

Abstract
We have investigated the cellular target of K+ channel blockers responsible for the inhibition of the EDHF-mediated relaxation in the rat mesenteric artery by studying their effects on tension, smooth muscle cell (SMC) membrane potential and endothelial cell Ca2+ signal ([Ca2+]endo).
In arteries contracted with prostaglandin F2α (2.5 – 10 μM), relaxation evoked by ACh (0.01 – 3 μM) was abolished by a combination of charybdotoxin (ChTX, 0.1 μM) plus apamin (Apa, 0.1 μM) and was inhibited by 68±6% (n=6) by 4-aminopyridine (4-AP, 5 mM).
ACh (0.001 – 3 μM) increased [Ca2+]endo and hyperpolarized SMCs with the same potency, the pD2 values were equal to 7.2±0.08 (n=4) and 7.2±0.07 (n=9), respectively. SMCs hyperpolarization to ACh (1 μM) was abolished by high K+ solution or by ChTX/Apa. It was decreased by 66±5% (n=6) by 4-AP.
The increase in [Ca2+]endo evoked by ACh (1 μM) was insensitive to ChTX/Apa but was depressed by 58±16% (n=6) and 27±4% (n=7) by raising external K+ concentration and by 4-AP, respectively.
The effect of 4-AP on [Ca2+]endo was not affected by increasing external K+ concentration. In Ca-free/EGTA solution, the transient increase in [Ca2+]endo evoked by ACh (1 μM) was abolished by thapsigargin (1 μM) and was decreased by 75±7% (n=5) by 4-AP.
These results show that inhibition of EDHF-evoked responses by 4-AP may be attributed to a decrease in the Ca2+ release activated by ACh in endothelial cells. The abolition of SMCs hyperpolarization to ACh by ChTX/Apa is not related to an interaction with the [Ca2+]endo.
Keywords: Acetylcholine, EDHF, calcium, endothelial cells, K+ channel, relaxation, mesenteric artery, rat
Introduction
Acetylcholine (ACh) stimulates an endothelium-dependent relaxation of pre-contracted arteries, which has been reported to be mediated by several factors as nitric oxide (NO) (Furchgott & Zawadzki, 1980) and prostacyclin (Moncada & Vane, 1978). In most arteries, the relaxation evoked by ACh is not completely abolished by nitric oxide synthase and cyclo-oxygenase inhibitors and is accompanied by the hyperpolarization of the smooth muscle cell membrane. These responses are attributed to the release of an endothelium-derived hyperpolarizing factor (EDHF) (Taylor & Weston, 1988; Feletou & Vanhoutte, 1988). Its chemical nature and its mechanism of action remain elusive.
It is known that the release of EDHF is activated by an increase of intracellular calcium concentration in the endothelial cells (ECs), which is initiated by the release of Ca2+ stores (Chen & Suzuki, 1990, Fukao et al., 1995) and maintained by the influx of Ca2+ from the extracellular space (Fukao et al., 1997). Although voltage-dependent Ca2+ channels have been described in ECs (Bossu et al., 1992), they do not contribute to the Ca2+ regulation (Himmel et al., 1993). The Ca2+ influx activated by agonists in ECs occurs through non-selective cation channels (Nilius, 1990). It is sensitive to the membrane potential (Lückhöff & Busse, 1990), which affects the driving force for Ca2+. The hyperpolarization of the membrane of ECs favours the Ca2+ influx (Lückhöff & Busse, 1990), while depolarization reduces the plateau phase of the Ca2+ signal induced by an agonist (for review Nilius et al., 1997).
In various arteries, apamin (an inhibitor of small conductance Ca2+-activated K+ channel, SKCa) alone or in combination with charybdotoxin (ChTX) (an inhibitor of large conductance Ca2+-activated K+ channel, BKCa, and intermediate conductance Ca2+-activated K+ channel, IKCa), inhibits the responses attributed to EDHF (Waldron & Garland, 1994a; Zygmunt & Höggestätt, 1996; Chataigneau et al., 1998; Quignard et al., 1999) whereas apamin plus iberiotoxin, a specific inhibitor of BKCa channels, do not affect EDHF-mediated relaxation (Waldron & Garland, 1994a; Zygmunt & Höggestätt, 1996). This suggests that activation of IKCa and SKCa channels could be responsible for the hyperpolarization of smooth muscle cells (SMCs). However, KCa channels are extensively expressed in ECs (Demirel et al., 1994; Groschner et al., 1994; Marchenko & Sage, 1996). It has been proposed that the inhibition of EDHF responses by the combination of ChTX plus apamin could result from their action at the level of IKCa and SKCa channels on ECs (Edwards et al., 1998; Doughty et al., 1999). Voltage-dependent K+ channels (Kv) have also been proposed to be involved in EDHF-mediated responses. Indeed, 4-aminopyridine (4-AP), a specific delayed rectifier channel blocker, inhibits ACh-induced endothelium-dependent hyperpolarization in coronary artery of the guinea-pig (Eckman et al., 1998), but the contribution of this channel subtype is not observed in all arteries (Zygmunt et al., 1997). The inhibition by 4-AP of the endothelium-dependent hyperpolarization evoked by ACh in the isolated carotid artery of guinea-pig and in rat hepatic artery has been shown to be associated with an inhibition of the hyperpolarization of ECs (Quignard et al., 2000).
The aim of this study was to investigate the site of action of K+ channel blockers involved in the inhibition of EDHF responses activated by ACh in the rat superior mesenteric artery. Simultaneous measurement of contractile responses and membrane potential was used to record the hyperpolarization and relaxation of SMCs. Since raising cytosolic Ca2+ concentration in ECs appears to be the first step in the EDHF pathway, the effect of K+ channel blockers was investigated on the Ca2+ signal in ECs by using front surface fluorimetry in indo-1-loaded artery.
Our results showed that Kv and KCa are involved in the relaxation induced by ACh in the presence of NO synthase and cyclo-oxygenase inhibitors. Inhibition by ChTX and apamin of the SMCs hyperpolarization evoked by ACh is not related to the interaction of the blockers with the increase in calcium signal in the endothelium. The inhibition of EDHF-evoked responses by 4-AP can be, at least partly, attributed to the inhibition of the Ca2+ release in ECs.
Methods
Normotensive Wistar-Kyoto (WKY) male rats (Iffa Credo, L'Arbresle, France) were used. All rats were killed by decapitation at 14 weeks. The superior mesenteric artery was rapidly removed and immersed in physiological solution (composition in mM): NaCl 122, KCl 5.9, NaHCO3 15, glucose 10, MgCl2 1.25 and CaCl2 1.25, gassed with a mixture of 95% O2-5% CO2. The superior mesenteric artery was carefully cleaned of all fat and connective tissue. All experiments were performed in the presence of Nω-nitro-L-arginine (L-NOARG) and indomethacin to block the nitric oxide synthase and the cyclo-oxygenase, respectively.
Simultaneous measurement of contractile tension and membrane potential
A segment of the superior mesenteric artery, about 2 mm in length, was inverted and mounted in a wire myograph (Model 500A, Danish Myo Technology A/S, Aarhus, Denmark) as described (Ghisdal et al., 1999). Briefly, two 40 μm wires were threaded through the lumen of the vessel segment. One wire was attached to a stationary support driven by a micrometer, while the other was attached to an isometric force transducer. Vessels were maintained under zero force for 60 min. A passive diameter-tension curve was constructed as described (Mulvany & Halpern, 1977). From this curve the effective transmural pressure was calculated. The vessel was set at a tension equivalent to that generated at 0.9 times the diameter of the vessel at 100 mmHg. The bath of the myograph was continuously perfused with physiological solution gassed with a mixture of 95% O2-5% CO2 and warmed at 37°C.
Measurement of the smooth muscle membrane potential was made with a glass microelectrode (Clark, Electromedical instruments, type GC 120F-15) filled with 1.5 M KCl and advanced through the luminal surface of the arterial segment with a micromanipulator (Leitz). The input resistance of the microelectrodes varied between 50 and 80 MΩ. Potential differences were measured with reference (reference electrode: Clark, Electromedical instruments, type E208) to the grounded bath by means of a Dagan amplifier (8100, Minneapolis, MN, U.S.A.). Electrical responses were monitored on an oscilloscope (Hitachi, oscilloscope V-252, 20 MHz). Membrane potential and tension were simultaneously recorded with a pen recorder (Kontron, 500 SP). Criteria for a successful impalement were (1) an abrupt drop in voltage on entry of microelectrode into the cell, (2) stable membrane potential for at least 2 min, and (3) a sharp return to zero on withdrawal of the electrode.
After being mounted in the organ chamber, the rings were maintained in gassed physiological solution (see above). Endothelium integrity was assessed at the beginning of each experiment by the application of 1 μM ACh on the plateau of the contraction evoked by noradrenaline (1 μM). When the rings relaxed with success, the preparation was maintained in physiological solution containing indomethacin (10 μM) and L-NOARG (100 μM) at 37°C. Drugs were applied in the perfusion solution. High KCl solutions were obtained by equimolar substitution of Na+ ions for K+ ions. When high-KCl solution or K+ channel blockers were used, the experiment was performed in the presence of phentolamine (1 μM) to rule out the contribution of α-adrenergic transmitter released by nervous ending. ACh concentration-response curves for the change in resting membrane potential were established by the successive application of different concentrations of the agonist with 30 min time intervals between two concentrations in order to avoid the development of tachyphylaxis. The inhibitors used were pre-incubated 10 – 15 min before application of ACh.
Measurement of endothelial cell calcium signal
A segment of the superior mesenteric artery, about 2.5 mm in length, was inverted and mounted between two hooks under a tension of 10 mN in a 3 ml cuvette continuously perfused with physiological solution (composition as above) gassed with a 95 – 5% mixture of O2 and CO2 and warmed at 37°C. An isometric force transducer measured the muscle tone.
Endothelium integrity was assessed at the beginning of each experiment by the application of 1 μM ACh on the plateau of the contraction evoked by 1 μM noradrenaline. Only the segments where the contraction was inhibited by 75% were used. Mesenteric artery rings were then incubated for 3 h at room temperature (22°C) in physiological solution containing 5 μM indo-1 acetoxymethyl ester (indo-1-AM) and 0.05% Cremophor EL. After the loading period, the rings were washed in physiological solution containing L-NOARG (100 μM), indomethacin (10 μM), phentolamine (1 μM) and nimodipine (1 μM) at 37°C for 30 min. Nimodipine, a voltage-dependent calcium channel blocker, was present in the physiological solution to rule out the contribution of smooth muscle Ca2+ signal when high KCl solution or K+ channel blockers were used. ACh concentration-response curves for the changes in cytosolic Ca2+ signal of ECs ([Ca2+]endo) were established by the cumulative application of increasing concentrations of the agonist. All physiological calcium-free solutions were supplied with 0.2 mM ethylene glycol-bis (b-amino ethyl ether) tetraacetic acid (EGTA).
The cuvette was part of a fluorimeter (CAF, JASCO, Tokyo, Japan) which allowed estimation of the calcium signal. After excitation at 340 nm, the fluorescence signals emitted at 405 nm (F405) and 500 nm (F500) were measured simultaneously with the contractile tension and recorded on a computer by using the data acquisition hardware MacLab and data recording software Chart (AD Instruments Pty Ltd., Castle Hill, Australia). At the end of each experiment, the autofluorescence of the tissue was measured at 405 and 500 nm by quenching the indo-1 fluorescence with MnCl2 (5 mM) and subtracted from F405 and F500. The [Ca2+]endo was estimated by the ratio of the fluorescence emitted at 405 and 500 nm.
Drugs
Indo 1-AM was from Calbiochem (EuroBiochem, Bierges, Belgium). Acetylcholine chloride (ACh), 4-aminopyridine (4-AP), apamin (Apa), cremophor EL, L-indomethacin, Nω-nitro-L-arginine (L-NOARG), phentolamine, prostaglandin F2α (PGF2α) and thapsigargin were obtained from Sigma. Stock solution of indomethacin was prepared in 2% Na2CO3. Charybdotoxin (ChTX) was from Latoxan (Rosans, France). Nimodipine was from Bayer (Leverkussen, Germany) and stock solution (10 mM) was prepared in ethanol. In the experiments performed with 4-AP, the physiological solution was buffered to pH 7.4 with tris(hydroxy-methyl)-aminomethane (Tris, 5 mM) and N-[2-hydroxy-ethyl]piperazine-N′-[2-ethanesulphonic acid] (HEPES).
Statistics
Results are given as mean±standard error (s.e.mean). Comparisons were made using Student's t-test or by analysis of variance followed by a Bonferroni test (one-way ANOVA), when more than two groups were involved in the comparison. P values lower than 0.05 indicated significant differences. EC50 values (concentration of an agonist that produces 50% of the maximal effect) were calculated by non-linear curve fitting of the experimental data of the concentration-response curves to the equation:
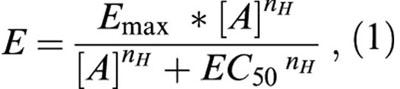 |
where Emax is the maximum amplitude of the effect produced by the agonist, [A] is the concentration of the agonist and nH is the Hill slope (Multifit, Day Computing, Cambridge, UK; KaleidaGraph, Synergy Software, Reading, PA, U.S.A.). The negative logarithm to base 10 of EC50 values (pD2) was used for the statistical analysis.
Results
Effect of K+ channel blockers on the EDHF-dependent relaxation induced by acetylcholine
In order to investigate the effect of K+ channel blockers on the relaxation evoked by ACh in rat mesenteric artery, we have performed a series of experiments where ACh (0.01 – 3 μM) was added cumulatively on the vessels pre-contracted by prostaglandin F2α (PGF2α). The PGF2α concentration (2.5 – 10 μM) was adapted to produce a contraction equivalent to that evoked by a 100 mM KCl solution (13.2±0.8 mN, n=26). In the presence of L-NOARG (100 μM), indomethacin (10 μM) and phentolamine (1 μM), maximum relaxation to ACh reached 75±2.4% of the contraction (ACh 3 μM); the pD2 value of ACh was equal to 6.6±0.08 (n=13) (Figure 1). The association of charybdotoxin (ChTX, 0.1 μM) plus apamin (Apa, 0.1 μM) to inhibit KCa channels produced a contraction of 2±0.3 mN (n=4). In the presence of ChTX/Apa, the concentration of PGF2α was reduced about two times to get a similar level of contraction as in the absence of the blockers. KCa channel blockers abolished the relaxation induced by ACh (Figure 1), which even produced a slight additional contraction to that evoked by PGF2α. 4-Aminopyridine (4-AP, 5 mM) was used to block Kv channels. It caused a small increase in tone of 0.3±0.08 mN (n=6) and did not affect the contraction to PGF2α significantly. The pre-exposure of the vessels to 4-AP depressed the relaxation to ACh 3 μM by 68±5.9% (n=6) (Figure 1).
Figure 1.
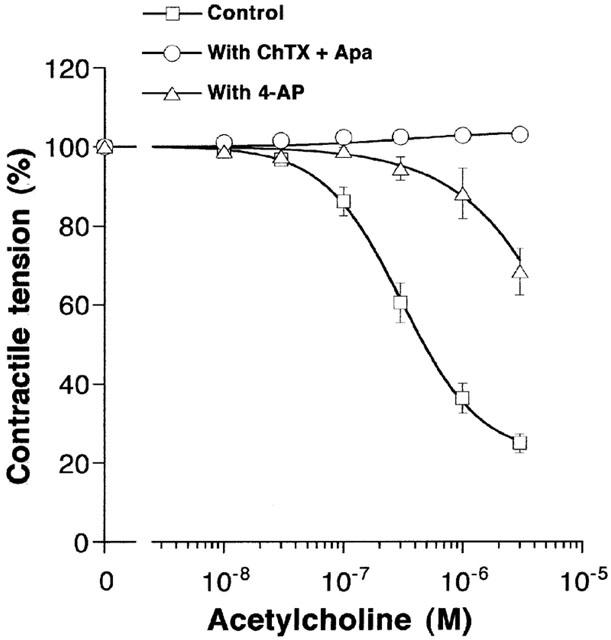
Effect of K+ channel blockers on the EDHF-dependent relaxation induced by acetylcholine in rat superior mesenteric artery. Concentration-response curves for the effects of acetylcholine on the contractile tension of mesenteric arteries stimulated by prostaglandin F2α in the absence (control, n=13) and in the presence of charybdotoxin and apamin (ChTX+Apa, 0.1 μM; n=3), or 4-aminopyridine (4-AP, 5 mM, n=6). All experiments were performed in the presence of Nω-nitro-L-arginine (100 μM), indomethacin (10 μM) and phentolamine (1 μM).
Effect of K+ channel blockers on the hyperpolarization evoked by acetylcholine in smooth muscle cells
In the presence of L-NOARG (100 μM), indomethacin (10 μM) and phentolamine (1 μM), the resting membrane potential (Em) of main mesenteric artery SMCs was averaged at −46.6±0.3 mV (n=34). Exposure of the vessels to ACh (0.001 – 1 μM) induced a concentration-dependent hyperpolarization of SMCs (Figure 2A). The pD2 value of ACh was equal to 7.2±0.07 (n=9). Increasing external KCl concentration depressed the hyperpolarization to ACh in a concentration-dependent manner (Figure 2B). Hyperpolarization was completely abolished in the presence of 40 mM KCl. The concentration of KCl producing 50% inhibition was equal to 12.4±2.2 mM (n=4) (Figure 2B). The incubation of artery rings with ChTX plus Apa depolarized SMCs by 7.5±1.5 mV (n=4) and abolished the hyperpolarization induced by 1 μM ACh (P<0.05; n=4) (Figure 3B). In artery rings pre-exposed to 5 mM 4-AP, SMCs were depolarized by 6.3±1.1 mV (n=6) and the hyperpolarization evoked by 1 μM ACh was inhibited by 66±5.2% (P<0.05; n=6) (Figure 3B). These results confirmed the involvement of Kv and KCa channels in the EDHF pathway (Corriu et al., 1996; Eckman et al., 1998).
Figure 2.
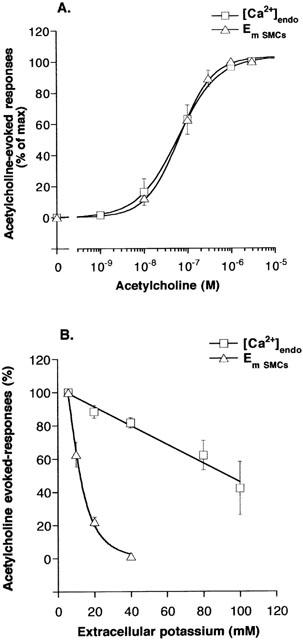
Effect of increase in extracellular K+ concentration on changes of endothelial cells Ca2+ and SMCs membrane potential evoked by ACh. (A) Concentration-response curves to acetylcholine (0.001 – 3 μM) were established in unstimulated mesenteric arteries. Change in membrane potential of smooth muscle cells (EmSMCs, n=9) and increase in Ca2+ signal in endothelial cells ([Ca2+]endo, n=4) are expressed as a percentage of the maximal responses to ACh (% of max). Data are presented as means±s.e.mean. (B) Effect of varying extracellular K+ concentration on the increase in Ca2+ signal in endothelial cells ([Ca2+]endo, n=6) and the hyperpolarization of SMCs (EmSMCs, n=4) induced by acetylcholine (ACh, 1 μM). Data are expressed as percent of the responses in the presence of 5.9 mM KCl and are presented as means±s.e.mean.
Figure 3.
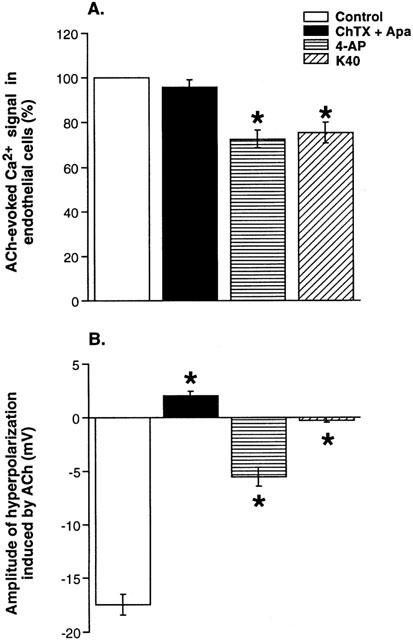
Comparison of the effects of K+ channel blockers on acetylcholine-evoked changes in endothelial cells Ca2+ signal (A) and SMCs membrane potential (B). Responses to ACh (1 μM) were measured in the absence (control) and in the presence of charybdotoxin and apamin (ChTX+Apa, 0.1 μM), 4-aminopyridine (4-AP, 5 mM) or in physiological solution containing 40 mM of KCl (K40). Endothelial cells Ca2+ signal was expressed as a percentage of the maximum amplitude of acetylcholine-evoked responses in the absence of test drugs. Data are presented as means±s.e.mean. Asterisks denote a statistically significant difference from control values (P<0.05).
Effect of acetylcholine on cytosolic Ca2+ signal in endothelial cells
The following experiments were designed in order to determine whether the inhibition by K+ channel blockers of EDHF-mediated relaxation and hyperpolarization could result from an action of the blockers on Ca2+ signal in ECs. Endothelial cells Ca2+ signal ([Ca2+]endo) was recorded in indo-1-loaded arteries incubated in the presence of nimodipine (1 μM) and phentolamine (1 μM). Under these conditions, high KCl solution (100 mM) did not increase calcium signal, as it would be expected if signal arised from SMCs. Figure 4A shows a typical record illustrating the effect of ACh. The muscarinic agonist induced a fast increase in the F405/F500 ratio, which was stable for about 2 min. The ratio then slightly decreased: after 6 min, the Ca2+ signal levelled at 43±2.4% (n=9) of its peak value (Figure 4A). All effects were corrected for the decrease in the Ca2+ signal with time. When the endothelium was mechanically removed, ACh did not affect the Ca2+ signal (Figure 4B), attesting of the EC specificity of the changes evoked by ACh.
Figure 4.
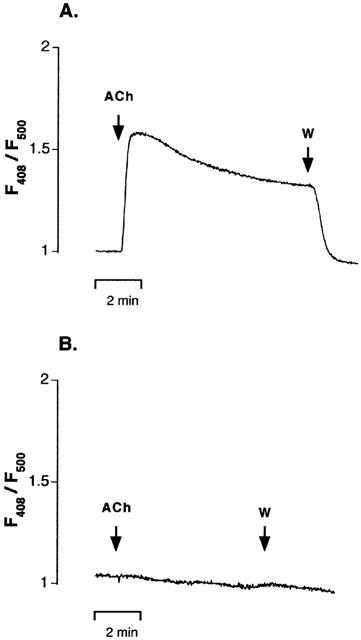
Endothelial cells Ca2+ signal in indo-1-loaded mesenteric artery. Representative experimental traces showing the increase in indo-1 fluorescence ratio (F405/F500) induced by acetylcholine (ACh, 1 μM) in the presence (A) and in the absence (B) of the endothelium. Fluorescence ratio obtained after subtracting the autofluorescence was normalised to the value measured before addition of ACh. In the presence of an intact endothelium, the muscarinic agonist induced a fast increase in the F405/F500 ratio, which returned to the basal level after wash (W). When the endothelium was mechanically removed, ACh did not affect Ca2+ signal. ACh was applied as indicated.
In intact artery rings, the cumulative application of ACh (0.001 – 3 μM) induced a concentration-dependent increase in [Ca2+]endo (Figure 2A). The maximum effect of ACh was obtained at the concentration of 1 – 3 μM. The pD2 value of ACh was equal to 7.2±0.08 (Figure 2A; n=4). It is worth noting that the concentration-response relations for the hyperpolarization and the elevation of [Ca2+]endo evoked by ACh were perfectly superimposed, with a correlation coefficient close to one (Figure 2A). The participation of EC membrane potential to the increase in [Ca2+]endo induced by ACh (1 μM) was examined by investigating the effect of enhanced extracellular K+ concentration. Elevation of extracellular K+ concentration from 5.9 to 100 mM inhibited the Ca2+ signal evoked by ACh by 58±16% (n=6; Figure 2B). Inhibition was concentration-dependent and was linear up to 100 mM KCl. The effect of K+ channel blockers on ACh-evoked [Ca2+]endo is summarized in Figure 3A. Neither the association ChTX plus Apa (0.1 μM) nor 4-AP (5 mM) did modify resting Ca2+ signal. The association of ChTX plus Apa had no effect on the response elicited by 1 μM ACh. In the presence of 4-AP, the Ca2+ signal evoked by ACh (1 μM) was decreased by 27±4% (n=7, P<0.05 vs control). In order to determine whether the effect of 4-AP could result from a depolarization of ECs, the external K+ concentration was elevated to 40 mM. Under this condition, the Ca2+ signal evoked by ACh (1 μM) was reduced by 24±4.6% (n=7) but it was still inhibited by 4-AP by 22±2% (n=6).
In the second series of experiments, we have tested the hypothesis that 4-AP could affect the intracellular calcium release process stimulated by acetylcholine. In this aim, the artery rings were incubated for 10 min in Ca2+-free/EGTA physiological solution containing 40 mM of KCl. This produced a decrease in Ca2+ signal of 10±1% (n=8). The addition of ACh (1 μM) then evoked a rapid but transient increase in [Ca2+]endo, which returned to the baseline values within 2 min (Figure 5A). The magnitude of the calcium peak represented 51±6% (n=8) of the maximum increase in Ca2+ signal evoked by ACh in the presence of Ca2+ and K+ 40 mM. It was completely inhibited by thapsigargin (1 μM) (Figure 5B; n=4), an inhibitor of the Ca2+-ATPase pump of the endoplasmic reticulum (Lytton et al., 1991). In the presence of 4-AP, the transient calcium peak elicited by ACh was inhibited by 75±7% (n=5, P<0.05 vs control). The readmission of Ca2+ in the medium produced a rapid and large increase in [Ca2+]endo (Figure 5A). The amplitude of the increase in Ca2+ signal evoked by the readmission of Ca2+ was enhanced in the presence of thapsigargin (Figure 5B) but was decreased by 22±4% in the presence of 4-AP (n=5, P<0.05 vs control) (Figure 5C).
Figure 5.
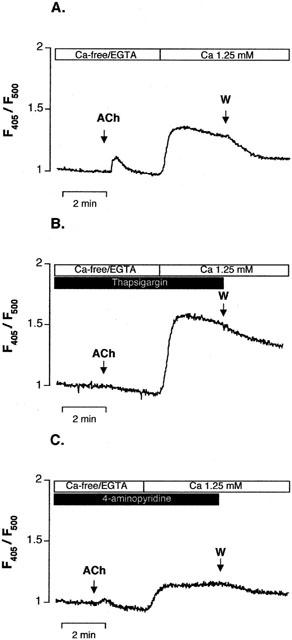
Effect of 4-aminopyridine on intracellular Ca2+ release stimulated by acetylcholine in indo-1-loaded endothelial cells. Representative experimental traces showing the increase in indo-1 fluorescence ratio (F405/F500) evoked by acetylcholine (ACh, 1 μM) in arteries pre-incubated for 10 min in Ca2+-free/EGTA solution in the presence of 40 mM of K+. Fluorescence ratio corrected for the autofluorescence was normalized to the value measured before addition of ACh. Readmission of Ca2+ (Ca 1.25 mM) to the solution evoked an increase in fluorescence ratio. (A) Control condition, (B) in the presence of the thapsigargin (1 μM) and (C) in the presence of 4-aminopyridine (5 mM). (W) Indicates the washout of the artery.
Discussion
The present results showed that inhibition of EDHF-mediated relaxation to ACh by the K+ channel blocker 4-AP results from the interaction of the blocker with the ECs Ca2+ signal. On the opposite, ChTX and Apa, which completely blocked SMCs hyperpolarization to ACh, did not affect the increase in [Ca2+]endo evoked by the muscarinic agonist.
EDHF mediated relaxation
In WKY superior mesenteric artery, as in several arteries, ACh induces an endothelium-dependent relaxation that is resistant to nitric oxide and cyclo-oxygenase inhibitors, and is associated with the hyperpolarization of SMCs (Waldron & Garland, 1994b; Ghisdal et al., 1999). Inhibition of hyperpolarization and relaxation to ACh by KCl (Chen & Suzuki, 1989; Adeagbo & Triggle, 1993) or a combination of apamin and charybdotoxin, but not apamin and iberiotoxin (Waldron & Garland, 1994a; for review Feletou & Vanhoutte, 1999) is considered as a finger print of EDHF-mediated responses.
Fluorescence studies in indo-1-loaded arteries confirmed the previous observations by Chen & Suzuki (1990) and by Fukao et al. (1997) that the hyperpolarization evoked by ACh is initiated by thapsigargin-sensitive Ca2+ release from intracellular Ca2+ pool and maintained by Ca2+ influx pathway distinct from L-type Ca2+ channels coupled to the depletion of intracellular stores. The Ca2+ influx in ECs is controlled by the membrane potential as indicated by the observation that increasing extracellular K+ concentration during agonist stimulation diminished the rise in [Ca2+]endo (Kamouchi et al., 1999; Wang & van Breemen, 1999; Knot et al., 1999). The close correlation that was found between the effect of ACh on [Ca2+]endo and Em of SMCs indicates that blunting the Ca2+ signal in ECs could cause a proportional reduction in the EDHF-evoked SMCs hyperpolarization. Thus, the different sensitivity to external K+ of ACh-evoked [Ca2+]endo and SMCs hyperpolarization suggests that inhibition of [Ca2+]endo signal could contribute to, but could not be the only determinant of the K+-sensitivity of the relaxation evoked by EDHF.
Involvement of endothelial cell Kv channels in the EDHF-mediated responses
The present results showed that 4-AP partially inhibited the endothelium-dependent relaxation and hyperpolarization of SMCs to ACh in rat mesenteric artery. Similar effect has been reported in guinea-pig coronary artery and in porcine coronary artery (Shimizu & Paul, 1997; Eckman et al., 1998). Fluorescence studies in indo-1-loaded artery revealed that 4-AP also inhibited ACh-evoked increase in [Ca2+]endo, suggesting that the inhibition of SMCs hyperpolarization by 4-AP could be caused by an effect of the blocker on the endothelium. ECs have Kv channels (Takeda et al., 1987; Hogg et al., 1999; Dittrich & Daut, 1999), which are involved in the EC hyperpolarization induced by ACh in the guinea-pig coronary artery (Chen & Cheung, 1992; Quignard et al., 2000). Inhibition by 4-AP of ACh-evoked increase in [Ca2+]endo could result from the inhibition by 4-AP of endothelium Kv channels and the consecutive depolarization of ECs. This hypothesis had to be rejected following the observation that clamping the membrane potential of ECs with high-KCl solution did not affect the inhibition of Ca2+ signal by 4-AP. In addition, ChTX/Apamin did not affect the endothelial cell Ca2+ response stimulated by ACh. Kv 1.2 and Kv 1.3 channels, present in mesenteric artery (Xu et al., 1999), are sensitive to ChTX (Grissmer et al., 1994). It is then highly likely that endothelial cell Kv 1.2 and Kv 1.3 channels do not contribute to the ACh response. Interestingly, experiments performed in Ca2+ free solution revealed that the pre-exposure of the arteries to 4-AP strongly inhibited the transient thapsigargin-sensitive increase in [Ca2+]endo stimulated by ACh, suggesting that 4-AP could interact with the intracellular calcium release process activated by ACh. It has been shown in bovine aortic ECs that Ca2+ release from IP3-sensitive stores is modulated by a K+ counter-ion system present in the membrane of endoplasmic reticulum (ER). An inward movement of K+ through the ER membrane could facilitate the sustained release of Ca2+ during IP3 -induced mobilisation from internal stores (Wood & Gillespie, 1998). These intracellular K+ channels can be blocked by K+ channel blockers like TEA, 4-AP or 3,4-DAP, which reduce the IP3 response to a level not significantly different to that of complete K+ replacement (Wood & Gillespie, 1998). After incubation in Ca2+ free condition and challenge with ACh, re-admission of Ca2+ in the solution evoked a rapid increase in [Ca2+]endo. This response also was depressed in the presence of 4-AP. At the opposite, emptying Ca2+ stores with thapsigargin led to an increased capacitative Ca2+ entry. The inhibition of the Ca2+ re-admission process by 4-AP is then not in the line of a thapsigargin-like action of 4-AP at the level of the Ca2+ pump of the ER, as has been suggested by Ishida & Honda (1993). Indeed, the latter effect would lead to the emptying of intracellular Ca2+ stores and the increase in the capacitative Ca2+ entry, as observed with thapsigargin.
Since EC hyperpolarization to ACh results from the activation by Ca2+ of KCa channels (Wang et al., 1996; Ohashi et al., 1999), the inhibition of endothelial cells Ca2+ signal by 4-AP explains the observation by Quignard et al. (2000) that 4-AP inhibits ACh-evoked hyperpolarization of ECs. The close relation between ACh-evoked SMCs hyperpolarization and increase in [Ca2+]endo suggests that inhibition of Ca2+ signal in ECs by 4-AP can be responsible for about 27 % inhibition of the SMCs hyperpolarization. Additional effect of 4-AP is thus required to justify the total 58% inhibition on EDHF-mediated SMCs hyperpolarization.
Involvement of endothelial cell KCa channels in the EDHF-mediated response
Involvement of KCa channels in the EDHF-mediated relaxation has been suggested by the effect of the KCa blockers ChTX/Apa, which abolish EDHF-mediated hyperpolarization in several arteries (Waldron & Garland, 1994a; Zygmunt & Höggestätt, 1996, Corriu et al., 1996; Prieto et al., 1998; Quignard et al., 1999; Doughty et al., 1999). The present results showed that, in rat superior mesenteric artery, ChTX/Apa inhibited the relaxation and the hyperpolarization to ACh but did not affect [Ca2+]endo.
Doughty et al. (1999) showed that, in third-order superior mesenteric artery of the rat, ChTX and apamin block EDHF-mediated relaxation only when they are applied intraluminally. ChTX/Apa-sensitive K+ channels are indeed present in vascular ECs (Marchenko & Sage, 1996) and are responsible for the hyperpolarization evoked by ACh in ECs (Wang et al., 1996; Ohashi et al., 1999). The present study ruled out the possibility that the combination of these toxins abolishes SMCs hyperpolarization by inhibiting Ca2+ signal in ECs, but cannot exclude that endothelial cells ChTX/Apa-sensitive-KCa channels are involved in the EDHF pathway, downstream the increase of Ca2+ concentration in ECs. Edwards et al. (1998) reported that in rat hepatic and mesenteric artery ACh opens ChTX and apamin-sensitive K+ channels in ECs, leading to the release of K+ in the myo-endothelial space. Accumulation of K+ in myo-endothelial space has been reported to hyperpolarize the endothelium by increasing outward current through inward rectifying K+ channels. Hyperpolarization could then be transmitted to SMCs through myo-endothelial gap junctions (Doughty et al., 2001). Activation by K+ of SMCs Na+/K+-ATPase could also be responsible for the hyperpolarization of SMCs (Doughty et al., 2000; Dora & Garland, 2001).
It is concluded that voltage- and Ca2+-dependent K+ channels are involved in EDHF-mediated relaxation evoked by acetylcholine in the rat superior mesenteric artery. Our results showed that inhibition of EDHF responses by 4-AP can be, at least partly, attributed to an inhibition of the intracellular Ca2+ release process activated by ACh in ECs. The present study ruled out the possibility that ChTX and apamin abolish the EDHF-dependent hyperpolarization and relaxation by acting on endothelial cell Ca2+ signal process.
Acknowledgments
This work was supported by a grant from the Ministère de l'Education et de la Recherche Scientifique (Action Concertée no 00/05-260) and from the FRSM (grant no 3.4534.98). Ph Ghisdal was supported by a grant from the ‘Fonds Spéciaux de Recherche - UCL'. The authors thank G. Leonardy for her excellent technical support.
Abbreviations
- ACh
acetylcholine
- ANOVA
analysis of variance
- 4-AP
4-aminopyridine
- Apa
apamin
- BKCa
large conductance Ca2+-activated K+ channel
- ChTX
charybdotoxin
- 3,4-DAP
3,4-diaminopyridine
- EC
endothelial cell
- EDHF
Endothelium-Derived Hyperpolarizing Factor
- Em
membrane potential
- ER
endoplasmic reticulum
- IKCa
intermediate conductance Ca2+-activated K+ channel
- Indo-1 AM
indo-1 acetoxymethylester
- IP3
inositol triphosphate
- L-NOARG
Nω-nitro-L-arginine
- NO
nitric oxide, PGF2α, prostaglandin F2α
- SKCa
small conductance Ca2+-activated K+ channel
- SMC
smooth muscle cell
- TEA
tetraethylammonium
- WKY
Wistar Kyoto rat
References
- ADEAGBO A.S.O., TRIGGLE C.R. Varying extracellular [K+]; a functional approach to separating EDHF- and EDNO-related mechanisms in perfused rat mesenteric arterial bed. J. Cardiovasc. Pharmacol. 1993;21:423–429. [PubMed] [Google Scholar]
- BOSSU J.L., ELHAMDANI A., FELTZ A. Voltage-dependent calcium entry in confluent bovine capillary endothelial cells. FEBS Lett. 1992;299:239–242. doi: 10.1016/0014-5793(92)80123-x. [DOI] [PubMed] [Google Scholar]
- CHATAIGNEAU T., FELETOU M., DUHAULT J., VANHOUTTE P.M. Epoxyeicosatrienoic acids, potassium channel blockers and endothelium-dependent hyperpolarization in the guinea-pig carotid artery. Br. J. Pharmacol. 1998;123:574–580. doi: 10.1038/sj.bjp.0701629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN G.F., SUZUKI H. Some electrical properties of the endothelium-dependent hyperpolarization recorded from rat arterial smooth muscle cells. J. Physiol. 1989;410:91–106. doi: 10.1113/jphysiol.1989.sp017522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN G.F., SUZUKI H. Calcium dependence of the endothelium-dependent hyperpolarization in smooth muscle cells of the rabbit carotid artery. J. Physiol. 1990;421:521–534. doi: 10.1113/jphysiol.1990.sp017959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN G.F., CHEUNG D.W. Characterization of acetylcholine-induced membrane hyperpolarization in endothelial cells. Circ. Res. 1992;70:257–263. doi: 10.1161/01.res.70.2.257. [DOI] [PubMed] [Google Scholar]
- CORRIU C., FELETOU M., CANET E., VANHOUTTE P.M. Endothelium-derived factors and hyperpolarization of the carotid artery of the guinea-pig. Br. J. Pharmacol. 1996;119:959–964. doi: 10.1111/j.1476-5381.1996.tb15765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEMIREL E., RUSKO J., LASKEY R.E., ADAMS D.J., VAN BREEMEN C. TEA inhibits ACh-induced EDRF release: endothelial Ca2+-dependent K+ channels contribute to vascular tone. Am. J. Physiol. 1994;267:H1135–H1141. doi: 10.1152/ajpheart.1994.267.3.H1135. [DOI] [PubMed] [Google Scholar]
- DITTRICH M., DAUT J. Voltage-dependent K+ current in capillary endothelial cells isolated from guinea pig heart. Am. J. Physiol. 1999;277:H119–H127. doi: 10.1152/ajpheart.1999.277.1.H119. [DOI] [PubMed] [Google Scholar]
- DORA K.A., GARLAND C.J. Properties of smooth muscle hyperpolarization and relaxation to K+ in the rat isolated mesenteric artery. Am. J. Physiol. 2001;280:H2424–H2429. doi: 10.1152/ajpheart.2001.280.6.H2424. [DOI] [PubMed] [Google Scholar]
- DOUGHTY J.M., PLANE F., LANGTON P.D. Charybdotoxin and apamin block EDHF in rat mesenteric artery if selectively applied to the endothelium. Am. J. Physiol. 1999;276:H1107–H1112. doi: 10.1152/ajpheart.1999.276.3.H1107. [DOI] [PubMed] [Google Scholar]
- DOUGHTY J.M., BOYLE J.P., LANGTON P.D. Potassium does not mimic EDHF in rat mesenteric arteries. Br. J. Pharmacol. 2000;130:1174–1182. doi: 10.1038/sj.bjp.0703412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOUGHTY J.M., BOYLE J.P., LANGTON P.D. Blockade of chloride channels reveals relaxation of rat small mesenteric arteries to raised potassium. Br. J. Pharmacol. 2001;132:293–301. doi: 10.1038/sj.bjp.0703769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., DORA K.A., GARDENER M.J., GARLAND C.J., WESTON A.H. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- ECKMAN D.M., HOPKINS N., MCBRIDE C., KEEF K.D. Endothelium-dependent relaxation and hyperpolarization in guinea-pig coronary artery: role of epoxyeicosatrienoic acid. Br. J. Pharmacol. 1998;124:181–189. doi: 10.1038/sj.bjp.0701778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FELETOU M., VANHOUTTE P.M. Endothelium-dependent hyperpolarization of canine coronary smooth muscle. Br. J. Pharmacol. 1988;93:515–524. doi: 10.1111/j.1476-5381.1988.tb10306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FELETOU M., VANHOUTTE P.M. The third pathway: endothelium-dependent hyperpolarization. J. Physiol. Pharmacol. 1999;50:525–534. [PubMed] [Google Scholar]
- FUKAO M., HATTORI Y., KANNO M., SAKUMA I., KITABATAKE A. Thapsigargin- and cyclopiazonic acid-induced endothelium-dependent hyperpolarization in rat mesenteric artery. Br. J. Pharmacol. 1995;115:987–992. doi: 10.1111/j.1476-5381.1995.tb15908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FUKAO M., HATTORI Y., KANNO M., SAKUMA I., KITABATAKE A. Sources of Ca2+ in relation to generation of acetylcholine-induced endothelium-dependent hyperpolarization in rat mesenteric artery. Br. J. Pharmacol. 1997;120:1328–1334. doi: 10.1038/sj.bjp.0701027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FURCHGOTT R.F., ZAWADZKI J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- GHISDAL P., GODFRAIND T., MOREL N. Effect of nitro-L-arginine on electrical and mechanical responses to acetylcholine in the superior mesenteric artery from stroke-prone hypertensive rat. Br. J. Pharmacol. 1999;128:1513–1523. doi: 10.1038/sj.bjp.0702947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRISSMER S., NGUYEN A.N., AIYAR J., HANSON D.C., MATHER R.J., GUTMAN G.A., KARMILOWICZ M.J., AUPERIN D.D., CHANDY K.G. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol. Pharmacol. 1994;45:1227–1234. [PubMed] [Google Scholar]
- GROSCHNER K., GRAIER W.F., KUKOVETZ W.R. Histamine induces K+, Ca2+, and Cl− currents in human vascular endothelial cells. Role of ionic currents in stimulation of nitric oxide biosynthesis. Circ. Res. 1994;75:304–314. doi: 10.1161/01.res.75.2.304. [DOI] [PubMed] [Google Scholar]
- HIMMEL H.M., WHORTON A.R., STRAUSS H.C. Intracellular calcium, currents, and stimulus-response coupling in endothelial cells. Hypertension. 1993;21:112–127. doi: 10.1161/01.hyp.21.1.112. [DOI] [PubMed] [Google Scholar]
- HOGG D.S., ALBARWANI S., DAVIES A.R., KOZLOWSKI R.Z. Endothelial cells freshly isolated from resistance-sized pulmonary arteries possess a unique K+ current profile. Biochem. Biophys. Res. Commun. 1999;263:405–409. doi: 10.1006/bbrc.1999.1338. [DOI] [PubMed] [Google Scholar]
- ISHIDA Y., HONDA H. Inhibitory action of 4-aminopyridine on Ca2+-ATPase of the mammalian sarcoplasmic reticulum. J. Biol. Chem. 1993;268:4021–4024. [PubMed] [Google Scholar]
- KAMOUCHI M., DROOGMANS G., NILIUS B. Membrane potential as a modulator of the free intracellular Ca2+ concentration in agonist-activated endothelial cells. Gen. Physiol. Biophys. 1999;18:199–208. [PubMed] [Google Scholar]
- KNOT H.J., LOUNSBURY K.M., BRAYDEN J.E., NELSON M.T. Gender differences in coronary artery diameter reflect changes in both endothelial Ca2+ and ecNOS activity. Am. J. Physiol. 1999;276:H961–H969. doi: 10.1152/ajpheart.1999.276.3.H961. [DOI] [PubMed] [Google Scholar]
- LÜCKHÖFF A., BUSSE R. Activators of potassium channels enhance calcium influx into endothelial cells as a consequence of potassium currents. Naunyn Schmiedebergs Arch. Pharmacol. 1990;342:94–109. doi: 10.1007/BF00178979. [DOI] [PubMed] [Google Scholar]
- LYTTON J., WESTLIN M., HANLEY M.R. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J. Biol. Chem. 1991;266:17067–17071. [PubMed] [Google Scholar]
- MARCHENKO S.M., SAGE S.O. Calcium-activated potassium channels in the endothelium of intact rat aorta. J. Physiol. 1996;492:53–60. doi: 10.1113/jphysiol.1996.sp021288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MONCADA S., VANE J.R. Pharmacology and endogenous roles of prostaglandin endoperoxides, thromboxane A2, and prostacyclin. Pharmacol. Rev. 1978;30:293–331. [PubMed] [Google Scholar]
- MULVANY M.J., HALPERN W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ. Res. 1977;41:19–25. doi: 10.1161/01.res.41.1.19. [DOI] [PubMed] [Google Scholar]
- NILIUS B. Permeation properties of a non-selective cation channel in human vascular endothelial cells. Pflügers Arch. 1990;416:609–611. doi: 10.1007/BF00382697. [DOI] [PubMed] [Google Scholar]
- NILIUS B., VIANA F., DROOGMANS G. Ion channels in vascular endothelium. Annu. Rev. Physiol. 1997;59:145–170. doi: 10.1146/annurev.physiol.59.1.145. [DOI] [PubMed] [Google Scholar]
- OHASHI M., SATOH K., ITOH T. Acetylcholine-induced membrane potential changes in endothelial cells of rabbit aortic valve. Br. J. Pharmacol. 1999;126:19–26. doi: 10.1038/sj.bjp.0702262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRIETO D., SIMONSEN U., HERNANDEZ M., GARCIA-SACRISTAN A. Contribution of K+ channels and ouabain-sensitive mechanisms to the endothelium-dependent relaxations of horse penile small arteries. Br. J. Pharmacol. 1998;123:1609–1620. doi: 10.1038/sj.bjp.0701780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUIGNARD J.F., FELETOU M., THOLLON C., VILAINE J.P., DUHAULT J., VANHOUTTE P.M. Potassium ions and endothelium-derived hyperpolarizing factor in guinea-pig carotid and porcine coronary arteries. Br. J. Pharmacol. 1999;127:27–34. doi: 10.1038/sj.bjp.0702493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUIGNARD J.F., FELETOU M., EDWARDS G., DUHAULT J., WESTON A.H., VANHOUTTE P.M. Role of endothelial cell hyperpolarization in EDHF-mediated responses in the guinea-pig carotid artery. Br. J. Pharmacol. 2000;129:1103–1112. doi: 10.1038/sj.bjp.0703175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIMIZU S., PAUL R.J. The endothelium-dependent, substance P relaxation of porcine coronary arteries resistant to nitric oxide synthesis inhibition is partially mediated by 4-aminopyridine-sensitive voltage-dependent K+ channels. Endothelium. 1997;5:287–295. doi: 10.3109/10623329709052593. [DOI] [PubMed] [Google Scholar]
- TAKEDA K., SCHINI V., STOECKEL H. Voltage-activated potassium, but not calcium currents in cultured bovine aortic endothelial cells. Pflügers Arch. 1987;410:385–393. doi: 10.1007/BF00586515. [DOI] [PubMed] [Google Scholar]
- TAYLOR S.G., WESTON A.H. Endothelium-derived hyperpolarizing factor: a new endogenous inhibitor from the vascular endothelium. Trends Pharmacol. Sci. 1988;9:272–274. doi: 10.1016/0165-6147(88)90003-x. [DOI] [PubMed] [Google Scholar]
- WALDRON G.J., GARLAND C.J. Effect of potassium channel blockers on the L-NAME insensitive relaxations in rat small mesenteric artery (Abstract) Can. J. Physiol. Pharmacol. 1994a;72:A115. [Google Scholar]
- WALDRON G.J., GARLAND C.J. Contribution of both nitric oxide and a change in membrane potential to acetylcholine-induced relaxation in the rat small mesenteric artery. Br. J. Pharmacol. 1994b;112:831–836. doi: 10.1111/j.1476-5381.1994.tb13154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG X., VAN BREEMEN C. Depolarization-mediated inhibition of Ca2+ entry in endothelial cells. Am. J. Physiol. 1999;277:H1498–H1504. doi: 10.1152/ajpheart.1999.277.4.H1498. [DOI] [PubMed] [Google Scholar]
- WOOD P.G., GILLESPIE J.I. In permeabilised endothelial cells IP3-induced Ca2+ release is dependent on the cytoplasmic concentration of monovalent cations. Cardiovasc. Res. 1998;37:263–270. doi: 10.1016/s0008-6363(97)00207-1. [DOI] [PubMed] [Google Scholar]
- WANG X., CHU W., VAN BREEMEN C. Potentiation of acetylcholine-induced responses in freshly isolated rabbit aortic endothelial cells. J. Vasc. Res. 1996;33:414–424. doi: 10.1159/000159170. [DOI] [PubMed] [Google Scholar]
- XU C., LU Y., TANG G., WANG R.E. Expression of voltage-dependent K+ channel genes in mesenteric artery smooth muscle cells. Am. J. Physiol. 1999;277:G1055–G1063. doi: 10.1152/ajpgi.1999.277.5.G1055. [DOI] [PubMed] [Google Scholar]
- ZYGMUNT P.M., HÖGGESTÄTT E.D. Role of potassium channels in endothelium-dependent relaxation resistant to nitroarginine in the rat hepatic artery. Br. J. Pharmacol. 1996;117:1600–1606. doi: 10.1111/j.1476-5381.1996.tb15327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZYGMUNT P.M., EDWARDS G., WESTON A.H., LARSSON B., HÖGGESTÄTT E.D. Involvement of voltage-dependent potassium channels in the EDHF-mediated relaxation of rat hepatic artery. Br. J. Pharmacol. 1997;121:141–149. doi: 10.1038/sj.bjp.0701108. [DOI] [PMC free article] [PubMed] [Google Scholar]