

Abstract
We have studied the effects of a novel agonist, solid-phase von Willebrand Factor (sVWF), on tumour cell-induced platelet aggregation (TCIPA).
Washed platelet suspensions were obtained from human blood and the effects of HT-1080 human fibrosarcoma cells and sVWF on platelets were studied using aggregometry, phase-contrast microscopy, and flow cytometry.
Incubation of platelets with sVWF (1.2 μg ml−1) and HT-1080 cells (5×103 ml−1) resulted in a two-phased reaction characterized first by the adhesion of platelets to sVWF, then by aggregation.
TCIPA in the presence of sVWF was inhibited by S-nitroso-glutathione (GSNO, 100 μM) and prostacyclin (PGI2, 30 nM).
Platelet activation in the presence of tumour cells and sVWF resulted in the decreased surface expression of platelet glycoprotein (GP)Ib and up-regulation of GPIIb/IIIa receptors.
Pre-incubation of platelets with PGI2 (30 nM) resulted in inhibition of sVWF-tumour cell-stimulated platelet surface expression of GPIIb/IIIa as measured by flow cytometry using antibodies directed against both non-activated and activated receptor. In contrast, GSNO (100 μM) did not affect sVWF-tumour cell-stimulated platelet surface expression of GPIIb/IIIa.
Flow cytometry performed with PAC-1 antibodies that bind only to the activated GPIIb/IIIa revealed that GSNO (100 μM) caused inhibition of activation of GPIIb/IIIa.
The inhibitors exerted no significant effects on TCIPA-mediated changes in GPIb.
Thus, sVWF potentiates the platelet-aggregatory activity of HT-1080 cells and these effects appear to be mediated via up-regulation of platelet GPIIb/IIIa.
Prostacyclin and NO inhibit TCIPA-sVWF-mediated platelet aggregation. The mechanisms of inhibition of this aggregation by PGI2 differ from those of NO.
Keywords: HT-1080 fibrosarcoma cells, platelets, adhesion, aggregation, solid-phase von Willebrand factor, glycoprotein Ib, glycoprotein IIb/IIIa, nitric oxide, prostacyclin
Introduction
During the metastatic cascade the haematogenous spread of cancer cells can be accomplished by tumour cell-containing thrombi. Cancer cells have the ability to aggregate platelets (Gasic et al., 1968; 1973), which are the major components of arterial thrombi, and this action correlates with the metastatic potential of tumour cells (Radomski et al., 1991). Tumour cell-induced platelet aggregation (TCIPA) confers a number of advantages to the survival of cancer cells in the vasculature and to metastasis. Once covered with a coat of platelets the tumour cell acquires the ability to elude the body's defence system (Shau et al., 1993; Philippe et al., 1993). In addition, the platelets may shield the cancer cells from the high shear forces seen in flowing blood that could potentially damage the tumour cell. Moreover, aggregated platelets release a number of growth factors that can be used by tumour cells for growth (Honn et al., 1992). Furthermore, tumour cell – platelet aggregates embolize in the microvasculature, allowing tumour cells to adhere to damaged endothelium at an extravasation site (Mehta, 1984; Crissman et al., 1985). The pathways and mechanisms that mediate TCIPA have now been studied and are likely to involve the release of thromboxane A2, ADP, and matrix metalloproteinase-2 from the aggregates (Bastida et al., 1982; Pearlstein et al., 1981; Honn et al., 1987; Steinert et al., 1993; Jurasz et al., 2001). However, little is known about the interactions of platelet – tumour cell aggregates with von Willebrand factor (VWF) and its platelet receptors glycoprotein (GP)Ib and GPIIb/IIIa (Weiss et al., 1974; Nurden & Caen, 1974; Caen & Rosa, 1995; Ruggeri, 1999). Von Willebrand factor is a major adhesion protein that mediates platelet attachment to the vascular wall and this protein can interact both with GPIb and GPIIb/IIIa receptors. VWF is a large multimeric protein found in plasma, platelets, endothelial cells, and in the subendothelium.
GPIIb/IIIa is a platelet specific surface transmembrane receptor that belongs to the integrin superfamily. It is the most abundant platelet receptor with roughly 80,000 copies on the surface of each platelet. In addition, platelets contain a pool of GPIIb/IIIa stored in platelet α granules (Wagner et al., 1996) that is released onto the platelet surface upon platelet activation. Furthermore, GPIIb/IIIa undergoes activation and a conformational change that allows binding to its primary ligands fibrinogen and VWF (Ruggeri et al., 1999) to mediate platelet – platelet and platelet – vessel wall interactions.
GPIb belongs to the leucine-rich superfamily of receptors. It complexes with glycoproteins IX and V and forms an active receptor. The major ligand of GPIb is VWF and its binding to the receptor mediates platelet adhesion and arrest at the damaged fragment of the vessel wall. However, shear forces generated under conditions of flowing blood may also result in VWF-mediated platelet aggregation (Ruggeri, 1999).
Agonist-induced platelet surface receptor expression can be modulated by a number of anti-platelet agents. Nitric oxide, a well-known inhibitor of platelet activation and aggregation (Radomski et al., 1987a,1987b; 1991), has been shown to affect GPIIb/IIIa receptor expression during platelet activation (Salas et al., 1994). Moreover, prostacyclin (PGI2), the most potent known inhibitor of platelet aggregation (Moncada et al., 1976), prevents the mobilization of platelet GPIIb/IIIa in thrombin-activated platelets (Graber & Hawiger, 1982). In addition, it has been shown that GPIb/IX/V can be modified by the phosphorylation of Ser166 by cyclic AMP-dependent kinase (Wyler et al., 1986; Wardell et al., 1989) that is activated by PGI2.
Recently, Stewart et al. (1997) reported that VWF when immobilized on polystyrene beads acts as a novel solid-phase agonist (sVWF) in human platelet-rich plasma. In the paper accompanying this work we have found that sVWF stimulates both adhesion and agonist-induced aggregation of human platelets and that these effects could be regulated by NO and PGI2 (Radomski et al., in preparation). Therefore, the aim of our present study was to investigate the effects of sVWF, on TCIPA. In addition, we studied the expression of platelet GPIIb/IIIa and GPIb during these reactions and the regulation of these two receptors by NO and PGI2.
Methods
Blood platelets
Blood was collected from healthy volunteers who had not taken any drugs for 14 days prior to the study. Washed platelet suspensions (2.5×1011 l−1) were prepared as previously described (Radomski & Moncada, 1983).
Tumour cell culture
A human tumour cell line HT-1080 fibrosarcoma was obtained from the American Type Culture Collection (Rockville, MD, U.S.A.) and prepared as previously described (Jurasz et al., 2001). The cells were resuspended in Tyrode's solution at a concentration of 5×105 ml−1 (Jurasz et al., 2001). All cell culture reagents were purchased from Sigma (Oakville, ONT, Canada).
Platelet aggregation
Washed platelets were pre-incubated for 2 min at 37°C in a Chronolog whole blood ionised calcium lumi-aggregometer. Platelet aggregation was then initiated by the addition of HT-1080 cells (5×103 cells ml−1) and monitored by Aggro-Link software (Jurasz et al., 2001). Platelet aggregation was measured as an extent of light transmittance and then expressed as a per cent of maximal stimulus taken at a time point when the maximal stimulus reached 50% transmittance (Jurasz et al., 2001). Platelet samples for flow cytometry were taken at this time point.
Flow cytometry
Platelet flow cytometry was performed using a Becton Dickinson flow cytometer (FACSCalibur) equipped with a 488 nm wavelength argon laser, 525 and 575 nm band pass filters for the detection of fluorescein isothiocyanate and R-phycoerythrin fluorescence, and with Cell Quest software. Flow cytometry was performed on both single and double stained platelet samples. To account for spectral overlap between fluorescein isothiocyanate and R-phycoerythrin labels software compensation was performed in experiments using double stained platelet samples. To minimize the presence of aggregates in samples of platelets (10 μl of suspension) and fluorescent-labelled antibodies (10 μl containing 1 μg of anti-GPIIb and GPIb antibodies, and 10 μl containing 0.25 μg of PAC-1 antibodies) were diluted 10 fold using physiological saline. Platelets were identified by forward and side scatter signals. Ten to twenty thousand platelet specific events were initially analysed by the cytometer. Non-activated and activated platelets were gated so as not to analyse platelet aggregates and microparticles. Furthermore, the gated whole platelet population was arbitrarily sub-gated into large-, medium-, and small-sized platelet populations (Radomski et al., in preparation). The gates were then analysed for mean fluorescence.
Microscopy
Tumour cell-platelet samples were viewed using phase-contrast microscopy equipped with a Nikon camera (Jurasz et al., 2001).
Reagents
Prostacyclin and S-nitroso-glutathione (GSNO) were obtained from Sigma (Oakville, ONT, Canada). These reagents were incubated with platelets for 2 min prior to the addition of tumour cells. Fluorescein isothiocyanate conjugated (FITC) monoclonal mouse anti-platelet GPIIb (human CD41) antibodies and R-phycoerythrin conjugated (RPE) monoclonal mouse anti-platelet GPIb (human CD42b) antibodies were obtained from DAKO (Mississauga, ONT, Canada). PAC-1 FITC monoclonal mouse anti-platelet GPIIb/IIIa antibodies were obtained from Becton Dickinson (San José, CA, U.S.A.). Von Willebrand factor coated polystyrene beads (Stewart et al., 1997) were provided by Thrombotics Inc (Edmonton, AB, Canada). The beads were washed and resuspended in equivalent volume of saline and used for the experiments.
Statistics
Statistics were performed using Graph Pad Software Prism 3.0 (San Diego, CA, U.S.A.). All means are reported with standard error. One-way analysis of variance (ANOVA), Tukey – Kramer multiple comparisons test, and paired Student t-tests were performed where appropriate, and a P-value of less than 0.05 was considered as significant.
Results
TCIPA in the presence of sVWF
Incubation of HT-1080 cells with platelets resulted in TCIPA (Figure 1). This effect was potentiated in the presence of sVWF (Figure 1), as evidenced by the faster onset of aggregation. Furthermore, TCIPA in the presence of sVWF was two-phased. The first phase marked platelet adhesion to the immobilized sVWF and the second phase platelet – platelet – tumour cell aggregation (Figure 1A,B).
Figure 1.
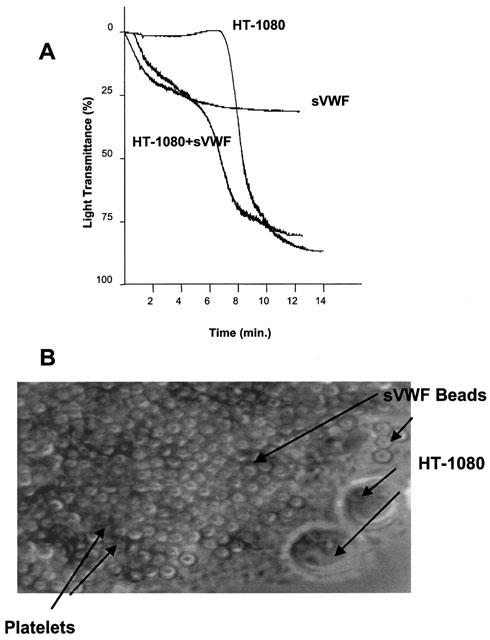
(A) Representative traces showing TCIPA in the presence or absence of sVWF (1.6 μg ml−1) (representative of six experiments). TCIPA was induced by HT-1080 cells (5×103 cells ml−1). (B) Phase-contrast microscopy of HT-1080 induced TCIPA in the presence of sVWF, ×400.
Effects of GSNO and PGI2
GSNO (100 μM) and PGI2 (30 nM) both significantly (P<0.01 and P<0.001, respectively; n=6) inhibited HT-1080 induced platelet aggregation (Figure 2A,B). Preincubation of platelets with GSNO (100 μM) and PGI2 (30 nM) also significantly (P=0.0016; n=6, and P=0.0003; n=3) inhibited TCIPA in the absence of sVWF. The latter observation is consistent with our previous findings (Jurasz et al., 2001).
Figure 2.
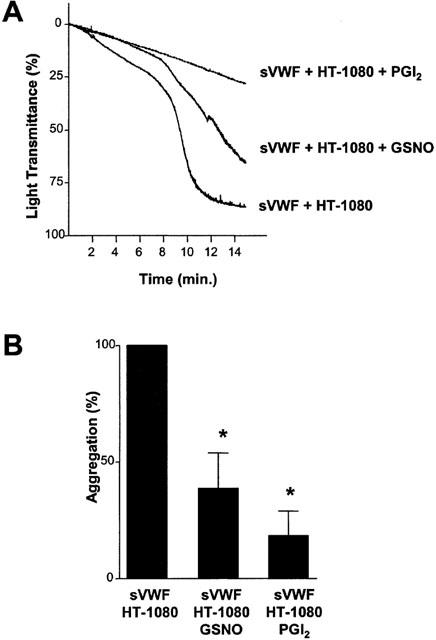
Representative traces (A) and the statistical analysis (B) showing the effects of GSNO (100 μM) and PGI2 (30 nM) on TCIPA in the presence of sVWF (1.6 μg ml−1). TCIPA was induced by HT-1080 cells (5×103 cells ml−1). Bars are mean±s.e.mean from six separate experiments. *P<0.05, treatments versus control.
GPIb and GPIIb/IIIa during the adhesion phase of TCIPA
In the absence of HT-1080 cells, sVWF caused an increase in the surface expression of platelet GPIb, as evidenced by a significant (P=0.0283; n=4) increase in mean fluorescence (Figure 3A). However, during the platelet adhesion phase of TCIPA in the presence of sVWF, HT-1080 cells caused a decrease in the platelet surface expression of GPIb, as compared to sVWF alone. This was evidenced by the significant (P=0.0231, n=4) decrease in mean fluorescence (Figure 3B).
Figure 3.
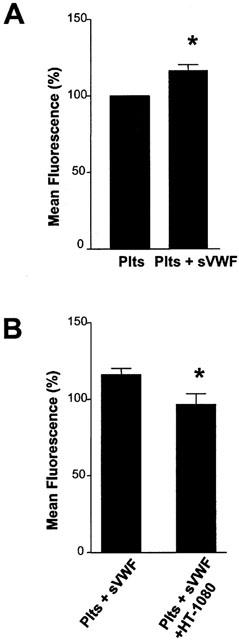
Effects of sVWF (1.6 μg ml−1) on platelet surface expression of GPIb in the absence (A) or presence (B) of HT-1080 cells. TCIPA was induced by HT-1080 cells (5×103 cells ml−1). Bars are mean±s.e.mean from four separate experiments. *P<0.05, treatment versus control.
No significant changes (P>0.05, n=3) in the surface expression of GPIIb/IIIa were detected during these experiments.
GPIIb/IIIa and GPIb during the aggregation phase of TCIPA
During the aggregation phase of TCIPA in the presence of sVWF GPIIb/IIIa expression significantly (P<0.01, n=4) increased (Figure 4A). Similar, during TCIPA in the absence of sVWF there was a significant (P=0.0010, n=9) increase in the platelet surface expression of GPIIb/IIIa. Platelet activation with sVWF in the absence of HT-1080 cells did not cause any changes in GPIIb/IIIa (Figure 4A).
Figure 4.
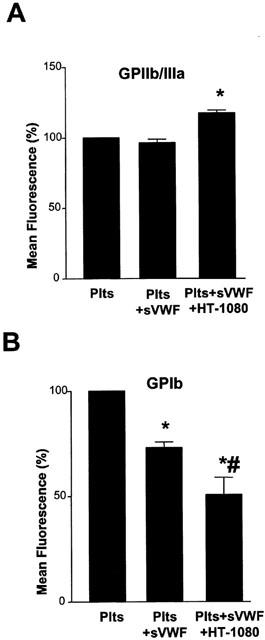
Effects of HT-1080 cells and sVWF (1.6 μg ml−1) on GPIIb/IIIa and GPIb expression during the aggregation phase of TCIPA (5×103 cells ml−1). Bars are mean±s.e.mean from four separate experiments. *P<0.05, treatments versus control. #P<0.05, platelets and VWF versus platelets and VWF and HT-1080 cells.
TCIPA in the presence of sVWF resulted in a significant (P<0.001, n=4) decrease in platelet surface GPIb (Figure 4B). TCIPA in the absence of sVWF also led to a significant (P=0.0001, n=9) decrease in platelet surface expression of GPIb (Figure 4B).
Effects of GSNO and PGI2 on both non-activated and activated GPIIb/IIIa
Flow cytometry performed on platelets pre-incubated with GSNO (100 μM) or PGI2 (30 nM) and then activated by HT-1080 cells and sVWF was analysed by examining different sized platelet populations. PGI2, but not GSNO, in large, medium and small platelet subpopulations significantly (P<0.01, P<0.05 and P<0.01, respectively, n=6) inhibited HT-1080-mediated increase in the surface number of platelet GPIIb/IIIa (Figure 5A – C). In contrast, GSNO failed to prevent the increase in platelet surface GPIIb/IIIa (P>0.05; n=6) (Figure 5A – C).
Figure 5.
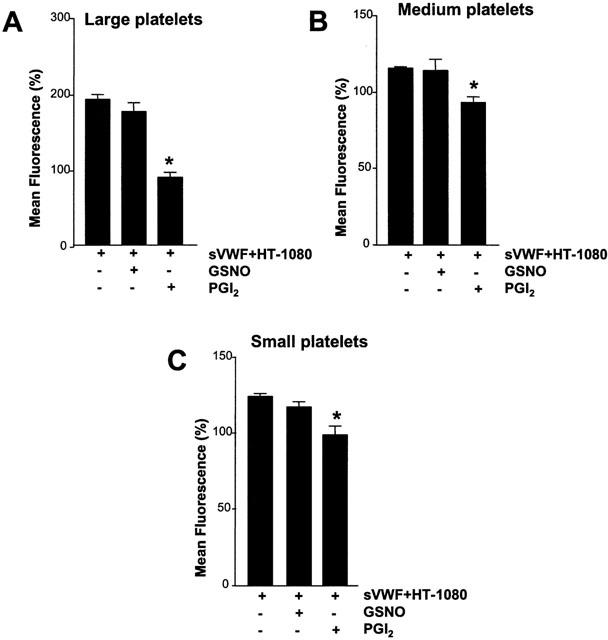
Effects of GSNO (100 μM) and PGI2 (30 nM) on expression of platelet GPIIb/IIIa in different-sized platelet populations during TCIPA. (A) Large platelets, (B) Medium platelets, and (C) Small platelets. TCIPA was induced by HT-1080 cells (5×103 cells ml−1) and sVWF (1.6 μg ml−1). Bars are mean±s.e.mean from six separate experiments. *P<0.05, treatments versus control. + and − denote the presence or absence of treatments, respectively.
Effects of GSNO on activated GPIIb/IIIa
Flow cytometry performed with the PAC-1 antibody that recognizes only the activated GPIIb/IIIa showed that in large, medium, and small platelet subpopulations GSNO (100 μM) significantly (P=0.0103, P=0.0176 and P=0.0110, respectively, n=3) inhibited the activation of GPIIb/IIIa during TCIPA (Figure 6A – C). GSNO (100 μM) caused also inhibition of activation of GPIIb/IIIa during TCIPA in the absence of VWF, as evidenced by the significant (P=0.0284, n=6) decrease in mean fluorescence.
Figure 6.
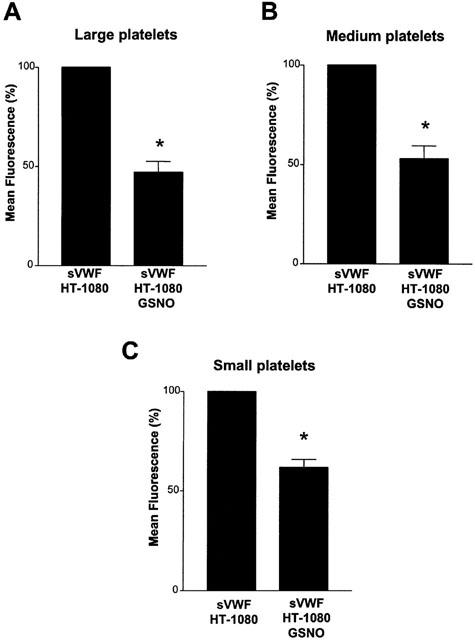
Effects of GSNO (100 μM) on the activation of platelet GPIIb/IIIa during TCIPA in the presence of SVWF measured with the PAC-1 antibody. TCIPA was induced by HT-1080 cells (5×103 cells ml−1) and sVWF (1.6 μg ml−1). Bars are mean±s.e.mean from three separate experiments. *P<0.05, treatment versus control.
Discussion
The objective of our investigation was to study TCIPA in the presence of sVWF, a novel solid-phase agonist (Stewart et al., 1997). As VWF is a major adhesion protein contributing to the interactions between platelets and the vascular wall (Ruggeri et al., 1999), the use of sVWF added yet another dimension to studies on TCIPA that have considered until now only the platelet – tumour cell relationship (Honn et al., 1981; Radomski et al., 1991; Jurasz et al., 2001). In addition, we investigated the effects of NO and PGI2, two known inhibitors of platelet aggregation (Radomski et al., 1987a,1987b; Moncada et al., 1976) and TCIPA (Radomski et al., 1991; Jurasz et al., 2001; Honn et al., 1981; Menter et al., 1984) on the TCIPA-sVWF-induced platelet activation.
As reported before, (Jurasz et al., 2001), human fibrosarcoma cells HT-1080 caused TCIPA. Preincubation of platelet samples with sVWF resulted in potentiation of TCIPA. This reaction consisted of two phases: the first dependent upon platelet adhesion to sVWF, and the second characterized by formation of tight sVWF – platelet – tumour cell aggregates. These data show that VWF appears to play a crucial role in the interactions between cancer cells and platelets in vitro, and most likely in vivo. In this context, it is interesting to note that numerous studies have shown that cancer patients have elevated levels of plasma VWF, in addition to other coagulation factors, which all may contribute to thrombotic risk. These include laryngeal (Paczuski et al., 1999), renal (Oleksowicz et al., 1999), colorectal (van duijnhoven et al., 1993), cervical (Gadducci et al., 1993), prostate (Ablin et al., 1988), and head and neck cancers (Sweeney et al., 1990). Moreover, Oleksowicz et al. (1999) have shown that patients with metastatic disease have elevated levels of highly polymeric forms of VWF due to a deficiency of VWF-processing protease. Furthermore, increased thrombotic readiness detected in cancer patients may be due to the elevated plasma levels of VWF (Green et al., 1997). In addition, patients with malignant breast cancer have increased VWF content in the cytosol of the malignant tissue (Pratt et al., 1989). Importantly, it has been demonstrated that the staging and tumour size correlate with disease progression whereby patients with higher plasma VWF have more advanced disease (Paczuski et al., 1999; Gadducci et al., 1993; Pratt et al., 1989). Experimentally, co-culture of human HRT-18 colon cancer cells with human umbilical vein endothelial cells leads to an increase in VWF release from the endothelial cells and this may lead to enhanced platelet adhesion (Morganti et al., 1996). Finally, platelet aggregation induced by human MCF breast cancer cells is increased in cancer patients with elevated levels of VWF (Oleksowicz et al., 1999). The mechanisms that lead to an increase in plasma VWF in cancer patients remain unclear; however, they may include damage to the endothelium, platelet activation and aggregation or tumour angiogenesis. Whatever the mechanisms underlie increased release of VWF, clinical and experimental evidence strongly indicate that this protein, along with others such as tissue factor, plays an integral role in haematogenous spread of cancer.
We have also studied the mechanisms of the activator effects of sVWF on TCIPA focusing upon two major platelet receptors GPIb and GPIIb/IIIa.
In agreement with our previous work (Radomski et al., in preparation), preincubation of platelets with sVWF alone caused increased surface expression of GPIb. This enhanced expression of GPIb is likely to support platelet adhesion to this ligand (Radomski et al., in preparation) in light of GPIbα/VWF-mediated tethering (Ruggeri, 1999). In contrast, TCIPA resulted in a significant reduction of GPIb expression on platelet surface both in the presence or absence of sVWF. Decreased surface expression of GPIb has been already reported as a result of thrombin-induced platelet aggregation or the exposure of platelet to the foreign surface (Keh et al., 1996; Michelson et al., 1996; Mellgren et al., 1995). This reduction could be a result of receptor internalization or proteolysis and shedding (Mellgren et al., 1995, Hughes et al., 2000; Bergmeier et al., 2000; Kinlough-Rathbone et al., 2000). The biological significance of reduced expression of GPIb during aggregatory reactions is unclear.
In contrast to GPIb, TCIPA-sVWF-mediated platelet activation was clearly associated with increased surface expression of GPIIb/IIIa emphasizing, once again, an important role of this receptor in platelet activation by tumour cells (Jurasz et al., 2001).
Platelet activation induced by tumour cells in the presence of sVWF was inhibited by NO and prostacyclin highlighting further the regulatory role of these mediators in the interactions between platelets, the vessel wall and tumour cells. Others and we have previously shown that these inhibitors down-regulate TCIPA (Honn et al., 1981; Radomski et al., 1991; Jurasz et al., 2001). Since the regulatory effects of NO and PGI2 on agonist-induced platelet aggregation are dependent on modulation of platelet receptor glycoprotein function, we investigated the effects of these inhibitors on TCIPA-sVWF-stimulated changes in GPIb and GPIIb/IIIa expression. PGI2 inhibited the surface expression of GPIIb/IIIa, while GSNO did not. Since both inhibitors reduced TCIPA-sVWF-stimulated platelet activation, differential effects of NO and PGI2 on GPIIb/IIIa expression were surprising. Therefore, we investigated whether GSNO had any effects on the activation of GPIIb/IIIa using the PAC-1 antibody, which recognizes the activated form of GPIIb/IIIa. The study revealed that NO inhibited the activation of GPIIb/IIIa caused by cancer cells. Recently, Keh et al. (1996) have shown that GSNO inhibits both the increased surface expression and activation of GPIIb/IIIa in platelets stimulated by thrombin. Our own data show that GSNO only inhibited GPIIb/IIIa activation. This discrepancy may be due to procedures used for sample preparation and/or the nature of the platelet activating agents. To measure total GPIIb/IIIa by flow cytometry Keh et al. (1996) fixed their platelet samples and stored them at 4°C for up to 4 h. Measurement of activated GPIIb/IIIa with the PAC-1 antibody, in our experiments, was performed on platelet samples that were not fixed. Fixation of platelets may alter platelet surface receptor expression and/or impair fluorescence intensity. The timing of flow cytometry assays of non-fixed samples is crucial; therefore, all our measurements were performed precisely at 5 min after sample acquisition. Furthermore, the discrepancy in results may arise from the differences between platelet activation by thrombin versus HT-1080 fibrosarcoma cells. Indeed, we have shown that platelet activation by HT-1080 cells is a complex process that involves the activation of the ADP, thromboxane, and MMP-2 pathways of platelet aggregation (Jurasz et al., 2001). Activation and aggregation of platelets by cancer cells is not only a complex process, but also a very potent one. Indeed, 103 cancer cells could induce aggregation of 108 platelets. Thus, NO may not be able to inhibit up-regulation of platelet surface GPIIb/IIIa induced by HT-1080 cells, but it does inhibit the conformational change in GP IIb/IIIa that precedes platelet aggregation. In contrast to NO, PGI2 down-regulated an increase in GPIIb/IIIa surface expression induced by TCIPA-sVWF.
Interestingly, it has been shown that tyrosine phosphorylation of platelet proteins associated with aggregation is inhibited by cyclic AMP-, but not cyclic GMP-elevating agents (Pumiglia et al., 1990). As tyrosine phosporylation is linked to GPIIb/IIIa function (Golden et al., 1990; Parise et al., 1990), a differential regulation of function of this receptor by NO and PGI2 may be explained by the differences in signal transduction mechanisms operated by the PGI2-cyclic AMP and NO-cyclic GMP systems. Neither PGI2 nor NO exerted any significant effects on reduction of GPIb surface expression caused by TCIPA-sVWF. This data indicates that inhibition of TCIPA-sVWF by NO and PGI2 is related to their interactions with GPIIb/IIIa rather than GPIb.
Platelet activation leads to the formation of platelet particles of various sizes. Therefore, in our flow cytometry assay we have analysed the effects of stimulators of aggregation (HT-1080 cells and sVWF) and inhibitors (PGI2 and GSNO) on large, medium, and small sized platelet populations. We found that these activators and inhibitors affected different sized platelet populations in the same manner. We feel that our model of TCIPA including sVWF may be of use to understand further the process of haematogenous metastasis of cancer. It is likely that a haematogenous metastasizing cancer cell should follow the path of least resistance (Cotmore & Carter, 1973). This means that the cell will arrest and extravasate at a site in the vasculature where vascular integrity has been disrupted. This site of vascular injury will undoubtedly already have platelets adhering and aggregating to it in an effort to control bleeding and to repair the damaged vessel. Therefore, our data suggests that the ability of the tumour cell to aggregate platelets and consequently upregulate platelet surface GPIIb/IIIa gives the tumour cell – platelet aggregate the advantage of engaging platelets already in the process of aggregation at the site of vascular injury.
In conclusion, we have demonstrated that sVWF amplifies TCIPA. The effects of sVWF are largely mediated via increased expression of GPIIb/IIIa in platelets. Finally, these actions of sVWF are modulated by NO and prostacyclin.
Acknowledgments
This work was supported by a grant from the Canadian Institutes of Health Research (CIHR grant 14074) to M.W. Radomski. P. Jurasz is a recipient of a fellowship from Canada Lung Association. M.W. Radomski is a CIHR Scientist. We are indebted to Mrs Concetta Carbanaro, Barbara Litwinowich and Margo Miller for their assistance in blood collection.
Abbreviations
- GPIb
Glycoprotein Ib
- GPIIb/IIIa
glycoprotein IIb/IIIa
- GSNO
S-nitroso-glutathione
- PGI2
prostacyclin
- sVWF
solid-phase von Willebrand factor
- TCIPA
tumour cell-induced platelet aggregation
References
- ABLIN R.J., BARTKUS J.M., GONDER M.J. Immunoquantitation of factor VIII-related antigen (von Willebrand factor antigen) in prostate cancer. Cancer Letts. 1988;40:283–289. doi: 10.1016/0304-3835(88)90087-0. [DOI] [PubMed] [Google Scholar]
- BASTIDA E., ORDINAS A., GIARDINA S.L., JAMIESON G.A. Differentiation of platelet-aggregating effects of human tumor cell lines based on inhibition of studies with apyrase, hirudin, and phospholipase. Cancer Res. 1982;42:4348–4352. [PubMed] [Google Scholar]
- BERGMEIER W., RACKENBRANDT K., SCHRODER W., ZIRNGIBL H., NIESWANDT E. Structural and functional characterization of the mouse von Willebrand factor receptor GPIb-IX with novel monoclonal antibodies. Blood. 2000;95:886–893. [PubMed] [Google Scholar]
- CAEN J.P., ROSA J.P. Platelet-vessel wall interaction: from the bedside to molecules. Thromb. Haemostas. 1995;74:18–24. [PubMed] [Google Scholar]
- COTMORE S.F., CARTER R.L. Mechanisms of enhanced intrahepatic metastasis in surfactant treated hampsters: an electron microscopy study. Int. J. Cancer. 1973;11:725–738. doi: 10.1002/ijc.2910110324. [DOI] [PubMed] [Google Scholar]
- CRISSMAN J.D., HATFIELD J., SCHALDENBRAND M., SLOANE B.F., HONN K.V. Arrest and extravasation of B16 amelanotic melanoma in murine lungs. A light and electron microscopic study. Lab Invest. 1985;53:470–478. [PubMed] [Google Scholar]
- GADDUCCI A., BAICCHI U., MARRAI R., FACCHINI V., DEL BRAVO B., FOSELLA P.V., FIORETTI P. Pretreatment plasma levels of fibrinopeptide-A (FPA), D-dimer (DD), and von Willebrand factor (VWF) in patients with operable cervical cancer: influence of surgical-pathological stage, tumor size, histologic type, and lymph node status. Gynecologic. Oncol. 1993;49:354–358. doi: 10.1006/gyno.1993.1139. [DOI] [PubMed] [Google Scholar]
- GASIC G.J., GASIC T.B., STEWART C.C. Antimetastatic effects associated with platelet reduction. Proc. Natl. Acad. Sci. U.S.A. 1968;48:1172–1177. doi: 10.1073/pnas.61.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GASIC G.J., GASIC T.B., GALANTI N., JOHNSON T., MURPHY S. Platelet-tumor cell interactions in mice. The role of platelets in the spread of malignant disease. Int. J. Cancer. 1973;11:704–718. doi: 10.1002/ijc.2910110322. [DOI] [PubMed] [Google Scholar]
- GOLDEN A., BRUGGE J.S., SHATTIL S.J. Role of platelet membrane glycoprotein IIb-IIIa in agonist induced tyrosine phosphorylation of platelet proteins. J. Cell. Biol. 1990;111:3117–3127. doi: 10.1083/jcb.111.6.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRABER S.E., HAWIGER J. Evidence that changes in platelet cyclic AMP levels regulate the fibrinogen receptor on human platelets. J. Biol. Chem. 1982;257:14606–14609. [PubMed] [Google Scholar]
- GREEN D., MALIEKEL K., SUSHKO E., AKHTAR R., SOFF G.A. Activated-protein-C resistance in cancer patients. Haemostasis. 1997;27:112–118. doi: 10.1159/000217442. [DOI] [PubMed] [Google Scholar]
- HONN K.V., CICONE B., SKOFF A. Prostacyclin: a potent antimetastatic agent. Science. 1981;212:1270–1272. doi: 10.1126/science.7015512. [DOI] [PubMed] [Google Scholar]
- HONN K.V., STEINERT B.W., MOIN K., ONODA J.M., TAYLOR J.D., SLOANE B.F. The role of platelet cyclooxygenase and lipoxygenase pathways in tumor cell induced platelet aggregation. Biochem. Biophysic. Res. Comm. 1987;145:384–389. doi: 10.1016/0006-291x(87)91333-7. [DOI] [PubMed] [Google Scholar]
- HONN K.V., TANG D.G., CHEN Y.Q. Platelets and cancer metastasis: more than an epiphenomenon. Sem. Thromb. Hemost. 1992;18:392–415. doi: 10.1055/s-2007-1002578. [DOI] [PubMed] [Google Scholar]
- HUGHES M., HAYWARD C.P., WARKENTIN T.E., HORSEWOOD P., CHORNEYKO K.A., KELTON G.J. Morphological analysis on microparticle generation in heparin-induced thrombocytopenia. Blood. 2000;96:188–194. [PubMed] [Google Scholar]
- JURASZ P., SAWICKI G., DUSZYK M., SAWICKA J., MIRANDA C., MAYERS I., RADOMSKI M.W. Matrix metalloproteinases 2 in tumor cell-induced platelet aggregation: Regulation by nitric oxide. Canc. Res. 2001;61:376–382. [PubMed] [Google Scholar]
- KEH D., GERLACH M., KURER I., SEILER S., KERNER T., FALKE K.J., GERLACH H. The effects of nitric oxide (NO) on platelet membrane receptor expression during activation with human α-thrombin. Blood Coagul. Fibrinol. 1996;7:615–624. doi: 10.1097/00001721-199609000-00007. [DOI] [PubMed] [Google Scholar]
- KINLOUGH-RATHBONE R.L., PERRY D.W., RAND M.L., PACKHAM M.A. Responses to aggregating agents after cleavage of GPIb of human platelets by the o-sialoglycoprotein endoprotease from Pasteurella haemolytica-potental surrogates for Bernard-Soulier platelets. Thromb. Res. 2000;99:165–172. doi: 10.1016/s0049-3848(00)00240-1. [DOI] [PubMed] [Google Scholar]
- MEHTA P. Potential role of platelets in the pathogenesis of tumor metastasis. Blood. 1984;63:55–63. [PubMed] [Google Scholar]
- MELLGREN K., FRIBERG L.G., HADNER T., MELLGREN G., WADENVIK H. Blood platelet activation and membrane glycoprotein changes during extracorporel life support (ECLS). In vitro studies. Int. J. Artific. Prot. 1995;18:315–321. [PubMed] [Google Scholar]
- MENTER D.G., ONODA J.M., TAYLOR J.D., HONN K.V. Effects of prostacyclin on tumor cell-induced platelet aggregation. Cancer Res. 1984;44:450–456. [PubMed] [Google Scholar]
- MICHELSON A.D., BENOIT S.E., FURMAN M.I., BRECKWOLDT W.L., ROHRER M.J., BARNARD M.R., LOSCALZO J. Effects of nitric oxide/EDRF on platelet surface glycoproteins. Am. J. Physiol. 1996;270:H1640–H1648. doi: 10.1152/ajpheart.1996.270.5.H1640. [DOI] [PubMed] [Google Scholar]
- MONCADA S., GRYGLEWSKI R.J., BUNTING J., VANE J.R. An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature. 1976;263:663–665. doi: 10.1038/263663a0. [DOI] [PubMed] [Google Scholar]
- MORGANTANI M., MITTERMAYER C., HEINZE U. , CAPRI A., SAGRIPANTI A. Expression of tissue type plasmonogen activator inhibitor and von Willebrand factor in the supernatant of endothelial cell culture in response to the seeding of adenocarcinoma cell line HRT-18. Biomed. Pharmacol. 1996;15:373–375. doi: 10.1016/s0753-3322(96)89671-5. [DOI] [PubMed] [Google Scholar]
- NURDEN A.T., CAEN J.P. An abnormal glycoprotein pattern in 3 cases of Glanzmann thrombasthemia. Br. J. Haematol. 1974;28:253–260. doi: 10.1111/j.1365-2141.1974.tb06660.x. [DOI] [PubMed] [Google Scholar]
- OLEKSOWICZ L., BHAGWATI N., DELEON–FERNANDEZ M. Deficiency of von Willebrand's Factor – cleaving protease in patients with disseminated malignancies. Cancer Res. 1999;59:2244–2250. [PubMed] [Google Scholar]
- PACZUSKI R., BIALOWSKA A., KOTSCHY M., BURDUK D., BETLEJEWSKI S. Von Willebrand Factor in plasma of patients with advanced stages of larynx cancer. Thrombosis Res. 1999;95:197–200. doi: 10.1016/s0049-3848(99)00041-9. [DOI] [PubMed] [Google Scholar]
- PARISE L.V., CRISS A.B., NANNIZZI L., WARDELL M.R. Glycoprotein IIIa is phosphorylated in intact human platelets. Blood. 1990;75:2363–2368. [PubMed] [Google Scholar]
- PEARLSTEIN E., AMBROGIO C., GASIC G., KARPATKIN S. Inhibition of the platelet-aggregating activity of two human adenocarcinomas of the colon and an anaplastic murine tumor with a specific thrombin inhibitor, dansylarginine N-(3-ethyl-1,5-pentanediyl)amide. Cancer Res. 1981;41:4535–4539. [PubMed] [Google Scholar]
- PHILIPPE C., PHILIPPE B., FOUQUERAY B., PEREZ J., LEBRET M., BAUD L. Protection from tumor necrosis factor-mediated cytolysis by platelets. Am. J. Pathol. 1993;143:1713–1723. [PMC free article] [PubMed] [Google Scholar]
- PRATT D.A., MILLER W.R., DAWES J. Thrombospondin in malignant and non-malignant breast tissue. Eur. J. Cancer Clin. Oncol. 1989;25:343–350. doi: 10.1016/0277-5379(89)90028-x. [DOI] [PubMed] [Google Scholar]
- PUMIGLIA K.M., HUANG C.K., FEINSTEIN M.B. Elevation of camp, but not cGMP, inhibits thrombin-stimulated tyrosine phosphorylation in human platelets. Biochem. Biophys. Res. Commun. 1990;171:738–745. doi: 10.1016/0006-291x(90)91208-a. [DOI] [PubMed] [Google Scholar]
- RADOMSKI M.W., JENKINS D.C., HOLMES L., MONCADA S. Human colorectal adenocarcinoma cells: differential nitric oxide synthesis determines their ability to aggregate platelets. Cancer Res. 1991;51:6073–6078. [PubMed] [Google Scholar]
- RADOMSKI M.W., MONCADA S. An improved method for washing platelets with prostacyclin. Thromb. Res. 1983;30:383–389. doi: 10.1016/0049-3848(83)90230-x. [DOI] [PubMed] [Google Scholar]
- RADOMSKI M.W., PALMER R.M.J., MONCADA S. The anti-aggregating properties of vascular endothelium: interactions between prostacyclin and nitric oxide. Br. J. Pharmacol. 1987A;92:639–646. doi: 10.1111/j.1476-5381.1987.tb11367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RADOMSKI M.W., PALMER R.M.J., MONCADA S. Comparative pharmacology of endothelium-derived relaxing factor, nitric oxide and prostacyclin in platelets. Br. J. Pharmacol. 1987B;92:181–187. doi: 10.1111/j.1476-5381.1987.tb11310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUGGERI Z.M. Structure and function of von Willebrand factor. Thromb. Haemostas. 1999;82:276–284. [PubMed] [Google Scholar]
- RUGGERI Z.M., DENT J.A., SALDIVAR E. Contribution of distinct adhesive interactions to platelet aggregation in flowing blood. Blood. 1999;94:172–178. [PubMed] [Google Scholar]
- SALAS E., MORO M.A., ASKEW S., HODSON H.F., BUTLER A.R., RADOMSKI M.W., MONCADA S. Comparative pharmacology of analogues of S-nitroso-N-acetyl-DL-penicillamine on human platelets. Br. J. Pharmacol. 1994;112:1071–1076. doi: 10.1111/j.1476-5381.1994.tb13192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHAU H., ROTH M.D., GOLUB S.H. Regulation of natural killer cell function by nonlymphoid cells. Nat. Immun. 1993;12:235–249. [PubMed] [Google Scholar]
- STEINERT B.W., TANG D.G., GROSSI I.M., UMBARGER L.A., HONN K.V. Studies on the role of platelet eicosanoid metabolism and integrin αIIbβ3 in tumor-cell induced platelet aggregation. Int. J. Cancer. 1993;54:92–101. doi: 10.1002/ijc.2910540116. [DOI] [PubMed] [Google Scholar]
- STEWART M.W., ETCHES W.S., BOSHKOV L.K., MANT M.J., GORDON P.A., SHAW A.R. Platelet activation by a novel solid-phase agonist: effects of VWF immobilized on polystyrene beads. Br. J. Haematol. 1997;97:321–329. doi: 10.1046/j.1365-2141.1997.372681.x. [DOI] [PubMed] [Google Scholar]
- SWEENEY J.D., KILLION K.M., PRUET C.F., SPAULDING M.B. von Willebrand factor in head and neck cancer. Cancer. 1990;66:2387–2389. doi: 10.1002/1097-0142(19901201)66:11<2387::aid-cncr2820661123>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- VAN DUIJNHOVEN E.M., LUSTERMANS F.A., VAN WERSCH J.W. Evaluation of the coagulation/fibrinolysis balance in patients with colorectal cancer. Haemostasis. 1993;23:168–172. doi: 10.1159/000216870. [DOI] [PubMed] [Google Scholar]
- WAGNER C.L., MASCELLI M.A., NEBLOCK D.S., WEISMAN H.F., COLLER B.S., JORDAN R.E. Analysis of GP IIb/IIIa receptor number by quantification of 7E3 binding to human platelets. Blood. 1996;88:907–914. [PubMed] [Google Scholar]
- WARDELL M.R., REYNOLDS C.C., BERNDT M.C., WALLACE R.W., FOX J.E. Platelet glycoprotein lbb is phosphorylated on serine 166 by cyclic AMP-dependent protein kinase. J. Biol. Chem. 1989;264:15656–15661. [PubMed] [Google Scholar]
- WEISS H.J., TSCHOPP T.B., BAUMGARTNER H.R., SUSSMAN I., JOHNSON M.M., EGAN J.J. Decreased adhesion of ginat (Bernard Soulier) platelets to subendothelium: further implications on the role of the von Willebrand factor in hemostasis. Am. J. Med. 1974;57:920–925. doi: 10.1016/0002-9343(74)90170-3. [DOI] [PubMed] [Google Scholar]
- WYLER B., BIENZ D., CLEMETSON K.J., LUSCHER E.F. Glycoprotein lbb is the only phosphorylated major membrane glycoprotein in human platelets. Biochem. J. 1986;234:373–379. doi: 10.1042/bj2340373. [DOI] [PMC free article] [PubMed] [Google Scholar]