

Abstract
Prostaglandins and the vasodilator neuropeptide, calcitonin-gene related peptide (CGRP), have both been implicated in the pathogenesis of migraine headache. We have used primary cultures of adult rat trigeminal neurones to examine the effects of prostanoids on CGRP release in vitro.
CGRP release was stimulated by prostaglandin E2 (PGE2) and the IP receptor agonist, carbaprostacyclin (cPGI2). These responses were extracellular calcium-dependent, and the PGE2-induced CGRP release was unaltered by inhibition of nitric oxide synthase (NOS), ATP receptor blockade, or the addition of adenosine deaminase.
Increases in CGRP levels were also observed in response to prostaglandin D2 (PGD2), and the EP2 receptor selective agonist, butaprost. No increases in CGRP release were observed in response to prostaglandin F2α (PGF2α) or the TP receptor selective agonist, U46619, or the EP3 receptor selective agonist, GR63799X.
The selective DP receptor antagonist, BWA868C, antagonized the PGD2-, but not PGE2- or cPGI2-induced release. Furthermore, the EP1 selective antagonist, ZM325802, failed to antagonize the PGE2-induced CGRP release from these cells.
These data indicate that activation of DP, EP and IP receptors can each cause CGRP release from trigeminal neurones, and suggest that the predominant EP receptor subtype involved may be the EP2 receptor. Together with evidence that the cyclo-oxygenase inhibitor, aspirin, particularly when administered intravenously is effective in treating acute migraine, these findings further suggest a role for prostaglandins in migraine pathophysiology.
Keywords: Prostanoids, prostaglandin E2, prostacyclin, migraine, calcitonin gene-related peptide, trigeminal ganglion, primary culture
Introduction
Prostaglandins and thromboxanes are metabolites of arachidonic acid that act as local mediators in both the central nervous system and the periphery. There are five naturally occurring prostanoids, prostaglandin D2 (PGD2), prostaglandin E2 (PGE2), prostaglandin F2α (PGF2α), prostacyclin (PGI2) and thromboxane A2 (TXA2). They exert their physiological actions through binding to membrane-bound receptors that are coupled to heterotrimeric guanine nucleotide-binding (G) proteins and belong to the class of seven transmembrane domain receptors. The scheme of prostanoid receptor classification system identifies specific receptors for each of the natural prostanoids, termed DP, EP, FP, IP and TP receptors, respectively (Kennedy et al., 1982; Coleman et al., 1994). At each of these receptors one of the natural prostanoids is at least one order of magnitude more potent than the other four. The EP receptor has been further classified into four subtypes, termed EP1, EP2, EP3 and EP4 receptors, based on studies using synthetic agonists and antagonists (Coleman et al., 1994).
The role of prostaglandins, particularly PGE2 and PGI2, in inflammatory pain is well accepted, partly because of the anti-nociceptive and anti-inflammatory effects of non-steroidal anti-inflammatory drugs (NSAIDS), and also from observations in several animal models of nociception that exogenous prostaglandins can cause hyperalgesia and allodynia (Vane, 1971; Bley et al., 1998). Prostaglandins have also been implicated in migraine headache. The cyclo-oxygenase inhibitor, aspirin, is effective in treating acute migraine, particularly when administered intravenously (Diener, 1999) and the effectiveness of this class of drug in migraine as well as other headaches has been comprehensively reviewed (Limmroth et al., 1999). It has also been reported that prostaglandins can cause migraine-like symptoms when administered to volunteers (see Coleman et al., 1990) and that levels of PGE2 are increased in the saliva of patients undergoing migraine attacks (Vardi et al., 1983; Tuca et al., 1989). Recently, Ebersberger et al. (1999) have also shown that PGE2 is released from rat dura mater encephali following electrical stimulation of the trigeminal ganglion and chemical stimulation with inflammatory mediators (5-hydroxytryptamine, histamine and bradykinin). This provides further evidence for a possible role for prostaglandins in facilitating meningeal nociceptor activation, and promoting inflammation and pain in headache.
Also implicated in migraine is the potent vasodilator neuropeptide, calcitonin gene-related peptide (CGRP), which is localized within trigeminal afferents that innervate intracranial, extracerebral blood vessels (Edvinsson et al., 1987). The cell bodies of these trigeminal neurones are located in the trigeminal ganglion (external to the brain at the level of the pons) and stimulation of the trigeminal ganglion in both cats and humans causes CGRP release that can be inhibited by the clinically effective anti-migraine agent, sumatriptan (Goadsby et al., 1988; Goadsby & Edvinsson, 1993). The levels of this peptide are also raised in the cephalic venous blood of migraineurs during the headache-phase, and are subsequently reduced by sumatriptan (Goadsby et al., 1990). In light of this evidence, suggesting roles for both prostaglandins and CGRP in migraine, we have investigated the receptors and mechanisms involved in prostanoid-induced CGRP release in cultured rat trigeminal neurones.
Methods
Preparation of primary cultures of adult rat trigeminal neurones
Cultures of adult rat trigeminal neurones were prepared as previously described (Carruthers et al., 2001). Briefly, adult Wistar rats (175 – 250 g; either sex) were killed by CO2 inhalation in strict accordance with U.K. Home Office regulations. Trigeminal ganglia were dissected out and placed in ice-cold calcium-, magnesium- and bicarbonate-free Hanks' balanced salt solution (CMF-Hanks'), before being chopped and incubated for 17 min at 37°C in 3 ml CMF-Hank's containing 20 u ml−1 papain. Cells were pelleted by centrifugation at 250×g for 3 min and the supernatant was replaced with 3 ml CMF-Hank's supplemented with 0.3% (w v−1) collagenase and 0.4% (w v−1) dispase II. After a further incubation at 37°C for 20 min, the cells were re-spun at 250×g and the pellet resuspended in 3 ml CMF-Hank's and 2 ml Liebowitz's L-15 medium supplemented with 5 mM Na+-HEPES, 5 mM D-Glucose, 10% (v v−1) heat-inactivated foetal bovine serum, 100 IU ml−1 penicillin, 100 μg ml−1 streptomycin and 0.15% (w v−1) deoxyribonuclease I. Clumps of cells were dissociated via trituration through a graded series of fire-polished Pasteur pipettes. After centrifugation at 250×g for 3 min, the cell pellet was resuspended in culture medium [Ham's F-12 (GlutaMAX-I) containing 10% heat-inactivated foetal bovine serum, 100 IU ml−1 penicillin, 100 μg ml−1 streptomycin, and nerve growth factor (m2.5S NGF; 50 ng ml−1)] before being plated down on Poly-D-lysine (150K+; 0.1 mg ml−1) and laminin (20 μg ml−1) pre-treated 12-well plates. Cells (200 – 500 per well) were incubated at 37°C in a 5% CO2/humidified air atmosphere for 4 – 6 days. After 24 h and every other day thereafter, the culture medium was replaced with F-12 medium further supplemented with the mitotic inhibitor cytosine-β-D-arabinofuranoside (20 μM) to limit the growth of non-neuronal cells.
CGRP release from trigeminal neuronal cultures
After 4 – 6 days in culture, the medium was gently aspirated and replaced with 1 ml CGRP release buffer (Vasko et al., 1994) (composition mM): Na+-HEPES 22.5, NaCl 135, KCl 3.5, MgCl2 1, CaCl2 2.5, D-glucose 3.3, 0.1% (w v−1) bovine serum albumin, 0.003% (w v−1) bacitracin, 1 μM phosphoramidon and 10 μM indomethacin (to inhibit tonic prostanoid synthesis), pH 7.4 at 37°C). Cells were incubated for 30 min at 37°C in release buffer, before this was replaced with 1 ml test agonist or vehicle (dimethylsuphoxide (DMSO), maximal concentration 0.1%) for a further 30 min. In someexperiments, cultures were incubated for the first 30 min period in either the nitric oxide synthase (NOS) inhibitor, NG-nitro-L-arginine (L-NAME; 10 μM), the P2 receptor antagonist, pyridoxalphosphate-6-azophenyl-2′,4′-disulphonic acid (PPADS; 30 μM), or with adenosine deaminase (1 u ml−1). To investigate whether the CGRP release observed was extracellular-calcium dependent, experiments were conducted in calcium-free release buffer (equimolar substitution of Ca2+ with Mg2+). Following each incubation, 0.8 ml samples were removed and assayed for CGRP content. A commercial enzyme immunometric assay (SPIbio, Massy, France) was used for quantitative analysis of the CGRP content of the eluates. CGRP levels were determined photometrically at 405 nm using a microplate reader (Packard SpectraCountTM). The antibody used in this assay is 100% cross-reactive between rat CGRPα and β and <0.01% cross-reactive with substance P (SPIbio, commercial information) and the minimum assay detection limit was approximately 10 pg ml−1. None of the compounds used in this study were found to cross-react non-specifically with the assay at the concentrations indicated.
Data analysis
The CGRP concentrations in the samples were quantified in pg ml−1. In order to account for differences in neuronal numbers and baseline CGRP concentrations between preparations and between individual wells, each well acted as its own control. The increase in immunoreactive CGRP concentrations following 30 min drug incubation was calculated as a percentage increase over the baseline concentration obtained in the 30 min prior to drug administration ((CGRP conc. after drug treatment – basal)/basal×100). Only one drug treatment was given to any single well. Unless otherwise stated, values from individual experiments were pooled and expressed as a mean±s.e.mean.
Individual concentration-effect curves were fitted to a logistic equation using Prism 3 (Graphpad Software Inc., San Diego, C.A., U.S.A.), with the maximum response for each agonist constrained to 100%. The mean pEC50 value was calculated by taking the average of the individual estimates. No estimate of pEC50 was made when a clearly defined maximum was not achieved. Statistical analysis between treatments was by one-way analysis of variance followed by the Bonferroni post hoc test, and P values of less than 0.05 were considered statistically significant.
Materials
All cell culture media was purchased from Gibco BRL (Paisley, U.K.) and 12-well plates were from Corning Costar (High Wycombe, U.K.). Collagenase (Type 2) and papain were from Worthington (Reading, U.K.) and dispase II was purchased from Roche (Lewes, U.K.). Bovine serum albumin (fraction V, protease-free), murine Engelbroth swarm laminin, bovine pancreas crude DNase I, cytosine-β-D-arabinofuranoside, poly-D-lysine (m.w. 150 K+) and L-NAME were obtained from Sigma (Poole, U.K.). Forskolin and adenosine deaminase (2326 u ml−1) were purchased from Calbiochem (Nottingham, U.K.). Nerve growth factor was from Alomone Labs (Botolph Clayton, U.K.). PGD2, PGE2, PGF2α, carbaprostacyclin (cPGI2), (15S)-hydroxy-11α,9α-(epoxymethano)prosta-5Z, 13E-dienoic acid (U46619), misoprostol methyl ester, sulprostone and indomethacin were all purchased from the Cayman Chemical Company (Ann Arbor, M.I., U.S.A.). Iloprost was obtained from Amersham (Little Chalfont, U.K.). Butaprost free acid, [1R-[1α(Z), 2β(R*),3α]]-4-(benzoylamino)phenyl-7-[3-hydroxy-2-(2-hydroxy-3-phenoxy-propoxy)-5 -oxocyclopentyl]-4-heptenoate (GR63799X), 3-benzyl-5-(6-carbohexyl)-1-(2-cyclohexyl-2-hydroxyethylamino)-hydantoin (BWA868C), ZM325802 (Shaw et al., 1999) were obtained from Glaxo SmithKline Research & Development (Stevenage, U.K.). PPADS was purchased from RBI (Poole, U.K.).
PGD2, PGE2, PGF2α, cPGI2, U46619, sulprostone, butaprost free acid, GR63799X, ZM325802, BWA868C, forskolin and indomethacin were dissolved with DMSO at 10 mM and immediately frozen at −20°C. Iloprost was supplied at 1 mM in Tris buffer and was diluted directly to 1 μM in release buffer. PPADS and L-NAME was prepared at 1 μM in water and serially diluted in release buffer.
Results
Effects of PGE2 and carbaprostacyclin (cPGI2) on CGRP release from trigeminal ganglion neuronal cultures
To investigate the effects of prostanoids on CGRP release from trigeminal neurones, we established primary cultures of adult trigeminal ganglion neurones. Incubation of the cultures in release buffer, containing indomethacin (10 μM) and phosphoramidon (1 μM), for 30 min resulted in an average baseline CGRP concentration of 112±8 pg ml−1 (range 23 – 251 pg ml−1; n=29 independent culture preparations). Incubation of cells with PGE2 (1 μM) or a stable analogue of prostacyclin, cPGI2 (1 μM) caused significant (P<0.001) increases in the concentration of immunoreactive CGRP of 199±25% and 177±26% respectively, compared with vehicle treated controls (7±10%; n=8). When cells were exposed to calcium-free release buffer (equimolar substitution of Ca2+ with Mg2+), the basal CGRP levels after 30 min were lower than in control solutions (36±2.3 pg ml−1 vs 67±12 pg ml−1), but the increases in CGRP release over baseline in response to PGE2 and cPGI2 were abolished (Figure 1).
Figure 1.
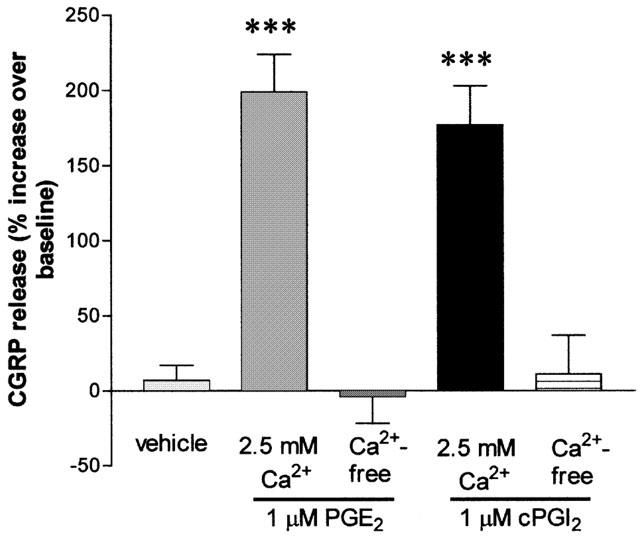
Effects of extracellular calcium on PGE2- and cPGI2-induced CGRP release. Following 4 – 6 days in culture, adult trigeminal ganglion cells were exposed to either PGE2 (1 μM) or cPGI2 (1 μM) in the presence or absence of 2.5 mM extracellular calcium (equimolar substitution of Ca2+ with Mg2+ in the release buffer). Data are expressed as the percentage increase in CGRP release and are presented as the mean±s.e.mean from 3 – 8 independent experiments. ***P<0.001, significantly different from controls.
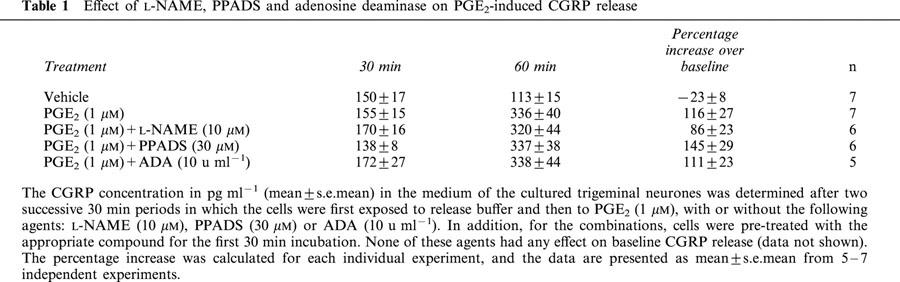
Effects of L-NAME, PPADS and adenosine deaminase on PGE2-mediated CGRP release
To further investigate the nature of the PGE2-mediated CGRP release, the effects of the non-specific NOS inhibitor, L-NAME (10 μM), the P2 receptor antagonist, PPADS (30 μM), and the adenosine metabolizing enzyme, adenosine deaminase (1 u ml−1), were tested. It was found that none of these agents modified baseline CGRP levels (data not shown), and, furthermore, that these agents did not modify CGRP release evoked by 1 μM PGE2 (Table 1).
Table 1.
Effect of L-NAME, PPADS and adenosine deaminase on PGE2-induced CGRP release
Effects of other prostanoid receptor agonists on CGRP release from cultured trigeminal neurones
Stimulation of the cultures with PGD2 (1 μM) significantly increased immunoreactive CGRP concentrations by 107±23% (n=5; P<0.05 compared to vehicle controls). In addition, the effect of cPGI2, indicative of IP receptor activation, was mimicked by another stable prostacyclin analogue, iloprost (1 μM) (168±26% increase in CGRP concentration; n=3). Neither PGF2α (1 μM), nor the TP receptor agonist, U46619 (1 μM) had any significant effect on CGRP concentrations following a 30 min incubation period (Figure 2A).
Figure 2.
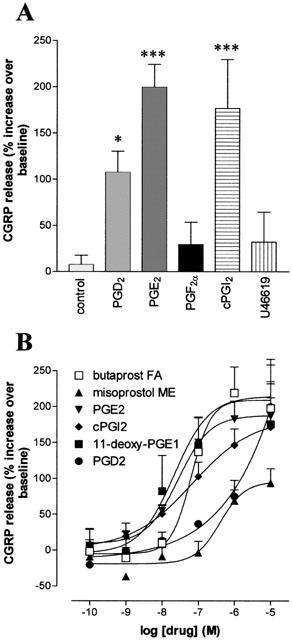
Effects of prostanoid agonists on CGRP release from cultured adult rat trigeminal neurones. After 4 – 6 days in culture, cells were incubated in release buffer (Vasko et al., 1994), in the presence of 10 μM indomethacin for 30 min, before being challenged for a further 30 min with either vehicle (DMSO, 0.1%) or test agonist. Data are expressed as the percentage increase in CGRP release over baseline (A) Effects PGD2, PGE2, PGF2α, cPGI2 and the TP receptor agonist, U46619 (each at 1 μM). *P<0.05, ***P<0.001 over controls. (B) Effects of increasing concentrations of PGD2, PGE2, cPGI2, the EP2 receptor agonist, butaprost free acid, the EP3>EP2 receptor agonist misoprostol and the EP4>EP2 receptor agonist, 11-deoxy PGE1. Data are presented as the percentage increase in CGRP secretion over baseline and represent the mean±s.e.mean of 3 – 8 experiments.
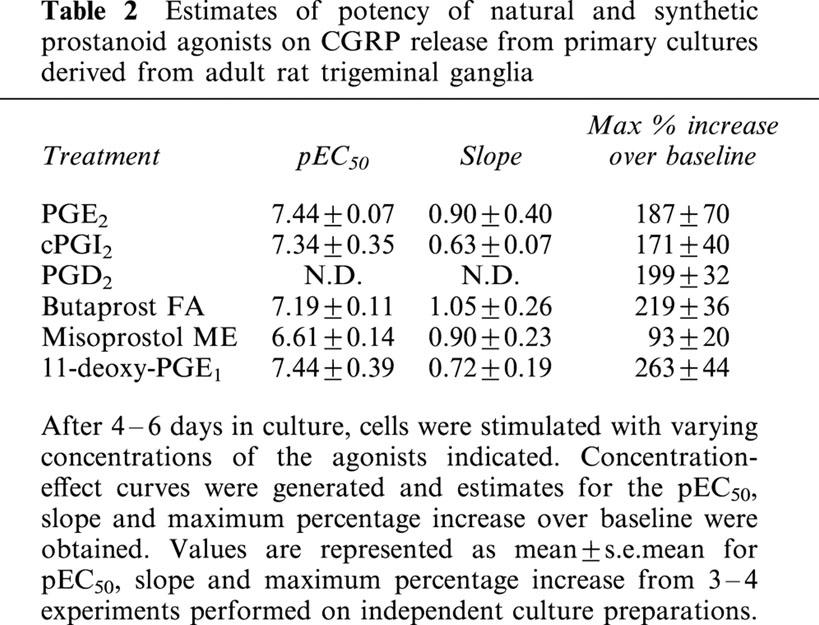
To further characterize the receptor types involved in prostaglandin-induced increases in CGRP release, concentration-effect curves were generated to PGE2, cPGI2, the EP2 receptor agonist butaprost (free acid), the EP3>EP2 receptor agonist misoprostol, and the EP4>EP2 receptor agonist, 11-deoxy-PGE1 (Figure 2B; Table 2). PGE2, 11-deoxy-PGE1, cPGI2 and butaprost all produced similar maximum increases in CGRP release, but the maximum response to misoprostol was lower. The overall rank order of agonist potency for these agonists was PGE2=11-deoxy-PGE1=cPGI2=butaprost>misoprostol. Neither the selective EP3 receptor agonist, GR63799X (1 μM), nor the EP3>EP1 receptor agonist, sulprostone (1 μM), had any significant effect on basal immunoreactive CGRP concentrations (both n=5). PGD2 also caused concentration dependent increases in immunoreactive CGRP release (Figure 2B) but a clearly defined maximum response was not achieved even with a concentration as high as 10 μM.
Table 2.
Estimates of potency of natural and synthetic prostanoid agonists on CGRP release from primary cultures derived from adult rat trigeminal ganglia
Effects of prostanoid receptor antagonists on PGD2 and PGE2-evoked CGRP release from trigeminal neurones
The specificity of the PGD2 mediated peptide release was investigated using the DP receptor antagonist, BWA868C (Giles et al., 1989). When cells were pre-incubated for 30 min with BWA868C, the PGD2-stimulated increases in CGRP release were abolished. In contrast, this antagonist had no effect on CGRP release stimulated by 1 μM PGE2 or cPGI2 (Figure 3A).
Figure 3.
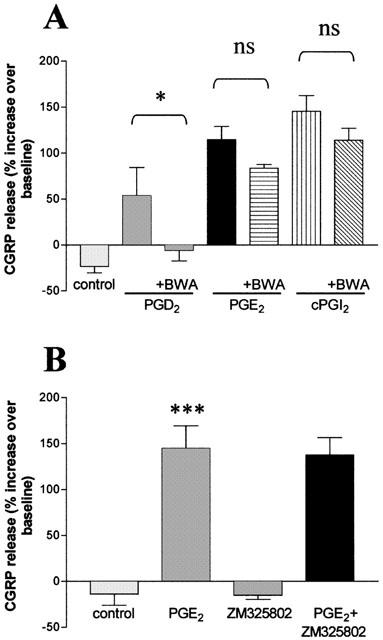
Effects of prostaglandin receptor antagonists on PGD2- and PGE2-induced CGRP release. (A) Cultured trigeminal ganglion neurones were incubated with release buffer for 30 min and then subsequently challenged with 1 μM PGD2, PGE2 or cPGI2 with or without the DP receptor antagonist, BWA 868C (1 μM). Co-incubation wells were pre-incubated with antagonist for 30 min before being incubated with the appropriate test agonist. *P<0.05 against PGD2 alone. (B) Rat trigeminal ganglion neurones (4 – 6 days in culture) were incubated with either release buffer for 30 min and then PGE2 alone or the EP1 receptor antagonist, ZM325802, (both 1 μM) for another 30 min or ZM325802 (1 μM) for 30 min and then PGE2 and ZM325802 (both 1 μM) for 30 min. Data are presented as the percentage increase in CGRP release over baseline and represent the mean±s.e.mean of 3 – 5 independent experiments. ***P<0.001 vs control.
The EP1 antagonist, ZM325802 (Figure 3B) and SC-19220 and SC-51322 (David W. Jenkins, unpublished observations) had no effect on PGE2 (1 μM) stimulated CGRP release. We were unable to use the weak EP4 receptor antagonist, AH23848B (pA2=5.4, Coleman et al., 1994), to determine the possible involvement of EP4 receptors in mediating PGE2-induced CGRP release as this antagonist itself caused a marked increase in baseline CGRP secretion (446 and 391% at 10 μM, n=2).
Effect of the EP3 receptor agonist, GR63799X, on forskolin mediated CGRP release
It has been reported that the EP3 receptor exists in several splice variants that can couple differentially to several downstream signalling pathways, including Gi/o to reduce cellular cyclic AMP levels and Gs to induce the opposite effect (see Narimuya et al., 1999). Therefore, although we observed no CGRP release per se in response to the specific EP3 receptor agonist, GR63799X (see above), we also tested the effects of this agonist against CGRP release stimulated by the adenylate cyclase activator forskolin. Incubation of the cultures in forskolin (1 μM) for 30 min increased basal immunoreactive CGRP concentrations by 102±9%. In the presence of GR63799X (1 μM), the forskolin-induced increase was 102±17%. Similar results were observed when the cultures were pre-incubated for 30 min in GR63799X (1 μM) before stimulation with forskolin (data not shown).
Discussion
The trigeminal ganglion contains the cell bodies of the afferent neurones of the fifth cranial nerve, thought to be responsible for the pain associated with migraine. Although we have previously shown, using reverse transcription polymerase chain reaction (RT – PCR) that whole excised trigeminal ganglia express mRNA for all four EP receptors and also the IP receptor (Jenkins et al., 2000), the effects of prostanoids have remained poorly characterized with respect to both receptor localization and function. We have therefore used cultured trigeminal neurones to investigate the receptors involved in prostanoid-induced CGRP release, since this is one of the major neuropeptides thought to be implicated in the pathophysiology of migraine.
All experiments in the present study were carried out on cultures of adult trigeminal neurones and conducted in the presence of indomethacin to inhibit de novo prostaglandin synthesis, thus avoiding the confounding influences of endogenous prostanoid release. We have demonstrated that, in cultured trigeminal neurones, CGRP release can be stimulated by PGD2, PGE2 and cPGI2, consistent with the activation of DP, EP, and IP receptors, respectively. The IP receptor agonist, iloprost, also caused CGRP release from these cells. In contrast, agonists at FP (PGF2α) and TP (U46619) receptors did not produce a significant effect. The release of CGRP by PGE2 and cPGI2 was not likely to have been compromised by a significant depletion of the releasable CGRP pool since release-induced by other agents, AH23848B (this study) and KCl (Carruthers et al., 2001) were substantially greater. In addition permeabilization of the trigeminal neurones with Triton X-100 (0.3%) causes an approximate 40 fold increase in the basal release of immunoreactive CGRP (unpublished observations).
Both the PGE2- and cPGI2-induced release of CGRP were highly dependent upon extracellular calcium entry, since removal of extracellular calcium effectively prevented agonist-induced release of CGRP. Furthermore, we also found that the non-specific NOS inhibitor, L-NAME, the non-selective ATP receptor antagonist, PPADS and the adenosine metabolizing enzyme, adenosine deaminase, did not modify the CGRP release induced by PGE2. Taken together, these data suggest that the PGE2 mediated effects are a direct consequence of EP receptor stimulation and not dependent on the secondary release of nitric oxide, ATP or adenosine. The lack of effect of L-NAME is particularly relevant since PGE2 can cause nitric oxide release from rat spinal cord (Sakai et al., 1998).
The ability of both cPGI2 and iloprost to release CGRP from cultured trigeminal neurones provides evidence for functional IP receptors in these neurones and is consistent with previous findings in DRGs (Hingtgen et al., 1995; Smith et al., 1998). Although several studies have demonstrated functional effects of PGE2 in DRGs, this study is the first to show a direct PGE2-induced release of CGRP from trigeminal neurones. This contrasts with findings in DRGs, which have shown PGI2, but not PGE2-induced CGRP release, although interestingly both agonists increased intracellular cyclic AMP (Vasko et al., 1994; Hingtgen et al., 1995; Hingtgen & Vasko, 1994). However, the present study was conducted in adult trigeminal neurones, rather than neonatal DRGs, and it would be necessary to carry out further studies to determine whether the differences in functional response observed were due to differences in source material (adult vs neonate) or tissue type (trigeminal ganglion vs DRG).
The response to PGE2 may be mediated by several different receptors and the effects of several EP-receptor selective agonists and antagonists were evaluated to identify the EP receptor subtype involved. The endogenous prostanoids also show some degree of cross-reactivity between receptor types (Coleman et al., 1994), so it is possible that responses to PGE2 may be mediated in part by the IP receptor. However, at present, no potent, selective IP antagonists are available to test this assertion. The EP1 antagonist, ZM325802 (Shaw et al., 1999) had no effect on PGE2-mediated CGRP release, suggesting that EP1 receptors are not involved. In addition, no attenuation of peptide release was observed using the less potent putative EP1 antagonists, SC-19220 and SC-51322 (D.W. Jenkins, unpublished observations). However, it has previously been reported that SC-19220 can block PGE2-induced substance P release from cultured rat DRGs (White, 1996).
There is a considerable body of evidence to suggest that the effects of PGE2 on sensory neurones are mediated by receptors that are coupled to increases in adenylate cyclase activity, implying the involvement of EP2 and EP4 receptors, and possibly an EP3 receptor splice variant, all of which may couple to Gs (Vasko et al., 1994; Cui & Nicol, 1995; Hingtgen et al., 1995; England et al., 1996; Smith et al., 1998; 2000). In this study, the EP2 selective agonist, butaprost, stimulated CGRP release, and the estimated potency was similar to previously reported values measuring the ability of this agonist to stimulate cyclic AMP accumulation in Chinese Hamster Ovary (CHO) cells, stably transfected with the recombinant rat EP2 receptor (Boie et al., 1997). Recently, the same agonist has also been reported to cause increases in cyclic AMP levels in neonatal DRG cultures that could be attenuated by an antisense oligonucletide directed at the EP2 receptor (Southall & Vasko, 2001). In addition to butaprost, we showed that the mixed EP3>EP2 receptor agonist, misoprostol, also caused a small, but significant increase in CGRP release, suggestive of activity at EP2 receptors. The EP4>EP2 receptor agonist, 11-deoxy-PGE1, also caused CGRP release, with an estimated potency consistent with EP2 receptor activation (Boie et al., 1997).
It seems unlikely that EP3 receptors are involved in PGE2-mediated CGRP release since the specific EP3 receptor agonist, GR63799X, or the mixed EP3>EP1 receptor agonist, sulprostone were without effect. In addition, GR63799X had no effect on forskolin-induced CGRP, an effect which we have previously shown to be inhibited by several agonists that couple to Gi/o (Carruthers et al., 2001). This lack of functional activity associated with EP3 receptors was somewhat surprising, since EP3 receptors have been localized in the trigeminal ganglion at both the mRNA level in the mouse (Sugimoto et al., 1994) and, recently, the protein level in the rat (Nakamura et al., 2000). There are no potent, selective EP4 receptor antagonists available at present, and the weak antagonist, AH23848B (pA2 ≅5.4; see Coleman et al., 1994) caused a large increase in CGRP release Since this release precluded further analysis, the potential significance of EP4 receptor activation is unknown.
In conclusion, we have shown that stimulation of cultured rat trigeminal ganglion neurones with either D, E or I series prostaglandins causes a Ca2+-dependent CGRP release. Since CGRP levels in jugular venous blood are known to be elevated during migraine (Goadsby et al., 1988) and electrical stimulation of the trigeminal ganglion is known to release PGE2 (Ebersberger et al., 1999), PGE2 may play an important role in the generation of migraine and in the peripheral sensitization thought to occur during migraine headache (see Burstein, 2001). Antagonists of prostanoid-induced CGRP release may provide novel therapeutic approaches to the treatment of migraine.
Abbreviations
- CGRP
calcitonin gene-related peptide
- cPGI2
carbaprostacyclin
- DMSO
dimethylsulphoxide
- FITC
fluorescein isothiocyanate
- L-NAME
NG-nitro-L-arginine
- NGF
nerve growth factor
- NGS
normal goat serum;
- NSAID
non-steroidal anti-inflammatory drug
- PBS
phosphate buffered saline
- PGD2
prostaglandin D2
- PGE2
prostaglandin E2
- PGF2α
prostaglandin F2α
- PGI2
prostaglandin I2
- PPADS
pyridoxalphosphate-6-axophenyl-2′,4′-disulphonic acid
- TXA2
thromboxane A2
References
- BLEY K.R., HUNTER J.C., EGLEN R.M., SMITH J.A.M. The role of IP prostanoid receptors in inflammatory pain. Trends Pharmacol. Sci. 1998;19:141–147. doi: 10.1016/s0165-6147(98)01185-7. [DOI] [PubMed] [Google Scholar]
- BOIE Y., STOCCO R., SAWYWE N., SLIPETS D.M., UNGRIN M.D., NEUSCHAFER-RUBE F., PUSCHEL G.P., METTERS K.M., ABRAMOWITZ M. Molecular cloning and characterization of the four rat prostaglandin E2 prostanoid receptor subtypes. Eur. J. Pharmacol. 1997;340:227–241. doi: 10.1016/s0014-2999(97)01383-6. [DOI] [PubMed] [Google Scholar]
- BURSTEIN R. Deconstructing migraine headache into peripheral and central sensitization. Pain. 2001;89:107–110. doi: 10.1016/s0304-3959(00)00478-4. [DOI] [PubMed] [Google Scholar]
- CARRUTHERS A.M., SELLERS L.A., JENKINS D.W., JARVIE E.M., FENIUK W., HUMPHREY P.P.A. Adenosine A1 receptor-mediated inhibition of PKA-induced calcitonin gene-related peptide release from rat trigeminal neurones. Mol. Pharm. 2001;59:1533–1541. doi: 10.1124/mol.59.6.1533. [DOI] [PubMed] [Google Scholar]
- COLEMAN R.A., KENNEDY I., HUMPHREY P.P.A., BUNCE K., LUMLEY P.Prostanoids and their receptors Comprehensive Medicinal Chemistry 1990Oxford: Pergamon Press; 643–714.Vol 3. ed Hansch, C., Sammes, P.G., Taylor, J.B. & Emmit, J.C. [Google Scholar]
- COLEMAN R.A., SMITH W.L., NARIMUYA S. VIII. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharm. Rev. 1994;46:205–229. [PubMed] [Google Scholar]
- CUI M., NICOL G.D. Cyclic AMP mediates the Prostaglandin E2-induced potentiation of bradykinin excitation in sensory neurons. Neurosci. 1995;66:459–466. doi: 10.1016/0306-4522(94)00567-o. [DOI] [PubMed] [Google Scholar]
- DIENER H.C. Efficacy and safety of intravenous acetylsalicylic acid lysinate compared to subcutaneous sumatriptan and parenteral placebo in the acute treatment of migraine. A double-blind, double-dummy, randomized, multicenter, parallel group study. Cephalagia. 1999;19:581–588. doi: 10.1046/j.1468-2982.1999.019006581.x. [DOI] [PubMed] [Google Scholar]
- EBERSBERGER A., AVERBECK B., MESSLINGER K., REEH P.W. Release of substance P, calcitonin gene-related peptide and prostaglandin E2 from rat dura mater encephali following electrical and chemical stimulation in vitro. Neurosci. 1999;89:901–907. doi: 10.1016/s0306-4522(98)00366-2. [DOI] [PubMed] [Google Scholar]
- EDVINSSON L., EKMAN R., JANSEN I., MCCULLOCH J., UDDMAN R. Calcitonin gene-related peptide and cerebral blood vessels: distribution and vasomotor effects. J. Cereb. Blood Flow Metab. 1987;7:720–728. doi: 10.1038/jcbfm.1987.126. [DOI] [PubMed] [Google Scholar]
- ENGLAND S., BEVAN S., DOCHERTY R.J. PGE2 modulates the tetrodotoxin-resistant sodium current in neonatal rat dorsal root ganglion neurones via the cyclic AMP-protein kinase A cascade. J. Physiol. (Lond.) 1996;495:429–440. doi: 10.1113/jphysiol.1996.sp021604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GILES H., LEFF P., BOLOF M.L., KELLY M.G., ROBERTSON A.D. The classification of prostaglandin DP-receptors in platelets and vasculature using BWA868C, a novel, selective and potent competitive antagonist. Br. J. Pharmacol. 1989;96:291–300. doi: 10.1111/j.1476-5381.1989.tb11816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOADSBY P.J., EDVINSSON L. The trigeminovascular system and migraine: studies characterising cerebrovascular and neuropeptide changes seen in man and cat. Ann. Neurol. 1993;33:48–56. doi: 10.1002/ana.410330109. [DOI] [PubMed] [Google Scholar]
- GOADSBY P.J., EDVINSSON L., EKMAN R. Release of vasoactive peptides in the extracerebral circulation of man and the cat during activation of the trigeminovascular system. Ann. Neurol. 1988;23:193–196. doi: 10.1002/ana.410230214. [DOI] [PubMed] [Google Scholar]
- GOADSBY P.J., EDVINSSON L., EKMAN R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann. Neurol. 1990;28:183–187. doi: 10.1002/ana.410280213. [DOI] [PubMed] [Google Scholar]
- HINGTGEN C.M., VASKO M.R. Prostacyclin enhances the release of substance P and calcitonin gene-related peptide from rat sensory neurons. Brain Res. 1994;655:51–60. doi: 10.1016/0006-8993(94)91596-2. [DOI] [PubMed] [Google Scholar]
- HINGTGEN C.M., WAITE K.J., VASKO M.R. Prostaglandins facilitate peptide release from rat sensory neurons by activating the adenosine 3′,5′-cyclic monophosphate transduction cascade. J. Neurosci. 1995;15:5411–5419. doi: 10.1523/JNEUROSCI.15-07-05411.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JENKINS D.W., SELLERS L.A., HUMPHREY P.P.A. Prostaglandins induce calcitonin gene-related peptide release from adult rat trigeminal neurones 2000C52Proceedings of the British Pharmacological Society University of Birmingham, 2000. Abstract Book
- KENNEDY I., COLEMAN R.A., HUMPHREY P.P.A., LEVY G.P., LUMLEY P. Studies on the characterization of prostanoid receptors: a proposed classification. Prostaglandins. 1982;24:667–689. doi: 10.1016/0090-6980(82)90036-3. [DOI] [PubMed] [Google Scholar]
- LIMMROTH V., KATSARAVA Z., DIENER H.C. Acetylsalicylic acid in the treatment of headache. Cephalagia. 1999;19:546–551. doi: 10.1046/j.1468-2982.1999.019006545.x. [DOI] [PubMed] [Google Scholar]
- NAKAMURA K., KANEKO T., YAMASHITA Y., HASEGAWA H., KATOH H., NEGISHI M. Immunohistochemical localization of prostaglandin EP3 receptor in the rat nervous system. J. Comp. Neurol. 2000;421:543–569. doi: 10.1002/(sici)1096-9861(20000612)421:4<543::aid-cne6>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- NARIMUYA S., SUGIMOTO Y., USHIKUBI F. Prostanoid receptors: structures, properties and functions. Physiol. Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- SAKAI M., MINAMI T., HARA N., NISHIHARA I., KITADE H., KAMIYAMA Y., OKUDA K., TAKAHASHI H., MORI H., ITO S. Stimulation of nitric oxide release from rat spinal cord by prostaglandin E2. Br. J. Phramacol. 1998;123:890–894. doi: 10.1038/sj.bjp.0701661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHAW J.S., BARKER A.J., BREAULT G., GRIFFEN E.J., HEAPY C.G., MCLAUGHLIN S.ZM325082: a potent and selective antagonist at the prostanoid EP1 receptor 1999p2729th World Congress on Pain, Vienna, 1999. Abstract Bookp
- SMITH J.A.M., AMAGUSU S.M., EGLEN R.M., HUNTER J.C., BLEY K.R. Characterization of prostanoid receptor-evoked responses in rat sensory neurones. Br. J. Pharmacol. 1998;124:513–523. doi: 10.1038/sj.bjp.0701853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMITH J.A.M, DAVIS C.L., BURGESS G.M. Prostaglandin E2-induced sensitization of bradykinin evoked responses in rat dorsal root ganglion neurons is mediated by cAMP-dependent protein kinase A. Eur. J. Neurosci. 2000;12:3250–3258. doi: 10.1046/j.1460-9568.2000.00218.x. [DOI] [PubMed] [Google Scholar]
- SOUTHALL M.D., VASKO M.R. Prostaglandin receptor subtypes, EP3C and EP4, mediate the Prostaglandin E2-induced cAMP production and sensitization of sensory neurons. J. Biol. Chem. 2001;276:16083–16091. doi: 10.1074/jbc.M011408200. [DOI] [PubMed] [Google Scholar]
- SUGIMOTO Y., SHIGEMOTO R., NAMBA T., NEGISHI M., MIZUNO N., NARIMUYA S., ICHIKAWA A. Distribution of the messenger RNA for the prostaglandin E receptor subtype EP3 in the mouse central nervous system. Neurosci. 1994;62:919–928. doi: 10.1016/0306-4522(94)90483-9. [DOI] [PubMed] [Google Scholar]
- TUCA J.O., PLANAS J.M., PARELLADA P.P. Increase in PGE2 and TXA2 in the saliva of common migraine patients. Action of calcium channel blockers. Headache. 1989;29:498–501. doi: 10.1111/j.1526-4610.1989.hed2908498.x. [DOI] [PubMed] [Google Scholar]
- VANE J.R. Inhibition of prostaglandin synthesis as a mechanism of action for the aspirin- like drugs. Nature. 1971;231:232–235. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]
- VARDI J., FLECHTER S., ALGUATI A., REGEV I., AYALON D. Prostaglandin-E2 levels in the saliva of common migrainous women. Headache. 1983;23:59–61. doi: 10.1111/j.1526-4610.1983.hed2302059.x. [DOI] [PubMed] [Google Scholar]
- VASKO M.R., CAMPBELL W.B., WAITE K.J. Prostaglandin E2 enhances bradykinin-stimulated release of neuropeptides from rat sensory neurons in culture. J. Neurosci. 1994;14:4987–4997. doi: 10.1523/JNEUROSCI.14-08-04987.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHITE D.M. Mechanism of Prostaglandin E2-induced substance P release from cultured sensory neurons. Neurosci. 1996;70:561–565. doi: 10.1016/0306-4522(95)00353-3. [DOI] [PubMed] [Google Scholar]