

Abstract
The objective of the present study was to analyse the peripheral effects of cannabinoids on adrenaline release from adrenal chromaffin cells.
In pithed rabbits with electrically stimulated sympathetic outflow, intravenous injection of the cannabinoid receptor agonists WIN55212-2 and CP55940 (5, 50 and 500 μg kg−1) markedly lowered the plasma adrenaline concentration. The effect of WIN55212-2 was attenuated by the selective CB1 cannabinoid receptor antagonist SR141716A (500 μg kg−1). WIN55212-3 (same doses as WIN55212-2), the enantiomer of WIN55212-2 lacking affinity for cannabinoid receptors, had no effect on the plasma adrenaline concentration.
In rabbit isolated adrenal glands, the release of adrenaline elicited by electrical stimulation was measured by fast cyclic voltammetry. Electrically-evoked adrenaline release was inhibited by WIN55212-2 (0.3, 1, 3 and 10 μM) and this effect was antagonized by SR141716A (1 μM). The non-cholinergic component of adrenaline release observed after blockade of nicotinic (by hexamethonium 100 μM) and muscarinic (by atropine 0.5 μM) acetylcholine receptors was not depressed by WIN55212-2. WIN55212-3 (10 μM) had no effect on adrenaline release.
No detectable specific CB1 receptor binding and mRNA expression were found in rabbit adrenal glands with autoradiography and in situ hybridization.
The results show that cannabinoids inhibit adrenaline secretion in rabbit isolated adrenal glands; the likely mechanism is a presynaptic CB1 receptor-mediated inhibition of acetylcholine release from preganglionic sympathetic neurons. The inhibition of adrenaline secretion in adrenal glands most probably accounts for the decrease in the plasma adrenaline concentration observed after cannabinoid administration in pithed rabbits.
Keywords: Adrenaline release, autoradiography, cannabinoid receptor, fast cyclic voltammetry, in situ hybridization, rabbit isolated adrenal gland, plasma adrenaline, pithed rabbit, sympathetic regulation
Introduction
Systemic administration of natural and synthetic cannabinoid agonists elicits prominent changes in cardiovascular homeostasis both in humans and in experimental animals. However, little is known on the mechanisms involved in these effects (for review, see Dewey, 1986; Compton et al., 1996; Wagner et al., 1998). Especially, the interaction of cannabinoids with the peripheral components of cardiovascular regulation has seldom been investigated.
Recent studies have shown that activation of CB1 cannabinoid receptors inhibits noradrenaline release from cardiac and vascular postganglionic sympathetic neurons (Ishac et al., 1996; Malinowska et al., 1997; Molderings et al., 1999; Niederhoffer & Szabo, 1999; Szabo et al., 2001), resulting in cardiovascular depression (Malinowska et al., 1997; Niederhoffer & Szabo, 1999).
In contrast, the effects of cannabinoids on catecholamine secretion from the adrenal medulla are not known. In humans, smoking or oral administration of Δ9-tetrahydrocannabinol induces no change or an increase in urinary adrenaline excretion (Hollister et al., 1970; Weiss et al., 1972; Messiha & Soskin, 1973). Effects of cannabinoids on the adrenaline concentration in plasma were, to our knowledge, investigated only in rats. In this species, systemically administered Δ9-tetrahydrocannabinol produced either no change or a decrease in the plasma adrenaline concentration (Kumar et al., 1984; Patel et al., 1985a, 1985b). In all the studies cited above, the primary site of action of Δ9-tetrahydrocannabinol – central nervous system or adrenal medulla – could not be identified since the drug was administered systemically. Altered adrenal medullary function could be the consequence of Δ9-tetrahydrocannabinol-induced changes in plasma levels of pituitary hormones (Rodríguez de Fonseca et al., 1991; 1992). Direct effects of cannabinoids in the adrenal gland are, however, also possible.
The objective of the present study was to analyse the peripheral effects of cannabinoids on adrenaline release. Three kinds of experiments were carried out. First, effects of cannabinoids on the plasma adrenaline concentration were evaluated in a whole animal preparation, the pithed rabbit with electrically stimulated sympathetic outflow. Second, effects on adrenaline release were studied in rabbit isolated adrenals; in these experiments, the release of adrenaline was evoked by electrical stimulation and measured by fast cyclic voltammetry. In both experimental models, the effects of the mixed CB1/CB2 cannabinoid receptor agonist WIN55212-2 were compared to those of WIN55212-3, an enantiomer of WIN55212-2 possessing very low affinity for cannabinoid binding sites (Kuster et al., 1993; Pertwee, 1999). The interaction between WIN55212-2 and the CB1 cannabinoid receptor antagonist SR141716A (Rinaldi-Carmona et al., 1994; Pertwee, 1999) was also studied. Finally, CB1 cannabinoid receptor protein and mRNA were sought for in rabbit adrenal glands using autoradiography and in situ hybridization, respectively.
Methods
Experiments were carried out on 53 rabbits of a local breed (derived from ‘Deutscher Riesenscheck') obtained from Ketterer, Reute, Germany.
Pithed rabbits with electrically stimulated sympathetic outflow
The methods were described in Niederhoffer & Szabo (1999). Briefly, rabbits were deeply anaesthetized with pentobarbitone (75 mg kg−1) and artificially ventilated. Both carotid arteries were cannulated (for recording arterial pressure and heart rate, and for blood sampling). The right jugular vein was also cannulated and served for administration of drugs. After relaxation of skeletal muscles with gallamine triethiodide (5 mg kg−1), animals were pithed using a 3-mm-thick and 30-cm-long stainless steel rod. The entire sympathetic outflow was continuously stimulated through the pithing rod (2 Hz, 100 – 140 mA, 0.5 ms square-wave pulses). Parameters were first determined 60 – 75 min (t=0 min in subsequent text) after pithing.
Either solvent (0.5 ml kg−1) or increasing doses of WIN55212-2 (5, 50 and 500 μg kg−1), WIN55212-3 (5, 50 and 500 μg kg−1) or CP55940 (5, 50 and 500 μg kg−1) were injected at t=19, 37 and 55 min (for protocol, see Figure 1). One group of animals was pretreated at t=−10 min with the CB1 cannabinoid receptor antagonist SR141716A (500 μg kg−1).
Figure 1.
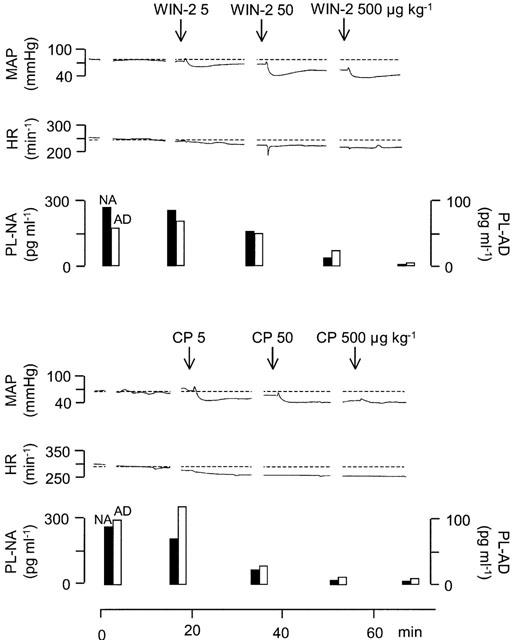
Effects of WIN55212-2 (WIN-2) and CP55940 (CP) on mean arterial pressure (MAP), heart rate (HR), and plasma noradrenaline (PL-NA) and adrenaline (PL-AD) concentrations in two pithed rabbits with electrically stimulated sympathetic outflow. WIN-2 (5, 50 and 500 μg kg−1) and CP (5, 50 and 500 μg kg−1) were injected i.v. as indicated by the arrows. Original tracings representing four (WIN-2) and three (CP) experiments with similar results (mean effects on mean arterial pressure, heart rate and the plasma noradrenaline concentration were shown in detail in Niederhoffer & Szabo, 1999).
Blood pressure and heart rate were read every 2 min from t=0 min onwards. Two-ml blood samples were taken at t=0, 14, 32, 50 and 68 min. The concentrations of noradrenaline and adrenaline in plasma were determined from these 2-ml-blood samples by high pressure liquid chromatography followed by electrochemical detection (Szabo & Schultheiss, 1990). In each experiment, values measured at t=0 and 14 min were averaged (PRE) and all values were expressed as percentages of PRE.
Values for mean arterial pressure, heart rate and the plasma noradrenaline concentration were already reported (see Niederhoffer & Szabo, 1999) and only two representative original tracings are shown in the present publication (see Figure 1). For clarity and because it was beyond the topic of the publication, values for the adrenaline plasma concentration were not displayed in our previous publication (Niederhoffer & Szabo, 1999) and are now shown.
Rabbit isolated adrenal glands
Rabbits were killed by decapitation. Both adrenal glands were rapidly dissected free and submerged in ice-cold buffer containing (mM): NaC 118, NaHCO3 25, KH2PO4 1.2, KCl 4.8, MgSO4 1.2, CaCl2 2, glucose 11 and Na2EDTA 0.03 and gassed with 95% O2/5% CO2. Glands were cut in half and each piece was placed in Perspex superfusion chambers and superfused with buffer (2 ml min−1; 34°C).
Changes in the extracellular concentration of adrenaline were evaluated by fast cyclic voltammetry, based on the method described for brain slices (Limberger et al., 1991; Stamford, 1995). A carbon fibre working electrode (tip diameter 8 μm) was inserted into the adrenal medulla approximately 50 μm below the surface of the slice. A voltammetric analyser (Millar Voltammetric Analyzer, PD Systems, West Molesey, Surrey, U.K.) delivered a W-form voltage to the electrode with a frequency of 4 Hz, recorded the resulting current and measured the adrenaline oxidation current at 0.6 V. Extracellular adrenaline concentration was calculated from the height of the peak of the oxidation current using a calibration curve obtained with standard adrenaline solutions.
Adrenaline secretion was elicited by electrical stimulation using a concentric bipolar stainless steel electrode inserted into the adrenal medulla within 200 μm of the working electrode. Electrical pulses (2 Hz, 20 V, 1 – 2 ms square-wave pulses) were generated by an isolated stimulator (Digitimer DS2, Welwyn Garden City, Herts, U.K.). A stimulation cycle included three pulse trains (consisting of 2, 5 and 10 pulses) delivered at 5 min intervals. The stimulation cycles were started 60 min after insertion of the stimulating electrode and repeated continuously until the end of the experiment. Stable electrically evoked adrenaline release could be monitored for more than 6 h (not shown).
In each experiment, the electrically evoked adrenaline release was first evaluated before drug application, i.e. during the first cycle of electrical stimulation (PRE-period); these values were taken as reference values (PRE). Drugs were then added to the superfusion medium. Effects of drugs on the electrically evoked adrenaline release were evaluated 30 min after the beginning of drug superfusion, and expressed as percentages of PRE. In a first series of experiments, effects of hexamethonium (100 μM), atropine (0.5 μM) and calcium removal on the electrically evoked adrenaline release were studied. In a second series of experiments, effects of either solvent, increasing concentrations of WIN55212-2 (0.3, 1, 3 and 10 μM), SR141716A (1 μM) or WIN55212-3 (10 μM) were determined. Effects of WIN55212-2 (same concentrations as above) in the presence of SR141716A (1 μM) or hexamethonium (100 μM) plus atropine (0.5 μM) were also tested.
Localization of CB1 cannabinoid receptors in rabbit adrenal glands
Rabbits were killed by decapitation. Brains and adrenal glands were removed and rapidly frozen by immersion in 2-methylbutane in dry ice. Samples were stored at −70°C until processed. Coronal 20-μm-thick sections of the cerebellum and sagittal 20-μm-thick sections of the adrenal gland were cut in a cryostat and used for analysis of CB1 receptor protein and mRNA expression.
CB1 receptor binding level was assessed according to the method described previously (Romero et al., 1998). In brief, sections were incubated at 37°C for 2.5 h in a buffer containing 10 nM [3H]-CP55940 (Dupont NEN, Boston, MA, U.S.A.), with or without 10 μM non-labelled CP55940 (to determine non-specific and total binding levels, respectively), then washed at 0°C for 2 h and dried. Slices were exposed on tritium-sensitive autoradiographic films with an autoradiographic tritium microscale standard (Amersham, Madrid, Spain) at 4°C for 10 days. The films were processed manually using the Kodak GBX kit (Eastman Kodak, Rochester, NY, U.S.A.). Developed films were analysed and quantitated using a computer-assisted densitometer (ImageQuant, Molecular Dynamics, Sunnyvale, U.S.A.) with calculation of CB1 receptor density using the standard curve generated from tritium microscale standards. The densitometric data were corrected for non-specific signals.
CB1 receptor mRNA levels were measured by in situ hybridization as previously described (Romero et al., 1998). Briefly, sections were fixed in formaldehyde, acetylated, dehydrated, delipidated and dried. The CB1 receptor transcript was hybridized with an equimolar mixture of three 48-mer oligonucleotide sequences complementary to bases no. 4 – 51, 349 – 396 and 952 – 999 of the rat CB1 receptor cDNA (DuPont NEN, Madrid, Spain). The oligonucleotide sequences were labelled at the 3′-end with [35S]-dATP (Amersham, Madrid, Spain) using terminal deoxynucleotidyl transferase (Boehringer Mannheim, Barcelona, Spain). An activity of 107 c.p.m. ml−1 was added to the hybridization buffer. Slices were incubated with the hybridization buffer in a humid chamber at 42°C for 20 h. Sections were then washed, dried and exposed on X-ray films (Hyperfilm β-Max, Amersham, Madrid, Spain) for 6 days. Following development, the intensity of the hybridization signal was assessed by measuring optical grain density using a computer-assisted densitometer (ImageQuant, Molecular Dynamics, Sunnyvale, CA, U.S.A.). The specificity of the signal was assessed by using an excess of cold probes.
Statistics
Means±s.e.mean of n experiments are given throughout. Statistical differences within groups (i.e., versus PRE values) were evaluated using the non-parametric two-tailed Wilcoxon signed rank test. Statistical differences between groups (i.e., versus solvent) were evaluated with the non-parametric two-tailed Mann-Whitney test. In all experiments, P<0.05 was taken as the limit of statistical significance and only this level is indicated even if P was <0.01 or <0.001.
Drugs
Drugs were obtained from the following sources: (−)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol (CP55940) from Pfizer (Groton, CT, U.S.A.); N-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-3-pyrazole-carboxamide (SR141716A) from Sanofi Recherche (Montpellier, France); R(+)-[2,3-dihydro-5-methyl-3-[(morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazinyl](1-naphthalenyl) methanone mesylate (WIN55212-2) and S(−)-[2,3-dihydro-5-methyl-3-[(morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazinyl]-(1-naphthalenyl) methanone mesylate (WIN55212-3) from RBI (Köln, Germany); 2-hydroxypropyl-β-cyclodextrin (β-CDX) from Fluka (Neu-Ulm, Germany); fatty acid free bovine serum albumin, (−)-adrenaline bitartrate, hexamethonium bromide and atropine sulphate from Sigma (Deisenhofen, Germany).
In experiments in pithed rabbits, WIN55212-2, WIN55212-3 and CP55940 were dissolved in 19% w v−1 solutions of β-CDX; further dilutions were made with the same solvent. SR141716A was dissolved in 66% v v−1 mixture of DMSO and water. All drugs were injected intravenously in a volume of 0.5 ml kg−1. Doses refer to the salts. In experiments in isolated adrenal glands, WIN55212-2, WIN55212-3 and SR141716A were dissolved in DMSO; further dilutions were made in buffer containing fatty acid free bovine serum albumin (1 g l−1). The final concentration of DMSO in buffer was 1% v v−1. Atropine and hexamethonium were dissolved in distilled water and further diluted with buffer.
Results
Pithed rabbits with electrically stimulated sympathetic outflow
After an initial stabilization period, baseline parameters were determined twice (at t=0 and 14 min), and the values were averaged to yield the PRE values. Mean PRE values for mean arterial pressure, heart rate and the plasma noradrenaline concentration were shown in detail in our previous publication (Niederhoffer & Szabo, 1999). The mean PRE value for the plasma adrenaline concentration in animals without pretreatment was 175±26 pg ml−1 (n=17); pretreatment with the cannabinoid receptor antagonist SR141716A (500 μg kg−1) had no significant effect on the PRE value for the plasma adrenaline concentration (116±21 pg ml−1; n=4).
Intravenous administration of cannabinoid receptor agonists markedly decreased mean arterial pressure and the plasma noradrenaline concentration whereas heart rate was not significantly changed; data were presented in detail in Niederhoffer & Szabo (1999) and only two representative original tracings are shown here (Figure 1).
Effects of cannabinoids on the plasma adrenaline concentration are shown in Figure 1 (original tracings) and Figure 2 (statistical evaluation). Intravenous administration of the solvent did not change the plasma adrenaline concentration. Intravenous injection of increasing doses of the cannabinoid agonists WIN55212-2 (5, 50 and 500 μg kg−1) and CP55940 (5, 50 and 500 μg kg−1) dose-dependently decreased the plasma adrenaline concentration. The inhibitory effect of WIN55212-2 was significantly attenuated by pretreatment with SR141716A (500 μg kg−1) and was not shared by WIN55212-3 (5, 50 and 500 μg kg−1, same doses as of WIN55212-2), the enantiomer of WIN55212-2 lacking affinity for cannabinoid receptors.
Figure 2.
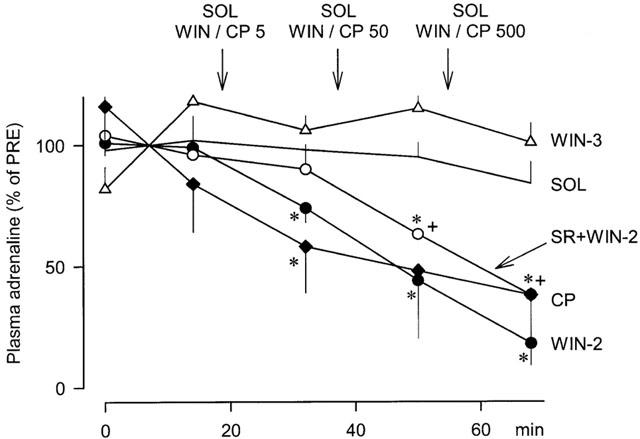
Effects of solvent (SOL), WIN55212-2 (WIN-2), WIN55212-3 (WIN-3) and CP55940 (CP) on the plasma adrenaline concentration in pithed rabbits with electrically stimulated sympathetic outflow. SOL (0.5 ml kg−1), WIN-2 (5, 50 and 500 μg kg−1), WIN-3 (5, 50 and 500 μg kg−1) and CP (5, 50 and 500 μg kg−1) were injected i.v. as indicated by the arrows. One of the two WIN-2-groups was pretreated at t=−10 min with SR141716A (SR; 500 μg kg−1). Values are given as percentages of PRE. Means±s.e.mean from six (SOL), four (WIN-2), four (WIN-3), four (SR+WIN-2) and three (CP) experiments. Differences versus SOL: *P<0.05; differences versus WIN-2:+P<0.05.
Rabbit isolated adrenal glands
Adrenaline release was stimulated electrically. A stimulation cycle included three pulse trains (consisting of 2, 5 and 10 pulses at 2 Hz) delivered at 5 min intervals. Stimulation cycles were repeated continuously until the end of the experiment. During the first stimulation cycle, the extracellular concentration of endogenous adrenaline increased by 0.5±0.1 μM, 1.4±0.4 μM and 2.8±0.7 μM after 2, 5 and 10 pulses (n=49), respectively (PRE).
Addition of the nicotinic acetylcholine receptor antagonist hexamethonium (100 μM) or the muscarinic acetylcholine receptor antagonist atropine (0.5 μM) to the superfusion medium significantly lowered the electrically evoked adrenaline release (Figure 3). When the two antagonists were combined in normal buffer, a slightly greater inhibition of the electrically evoked adrenaline release was observed (Figure 3). When they were combined in calcium-free buffer, the adrenaline release was almost completely abolished (Figure 3).
Figure 3.
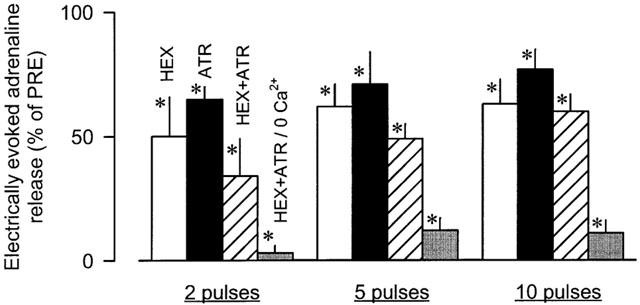
Effects of hexamethonium (HEX), atropine (ATR) and the combination of both (HEX+ATR) in normal and calcium-free buffer (0 Ca2+) on electrically evoked adrenaline release in rabbit isolated adrenal glands. Adrenaline release was elicited by trains of 2, 5 and 10 electrical pulses at 2 Hz. After an initial PRE-period, HEX (100 μM), ATR (0.5 μM) or HEX+ATR (same concentrations) were added to the superfusion medium. In one further group, HEX+ATR were superfused in calcium-free buffer. Effects of the drugs on electrically evoked adrenaline release were evaluated 30 min after the beginning of drug superfusion. Values are given as percentages of PRE. Means±s.e.mean from six (HEX), six (ATR), six (HEX+ATR) and six (HEX+ATR/0 Ca2+) experiments. Differences versus PRE: *P<0.05.
The electrically evoked adrenaline release was not modified by the solvent (92±12, 83±7 and 88±7% of PRE after 2, 5 and 10 pulses, respectively). Effects of WIN55212-2 are shown in Figure 4 (original tracings) and Figure 5 (statistical evaluation). Superfusion of increasing concentrations of WIN55212-2 (0.3, 1, 3 and 10 μM) significantly attenuated the electrically evoked adrenaline release. The maximal effect was observed during superfusion of WIN55212-2 10 μM (40 – 60% inhibition). In contrast, superfusion of the inactive enantiomer WIN55212-3 (10 μM) did not change the electrically evoked adrenaline release (89±6, 95±7 and 83±11% of PRE after 2, 5 and 10 pulses, respectively). The inhibitory effect of WIN5212-2 on the electrically evoked adrenaline release was antagonized by the CB1 receptor antagonist SR141716A (1 μM). The antagonist alone had no effect (84±11, 74±9 and 93±12% of PRE after 2, 5 and 10 pulses, respectively). The non-cholinergic component of the electrically evoked adrenaline release, measured in the presence of hexamethonium (100 μM) plus atropine (0.5 μM), was not depressed by WIN55212-2.
Figure 4.
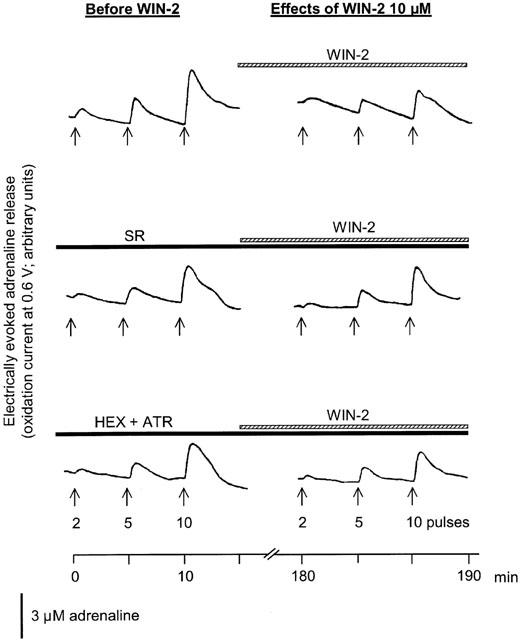
Effects of WIN55212-2 (WIN-2) on electrically evoked adrenaline release in rabbit isolated adrenal glands. Adrenaline release was elicited by trains of 2, 5 and 10 electrical pulses at 2 Hz as indicated by the arrows. After an initial PRE-period, increasing concentrations of WIN-2 (0.3, 1, 3 and 10 μM) were added to the superfusion medium. In two further experiments, WIN-2 was superfused in the presence of SR141716A (SR; 1 μM) or hexamethonium (HEX; 100 μM) plus atropine (ATR; 0.5 μM). Left panels show the electrically evoked adrenaline release before application of WIN-2. Right panels show the electrically evoked adrenaline release after 30 min of superfusion of WIN-2 10 μM. The vertical bar denotes the height of the oxidation current produced by superfusion of a 3 μM-adrenaline solution. Original tracings representing seven (WIN-2), six (SR+WIN-2) and five (HEX+ATR+WIN-2) experiments with similar results.
Figure 5.
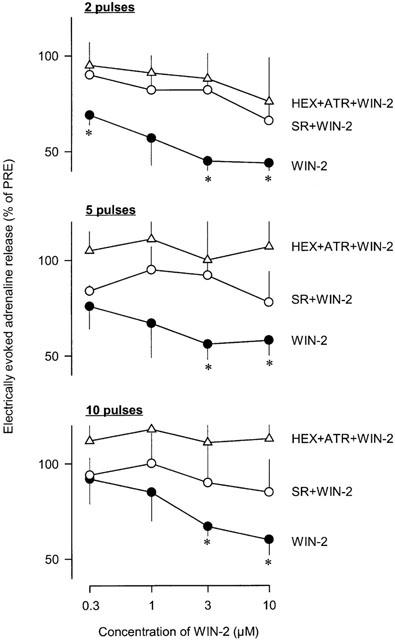
Effects of WIN55212-2 (WIN-2) on electrically evoked adrenaline release in rabbit isolated adrenal glands. Adrenaline release was elicited by trains of 2, 5 and 10 electrical pulses at 2 Hz. After an initial PRE-period, increasing concentrations of WIN-2 (0.3, 1, 3 and 10 μM) were added to the superfusion medium. In two further groups, WIN-2 was superfused in the presence of SR141716A (SR; 1 μM) or hexamethonium (HEX; 100 μM) plus atropine (ATR; 0.5 μM). Effects of WIN-2 on electrically evoked adrenaline release were evaluated 30 min after the beginning of superfusion of a given drug concentration. Values are given as percentages of PRE. Means±s.e.mean from seven (WIN-2), six (SR+WIN-2) and five (HEX+ATR+WIN-2) experiments. Differences versus PRE: *P<0.05.
Localization of CB1 cannabinoid receptors in rabbit adrenal glands
The presence of cannabinoid receptor protein in rabbit adrenal glands was studied with autoradiography using the mixed CB1/CB2 receptor agonist [3H]-CP55940 as radioligand. Slices obtained from rabbit cerebellum served as positive control. In the cerebellum, dense specific binding was found and the distribution pattern was comparable to that described in other species (not shown).
The total binding in the adrenal medulla and cortex was weak and non-specific in nature (Figure 6).
Figure 6.
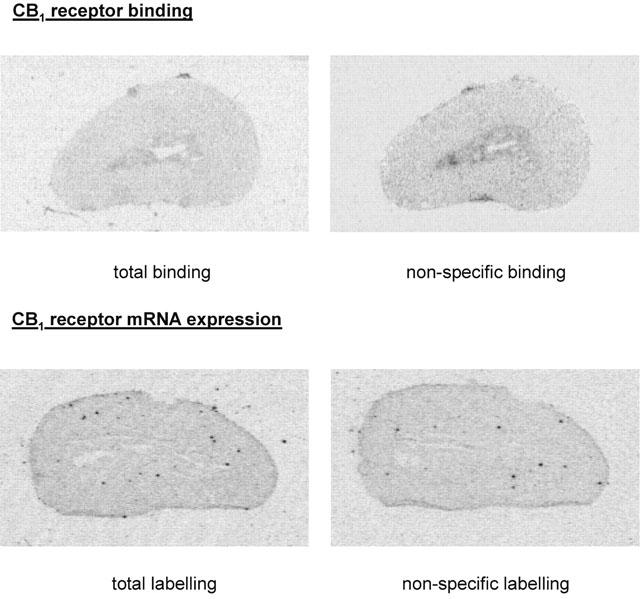
Representative autoradiograms for CB1 cannabinoid receptor binding (upper panels) and mRNA expression (lower panels) obtained from 20 μm-thick sagittal slices of rabbit adrenal glands. CB1 receptor binding was assessed using autoradiography with [3H]-CP55940; non-specific binding was determined in the presence of excess of non-labelled CP55940. CB1 receptor mRNA expression was assessed using in situ hybridization with rat CB1 antisense probes; non-specific labelling was determined in the presence of excess of cold probe. The figures represent five (CB1 receptor binding) and five (CB1 mRNA expression) experiments with similar results.
CB1 cannabinoid receptor mRNA expression in rabbit adrenal glands was assessed by in situ hybridization with rat CB1 antisense probes. We first verified that these probes worked also in the rabbit. In slices prepared from rabbit cerebellum, specific hybridization was obtained, with a distribution pattern similar to that observed in other species (not shown).
In the adrenal medulla and cortex, the total labelling was low and there was no difference when cold probes were added in excess (Figure 6).
Discussion
The results show that cannabinoids inhibit adrenaline release in a whole animal preparation and in isolated adrenal glands; the likely mechanism is a presynaptic CB1 receptor-mediated inhibition of acetylcholine release from preganglionic sympathetic neurons of the adrenal medulla.
In pithed rabbits with electrically stimulated sympathetic outflow, the synthetic CB1/CB2 cannabinoid receptor agonist WIN55212-2 markedly and dose-dependently decreased the plasma adrenaline concentration. This inhibitory effect was shared by CP55940, another cannabinoid receptor agonist with a chemical structure different from that of WIN55212-2. In contrast, WIN55212-3, the enantiomer of WIN55212-2 which possesses very low affinity for cannabinoid receptors, had no effect. These observations support involvement of specific cannabinoid receptors in the decrease in plasma adrenaline concentration.
Since adrenaline in plasma originates exclusively from adrenal chromaffin cells, effects of cannabinoids on adrenaline release were studied in isolated adrenal glands. In vivo, catecholamine exocytosis from chromaffin cells is regulated by the activity of presynaptic splanchnic nerves innervating the adrenal medulla. Stimulation of the nerve terminals causes release of acetylcholine and non-cholinergic neurotransmitters (e.g., opioid peptides and vasoactive intestinal peptide), which then activate postsynaptic receptors to evoke catecholamine secretion (Malhotra & Wakade, 1987; for review, see Marley, 1987 and Aunis, 1998). In our experimental preparation, adrenaline secretion was elicited by electrical stimulation through an electrode inserted into the adrenal medulla. The electrically evoked adrenaline release was only partly abolished by blockade of the nicotinic and muscarinic acetylcholine receptors and the residual non-cholinergic component of adrenaline release was largely dependent on the presence of calcium in the superfusion medium. These observations strongly suggest that the electrical stimulation mimicked the physiological nerve-mediated stimulation of catecholamine secretion.
Changes in the extracellular concentration of endogenous adrenaline were measured using fast cyclic voltammetry. In principle, the method does not allow distinction between adrenaline, noradrenaline and ascorbate, all of which can be released by chromaffin cells and are oxidized at the same potential (0.6 V; see Cahill & Wightman, 1995). However, several observations indicate that the contribution of noradrenaline and ascorbate to the oxidation current monitored in our experiments was minimal, i.e., changes in oxidation current reflected almost exclusively adrenaline secretion. First, the vast majority (>95%) of chromaffin cells in the rabbit adrenal medulla are adrenaline-containing cells and only few noradrenaline-containing cells are found (Palkama, 1962). Second, ascorbate is secreted from chromaffin cells through a nonexocytotic process independent of extracellular calcium (Cahill & Wightman, 1995) and we obtained no release under this experimental condition (see above). Third, the oxidation current resulting from ascorbate release is abolished in the presence of ascorbate oxidase (Boutelle et al., 1989) and we observed no change in the electrically evoked oxidation current when this enzyme was added to the superfusion medium (results not shown).
The major finding of the experiments in rabbit isolated adrenal glands is that the cannabinoid agonist WIN55212-2 concentration-dependently inhibited adrenaline secretion. The inhibitory effect of WIN55212-2 was not shared by its inactive enantiomer WIN55212-3, supporting involvement of specific cannabinoid receptors. Importantly, WIN55212-2 did not decrease the non-cholinergic component of the electrically evoked adrenaline secretion measured in the presence of nicotinic and muscarinic acetylcholine receptor blockade. This latter result strongly suggests that in the adrenal medulla, cannabinoids do not act at the level of chromaffin cells to inhibit adrenaline secretion (e.g., on voltage dependent calcium channels) but rather interact presynaptically with the cholinergic component of the nerve-mediated adrenaline secretion. A likely mechanism for this effect could be that activation of presynaptic cannabinoid receptors inhibits acetylcholine release from preganglionic sympathetic nerve terminals. Such an inhibition of electrically evoked acetylcholine release has already been reported in peripheral tissues (Pertwee et al., 1996; Coutts & Pertwee, 1997) and in the brain (Gifford & Ashby, 1996; Kathmann et al., 2001).
Complete blockade of the cannabinoid evoked inhibition of adrenaline release in isolated adrenal glands strongly suggests that CB1 cannabinoid receptors are involved in this effect. In pithed rabbits, the counteracting effect of SR141716A was only partial. This is most probably due to insufficient tissue concentration of the antagonist during the late phase of the experiment (the two higher doses of WIN55212-2 were given 47 and 65 min after administration of SR141716A). Accordingly, the concomitant decreases in plasma noradrenaline concentration and blood pressure elicited by WIN55212-2 were also only partly antagonized (see Niederhoffer & Szabo, 1999). However, since the antagonism was incomplete, and since CB2-like receptors have also been implicated in the inhibition of neurotransmitter release produced by cannabinoids in the mouse vas deferens (Griffin et al., 1997), a role for CB2-like receptors in the cannabinoid-induced inhibition of adrenaline secretion cannot be definitely ruled out.
The absence of CB1 cannabinoid receptor mRNA in the rabbit adrenal gland as demonstrated by in situ hybridization shows that the receptor is not synthetized in chromaffin cells (i.e., postsynaptically). This further suggests that cannabinoids did not act postsynaptically to inhibit adrenaline release and supports the hypothesis of a presynaptic localization of CB1 receptors on preganglionic sympathetic neurons of the adrenal medulla. However, no specific CB1 receptor binding could be detected using autoradiography. A possible explanation for this discrepancy is that the autoradiographic technique is not sensitive enough to visualize low quantities of receptors in the tissue. In agreement with this assumption, no specific [3H]-CP55940 binding was detected in rat atria and vas deferens using autoradiography (Lynn & Herkenham, 1994), although in functional studies, activation of presynaptic CB1 cannabinoid receptors decreases transmitter release in these tissues (Ishac et al., 1996). However, it cannot be excluded that the lack of detectable CB1 receptors in the adrenal gland indicates that the observed functional effects were mediated by a not yet identified cannabinoid receptor subtype.
In conclusion, cannabinoids lowered the electrically evoked adrenaline release in rabbit isolated adrenal glands; the likely mechanism is a presynaptic CB1 receptor-mediated inhibition of acetylcholine release from preganglionic sympathetic neurons innervating the adrenal medulla. This decrease in adrenaline secretion most probably accounts for the marked decrease in plasma adrenaline concentration observed after cannabinoid administration in the pithed rabbit. To our knowledge, our results are the first demonstrating an inhibitory action of cannabinoids on catecholamine secretion at the level of the adrenal medulla.
Acknowledgments
The study was supported by the Deutsche Forschungsgemeinschaft (Sz 72/2-3). The support and advice of Klaus Starke are gratefully acknowledged. We thank Claudia Schurr and Jens Scheibner for their contribution to this work. We also thank Pfizer (Groton, CT, U.S.A.), Pfizer (Madrid, Spain) and Sanofi Recherche (Montpellier, France) for generous supply of CP55940 and SR141716A, respectively.
Abbreviations
- CB
cannabinoid
- PRE
average of initial values (before drug application)
References
- AUNIS D. Exocytosis in chromaffin cells of the adrenal medulla. Int. Rev. Cytol. 1998;181:213–320. doi: 10.1016/s0074-7696(08)60419-2. [DOI] [PubMed] [Google Scholar]
- BOUTELLE M.G., SVENSSON L., FILLENZ M. Rapid changes in striatal ascorbate response to tail-pinch monitored by constant potential voltammetry. Neuroscience. 1989;30:11–17. doi: 10.1016/0306-4522(89)90349-7. [DOI] [PubMed] [Google Scholar]
- CAHILL P.S., WIGHTMAN R.M. Simultaneous amperometric measurement of ascorbate and catecholamine secretion from individual bovine adrenal medullary cells. Anal. Chem. 1995;67:2599–2605. doi: 10.1021/ac00111a017. [DOI] [PubMed] [Google Scholar]
- COMPTON D.R., HARRIS L.S., LICHTMAN A.H., MARTIN B.R.Marihuana Pharmacological Aspects of Drug Dependence. Handbook of Experimental Pharmacology 1996Springer: Heidelberg; 83–158.eds. Schuster, C.R. & Kuhar, M.J. pp [Google Scholar]
- COUTTS A.A., PERTWEE R.G. Inhibition by cannabinoid receptor agonists of acetylcholine release from the guinea-pig myenteric plexus. Br. J. Pharmacol. 1997;121:1557–1566. doi: 10.1038/sj.bjp.0701301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEWEY W.L. Cannabinoid pharmacology. Pharmacol. Rev. 1986;38:151–178. [PubMed] [Google Scholar]
- GIFFORD A.N., ASHBY C.R. Electrically-evoked acetylcholine release from hippocampal slices is inhibited by the cannabinoid receptor agonist, WIN55212-2, and is potentiated by the cannabinoid receptor antagonist, SR141716A. J. Pharmacol. Exp. Ther. 1996;277:1431–1436. [PubMed] [Google Scholar]
- GRIFFIN G., FERNANDO S.R., ROSS R.A., MCKAY N.G., ASHFORD M.L.J., SHIRE D., HUFFMAN J.W., YU S., LAINTON J.A.H., PERTWEE R.G. Evidence for the presence of CB2-like cannabinoid receptors on peripheral nerve terminals. Eur. J. Pharmacol. 1997;339:53–61. doi: 10.1016/s0014-2999(97)01336-8. [DOI] [PubMed] [Google Scholar]
- HOLLISTER L.E., MOORE F., KANTER S., NOBLE E. 1-tetrahydrocannabinol, synhexyl and Marihuana extract administered orally in man: catecholamine excretion, plasma cortisol levels and platelet serotonin content. Psychopharmacologia. 1970;17:354–360. doi: 10.1007/BF00404241. [DOI] [PubMed] [Google Scholar]
- ISHAC E.J.N., JIANG L., LAKE K.D., VARGA K., ABOOD M.E., KUNOS G. Inhibition of exocytotic noradrenaline release by presynaptic cannabinoid CB1 receptors on peripheral sympathetic nerves. Br. J. Pharmacol. 1996;118:2023–2028. doi: 10.1111/j.1476-5381.1996.tb15639.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KATHMANN M., WEBER B., SCHLICKER E. Cannabinoid CB1 receptor-mediated inhibition of acetylcholine release in the brain of NMRI, CD-1 and C57BL/6J mice. Naunyn-Schmiedeberg's Arch. Pharmacol. 2001;363:50–56. doi: 10.1007/s002100000304. [DOI] [PubMed] [Google Scholar]
- KUMAR M.S.A., PATEL V., MILLARD W.J. Effect of chronic administration of Δ9-tetrahydrocannabinol on the endogenous opioid peptide and catecholamine levels in the diencephalon and plasma of the rat. Subst. Alcohol Actions Misuse. 1984;5:201–210. [PubMed] [Google Scholar]
- KUSTER J.E., STEVENSON J.I., WARD S.J., D'AMBRA T.E., HAYCOCK D.A. Aminoalkylindole binding in rat cerebellum: selective displacement by natural and synthetic cannabinoids. J. Pharmacol. Exp. Ther. 1993;264:1352–1363. [PubMed] [Google Scholar]
- LIMBERGER N., TROUT S.J., KRUK Z.L., STARKE K. ‘Real time' measurements of endogenous dopamine release during short trains of pulses in slices of rat neostriatum and nucleus accumbens: role of autoinhibition. Naunyn-Schmiedeberg's Arch. Pharmacol. 1991;344:623–629. doi: 10.1007/BF00174745. [DOI] [PubMed] [Google Scholar]
- LYNN A.B., HERKENHAM M. Localization of cannabinoid receptors and nonsaturable high-density cannabinoid binding sites in peripheral tissues of the rat: implications for receptor-mediated immune modulation by cannabinoids. J. Pharmacol. Exp. Ther. 1994;268:1612–1623. [PubMed] [Google Scholar]
- MALHOTRA R.K., WAKADE A.R. Non-cholinergic component of rat splanchnic nerves predominates at low neuronal activity and is eliminated by naloxone. J. Physiol. 1987;383:639–652. doi: 10.1113/jphysiol.1987.sp016434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MALINOWSKA B., GODLEWSKI G., BUCHER B., SCHLICKER E. Cannabinoid CB1 receptor-mediated inhibition of the neurogenic vasopressor response in the pithed rat. Naunyn-Schmiedeberg's Arch. Pharmacol. 1997;356:197–202. doi: 10.1007/pl00005041. [DOI] [PubMed] [Google Scholar]
- MARLEY P.D. New insights into the non-nicotinic regulation of adrenal medullary function. T.I.P.S. 1987;8:411–413. [Google Scholar]
- MESSIHA F.S., SOSKIN R.A. Effect of Cannabis sativa administered by smoking on biogenic amine excretion in man. Res. Comm. Chem. Pathol. Pharmacol. 1973;6:325–328. [PubMed] [Google Scholar]
- MOLDERINGS G.J., LIKUNGU J., GÖTHERT M. Presynaptic cannabinoid and imidazoline receptors in the human heart and their potential relationship. Naunyn-Schmiedeberg's Arch. Pharmacol. 1999;360:157–164. doi: 10.1007/s002109900043. [DOI] [PubMed] [Google Scholar]
- NIEDERHOFFER N., SZABO B. Effect of the cannabinoid receptor agonist WIN55212-2 on sympathetic cardiovascular regulation. Br. J. Pharmacol. 1999;126:457–466. doi: 10.1038/sj.bjp.0702337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PALKAMA A. Distribution of adrenaline, noradrenaline, acid phosphatase, cholinesterases and non-specific esterases in the adrenal medulla of some mammals. A comparative histochemical study. Ann. Med. Exp. Biol. Fenn. 1962;40 Suppl. 3:1–25. [PubMed] [Google Scholar]
- PATEL V., BORYSENKO M., KUMAR M.S.A., MILLARD W.J. Effects of acute and subchronic Δ9-tetrahydrocannabinol administration on the plasma catecholamine, β-endorphin, and corticosterone levels and splenic natural killer cell activity in rats. Proc. Soc. Exp. Biol. Med. 1985a;180:400–404. doi: 10.3181/00379727-180-42195. [DOI] [PubMed] [Google Scholar]
- PATEL V., BORYSENKO M., KUMAR M.S. Effect of delta 9-THC on brain and plasma catecholamine levels as measured by HPLC. Brain Res. Bull. 1985b;14:85–90. doi: 10.1016/0361-9230(85)90179-0. [DOI] [PubMed] [Google Scholar]
- PERTWEE R.G. Pharmacology of cannabinoid receptor ligands. Curr. Med. Chem. 1999;6:635–664. [PubMed] [Google Scholar]
- PERTWEE R.G., FERNANDO S.R., NASH J.E., COUTTS A.A. Further evidence for the presence of cannabinoid CB1 receptors in guinea-pig small intestine. Br. J. Pharmacol. 1996;118:2199–2205. doi: 10.1111/j.1476-5381.1996.tb15663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RINALDI-CARMONA M., BARTH F., HEAULME M., SHIRE D., CALANDRA B., CONGY C., MARTINEZ S., MARUANI J., NELIAT G., CAPUT D., FERRARA P., SOUBRIE P., BRELIERE J.C., LE FUR G. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Letters. 1994;350:240–244. doi: 10.1016/0014-5793(94)00773-x. [DOI] [PubMed] [Google Scholar]
- RODRÍGUEZ DE FONSECA F., FERNÁNDEZ-RUIZ J., MURPHY L., ELDRIDGE J.C., STEGER R.W., BARTKE A. Effects of Δ9-tetrahydrocannabinol exposure on adrenal medullary function: evidence of an acute effect and development of tolerance in chronic treatments. Pharmacol. Biochem. Behav. 1991;40:593–598. doi: 10.1016/0091-3057(91)90368-c. [DOI] [PubMed] [Google Scholar]
- RODRÍGUEZ DE FONSECA F., MURPHY L.L., BONNIN A., ELDRIDGE J.C., BARTKE A., FERNÁNDEZ-RUIZ J.J. Δ9-tetrahydrocannabinol administration affects anterior pituitary, corticoadrenal and adrenomedullary functions in rats. Life Sci. Adv. 1992;11:147–156. [Google Scholar]
- ROMERO J., BERRENDERO F., GARCÍA-GIL L., DE LA CRUZ P., RAMOS J.A., FERNÁNDEZ-RUIZ J.J. Loss of cannabinoid receptor binding and messenger RNA levels and cannabinoid agonist-stimulated [35S]guanylyl-5′-O-(thio)-triphosphate binding in the basal ganglia of aged rats. Neuroscience. 1998;84:1075–1083. doi: 10.1016/s0306-4522(97)00552-6. [DOI] [PubMed] [Google Scholar]
- STAMFORD J.A.Voltammetry in brain slices Brain Slices in Basic and Clinical Research 1995CRC Press: Florida; 65–88.eds. Schurr, A. & Rigor, B. pp [Google Scholar]
- SZABO B., SCHULTHEISS A. Desipramine inhibits sympathetic nerve activity in the rabbit. Naunyn-Schmiedeberg's Arch. Pharmacol. 1990;342:469–476. doi: 10.1007/BF00169466. [DOI] [PubMed] [Google Scholar]
- SZABO B., NORDHEIM U., NIEDERHOFFER N. Effects of cannabinoids on sympathetic and parasympathetic neuro-effector transmission in the rabbit heart. J. Pharmacol. Exp. Ther. 2001;297:819–826. [PubMed] [Google Scholar]
- WAGNER J.A., VARGA K., KUNOS G. Cardiovascular actions of cannabinoids and their generation during shock. J. Mol. Med. 1998;76:824–836. doi: 10.1007/s001090050287. [DOI] [PubMed] [Google Scholar]
- WEISS J.L., WATANABE A.M., LEMBERGER L., TAMARKIN N.R., CARDON P.V. Cardiovascular effects of delta-9-tetrahydrocannabinol in man. Clin. Pharmacol. Ther. 1972;13:671–684. doi: 10.1002/cpt1972135part1671. [DOI] [PubMed] [Google Scholar]