [DOI] [PubMed] [Google Scholar]
- GREEN D.R., BISSONNETTE R.P., COTTER T.G. Apoptosis and cancer. Principles Practice Oncol. 1994;8:1–14. [PubMed] [Google Scholar]
- HSU S.L., YIN S.C., LIU M.C., REICHERT U., HO W.L. Involvement of cyclin-dependent kinase activities in CD437-induced apoptosis. Exp. Cell. Res. 1999;252:332–341. doi: 10.1006/excr.1999.4625. [DOI] [PubMed] [Google Scholar]
- JAO H.C., YANG R.C., HSU H.K., HSU C. The decrease of PKCalpha is associated with hepatic apoptosis at early and late phases of polymicrobial sepsis. Shock. 2001;15:130–134. doi: 10.1097/00024382-200115020-00009. [DOI] [PubMed] [Google Scholar]
- JUN C.D., OH C.D., KWAK H.J., PAE H.O., YOO J.C., CHOI B.M., CHUN J.S., PARK R.K., CHUNG H.T. Overexpression of protein kinase C isoforms protects RAW 264.7 macrophages from nitric oxide-induced apoptosis: Involvement of c-jun N-terminal kinase/stress-activated protein kinase, p38 kinase, and cpp-32 protease pathways. J. Immunol. 1999;162:3395–3401. [PubMed] [Google Scholar]
- KLUCK R.M., BOSSY-WETZEL E., GREEN D.R., NEWMEYER D.D. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- KORIYAMA H., KOUCHI Z., UMEDA T., SAIDO T.C., MOMOI T., ISHIURA S., SUZUKI K. Proteolytic activation of protein kinase C delta and epsilon by caspase-3 in U937 cells during chemotherapeutic agent-induced apoptosis. Cell. Signal. 1999;11:831–838. doi: 10.1016/s0898-6568(99)00055-8. [DOI] [PubMed] [Google Scholar]
- LEE H.Z., WU C.H. Serotonin-induced protein kinase C activation in cultured rat heart endothelial cells. Eur. J. Pharmacol. 2000;403:195–202. doi: 10.1016/s0014-2999(00)00495-7. [DOI] [PubMed] [Google Scholar]
- LI L., LORENZO P.S., BOGI K., BLUMBERG P.M., YUSPA S.H. Protein kinase Cdelta targets mitochondria, alters mitochondrial membrane potential, and induces apoptosis in normal and neoplastic keratinocytes when overexpressed by an adenoviral vector. Mol. Cell. Biol. 1999;19:8547–8558. doi: 10.1128/mcb.19.12.8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI W., MISCHAK H., YU J.C., WANG L.M., MUSHINSKI J.F., HEIDARAN M.A., PIERCE J.H. Tyrosine phosphorylation of protein kinase C-delta in response to its activation. J. Biol. Chem. 1994;269:2349–2352. [PubMed] [Google Scholar]
- LI P., NIJHAWAN D., BUDIHARDJO I., SRINIVASULA S., AHMAD M., ALNEMRI E., WANG X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- LIANG J.W., HSIU S.L., HUANG H.C., LEE-CHAO P.D. HPLC analysis of emodin in serum, herbs and Chinese herbal prescriptions. J. Food Drug Anal. 1993;1:251–257. [Google Scholar]
- LIU X., KIM C.N., YANG J., JEMMERSON R., WANG X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- PAE H.O., YOO J.C., CHOI B.M., LEE E.J., SONG Y.S., CHUNG H.T. 12-O-tetradecanoyl phorbol 13-acetate, protein kinase C (PKC) activator, protects human leukemia HL-60 cells from taxol-induced apoptosis: possible role for extracellular signal-regulated kinase. Immunopharmacol. Immunotoxicol. 2000;22:61–73. doi: 10.3109/08923970009016406. [DOI] [PubMed] [Google Scholar]
- PECERE T., GAZZOLA M.V., MUCIGNAT C., PAROLIN C., VECCHIA F.D., CAVAGGIONI A., BASSO G., DIASPRO A., SALVATO B., CARLI M., PALU G. Aloe-emodin is a new type of anticancer agent with selective activity against neuroectodermal tumor. Cancer Res. 2000;60:2800–2804. [PubMed] [Google Scholar]
- REED J.C. Cytochrome c: Can't live with it; Can't live without it. Cell. 1997;91:559–562. doi: 10.1016/s0092-8674(00)80442-0. [DOI] [PubMed] [Google Scholar]
- SHANMUGAM M., KRETT N.L., PETERS C.A., MAIZELS E.T., MURAD F.M., KAWAKATSU H., ROSEN S.T., HUNZICKER-DUNN M. Association of PKC delta and active Src in PMA-treated MCF-7 human breast cancer cells. Oncogene. 1998;16:1649–1654. doi: 10.1038/sj.onc.1201684. [DOI] [PubMed] [Google Scholar]
- SHINOURA N., YOSHIDA Y., NISHIMURA M., MURAMATSU Y., ASAI A., KIRINO T., HAMADA H. Expression level of Bcl-2 determines anti-or proapoptotic function. Cancer Res. 1999;59:4119–4128. [PubMed] [Google Scholar]
- SONG J.S., SWANN P.G., SZALLASI Z., BLANK U., BLUMBERG P.M., RIVERA J. Tyrosine phosphorylation-dependent and -independent associations of protein kinase C-delta with Src family kinases in the RBL-2H3 mast cell line: regulation of Src family kinase activity by protein kinase C-delta. Oncogene. 1998;16:3357–3368. doi: 10.1038/sj.onc.1201886. [DOI] [PubMed] [Google Scholar]
- STABEL S., PARKER P.J. Protein kinase C. Pharmacol. Ther. 1991;51:71–95. doi: 10.1016/0163-7258(91)90042-k. [DOI] [PubMed] [Google Scholar]
- SUN X.M., MACFARLANE M., ZHUANG J., WOLF B.B., GREEN D.R., COHEN G.M. Distinct caspase cascades are initiated in receptor-mediated and chemical-induced apoptosis. J. Biol. Chem. 1999;274:5053–5060. doi: 10.1074/jbc.274.8.5053. [DOI] [PubMed] [Google Scholar]
- TANAKA T., TSUJIO I., NISHIKAWA T., SHINOSAKI K., KUDO T., TAKEDA M. Significance of tau phosphorylation and protein kinase regulation in the pathogenesis of Alzheimer disease. Alzheimer Dis. Assoc. Disorders. 2000;14:S18–S24. doi: 10.1097/00002093-200000001-00004. [DOI] [PubMed] [Google Scholar]
- TSAI T.H., CHEN C.F. Ultraviolet spectrum identification of emodin in rabbit plasma by HPLC and its pharmacokinetics application. Asia Pac. J. Pharmacol. 1992;7:53–56. [Google Scholar]
- TSUJIO I., TANAKA T., KUDO T., NISHIKAWA T., SHINOZAKI K., GRUNDKE-IQBAL I., IQBAL K., TAKEDA M. Inactivation of glycogen synthase kinase-3 by protein kinase C delta; implications for regulation of tau phosphorylation. FEBS Letts. 2000;469:111–117. doi: 10.1016/s0014-5793(00)01234-5. [DOI] [PubMed] [Google Scholar]
- YANG F., ZHANG T., TIAN G., CAO H., LIU Q., ITO Y. Preparative isolation and purification of hydroxyanthraquinones from Rheum officinale Baill by high-speed counter-current chromatography using pH-modulated stepwise elution. J. Chromatogr. A. 1999;858:103–107. doi: 10.1016/s0021-9673(99)00827-4. [DOI] [PubMed] [Google Scholar]
- ZHANG L., CHANG C.J., BACUS S.S., HUNG M.C. Suppressed transformation and induced differentiation of HER-2/neu-overexpressing breast cancer cells by emodin. Cancer Res. 1995;55:3890–3896. [PubMed] [Google Scholar]
- ZHANG L., HUNG M.C. Sensitization of HER-2/neu-overexpressing non-small cell lung cancer cells to chemotherapeutic drugs by tyrosine kinase inhibitor emodin. Oncogene. 1996;12:571–576. [PubMed] [Google Scholar]
- ZHANG L., LAU Y.K., XI L., HONG R.L., KIM D.S., CHEN C.F., HORTOBAGYI G.N., CHANG C.J., HUNG M.C. Tyrosine kinase inhibitor, emodin and its derivative repress HER-2/neu-induced cellular transformation and metastasis-associated properties. Oncogene. 1998;16:2855–2863. doi: 10.1038/sj.onc.1201813. [DOI] [PubMed] [Google Scholar]