

Abstract
By performing microdialysis, this study investigated the pharmacokinetics of unbound camptothecin in rat blood, brain and bile in the presence of P-glycoprotein mediated transport modulators (cyclosporin A, berberine, quercetin, naringin and naringenin). Pharmacokinetic parameters of camptothecin were assessed using a non-compartmental model.
Camptothecin rapidly crosses the blood-brain barrier (BBB) within 20 min after camptothecin administration. The disposition of camptothecin in rat bile appeared to have a slow elimination phase and a peak concentration after 20 min of camptothecin administration. The area under the concentration versus time curve (AUC) for camptothecin in bile significantly surpassed that in blood, suggesting active transport of hepatobiliary excretion.
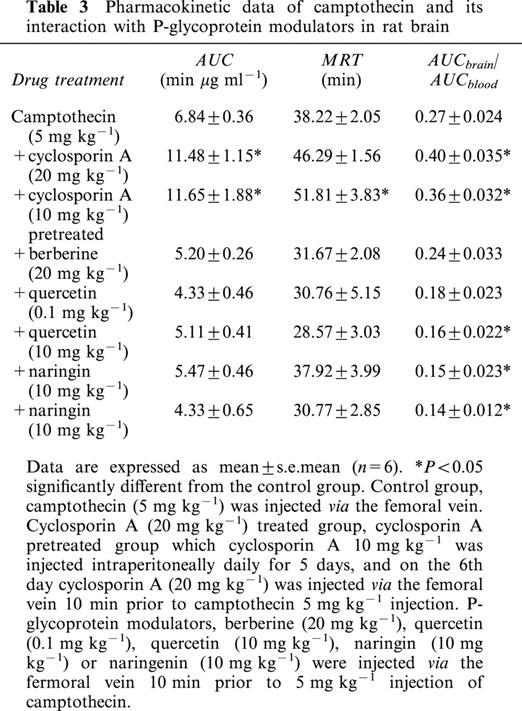
In the presence of cyclosporin A camptothecin AUC, in the brain, was significantly elevated but no significant change in the presence of berberine, quercetin, naringin and naringenin.
With treatment by smaller doses of quercetin (0.1 mg kg−1), naringin (10 mg kg−1) and naringenin (10 mg kg−1), they significantly diminished the camptothecin AUC in bile, but was not altered by the treatment of berberine (20 mg kg−1), a higher dose of quercetin (10 mg kg−1), and cyclosporin A treated (20 mg kg−1) and pretreated groups.
The distribution ratio (AUCbile/AUCblood) of camptothecin in bile was decreased in the cyclosporin A, quercetin, naringin and naringenin treated groups. However, the distribution ratio in the brain was increased in the cyclosporin A groups, but was decreased in the groups treated with quercetin, naringin and naringenin. These results revealed that P-glycoprotein might modulate hepatobiliary excretion and BBB penetration of camptothecin.
Keywords: Camptothecin, cyclosporin A, hepatobiliary excretion, microdialysis, P-glycoprotein, pharmacokinetics
Introduction
Camptothecin, a plant alkaloid, was first isolated from Camptotheca acuminata by Wall et al. (1966); Shamma & St. Georgiev (1974); Wall & Wani (1996) and Wall (1998). However, the major dose-limited toxicity led to the abandonment of further clinical testing during 1970 – 1980 (Moertel et al., 1972; Schultz, 1973). Subsequently the cytotoxic effect of camptothecin has been demonstrated to target the topoisomerase I (Hsiang et al., 1985; Rivory & Robert, 1995), which is important for cancer chemotherapy (D'arpa & Liu, 1989; Darzynkiewicz et al., 1996; Liu et al., 1996). Several anticancer drugs have been developed based on camptothecin, including irinotecan (CPT-11) and its metabolite 7-ethyl-10-hydroxycamptothecin (SN-38), as well as topotecan, 9-aminocamptothecin (NSC 603071) and 9-nitrocamptothecin (O'leary & Muggia, 1998; Muggia et al., 1996). In 1989 camptothecin's extraordinary anticancer activity against human cancer xenografts in nude mice and some of its semisynthetic derivatives were revealed (Giovanella et al., 1989).
Despite numerous studies describing the pharmacokinetics of camptothecin and its active derivatives, no researchers have described the simultaneous determination of the pharmacokinetics of camptothecin in rat blood, brain and bile employing microdialysis. However, a camptothecin derivative has been demonstrated to penetrate into the cerebrospinal fluid (Blaney et al., 1998). In addition, the average cerebrospinal fluid (CSF) to plasma distribution ratio of a camptothecin semisynthetic derivative, topotecan, has been found to be 0.3 in nonhuman primates (Blaney et al., 1993). In this respect, some factors may regulate the camptothecin penetration of BBB. Studying the effects of P-glycoprotein modulators on camptothecin distribution into the brain is therefore of particular interest.
P-glycoprotein is a product of the MDR1 gene (multidrug resistance), and it is well established that overexpression at the surface of cancer cells of the P-glycoprotein produces the classical type of multidrug resistance (Kartner et al., 1983; Roninson, 1992). This multidrug resistance also corresponds to diminished intracellular accumulation of the drugs, resulting in increased energy dependent drug efflux in non-cancerous tissues (Fardel et al., 1996). P-glycoprotein contributes to the barrier functions of the brain by extruding compounds that are potentially toxic to the brain from the capillary endothelia cells. In addition, P-glycoprotein offers a defence mechanism in the BBB function (Fromm, 2000; Tatsuta et al., 1992; Pardridge, 1998). Moreover, P-glycoprotein is expressed in the apical membrane of cells in excretory organs such as the liver, kidney and small intestine (Cordon-Cardo et al., 1990). The biliary excretion of xenobiotics is a complex process involving uptake into hepatocytes, internal sequestration and biotransformation, and transport into bile. This function has been demonstrated for excretion of xenobiotics via the canalicular membrane of hepatocytes into the bile (Hebert, 1997; Riley et al., 2000). In addition, biliary elimination has been recognized not only to depend on bile salt secretion, but also to require the active participation of a protein, the P-glycoprotein (Frijters et al., 1997).
Several grapefruit juice bioflavonoids, such as naringin, naringenin, quercetin, have been reported to regulate P-glycoprotein mediated drug efflux (Mitsunaga et al., 2000). In addition, since flavonoids are widespread in food and beverages, understanding their effect on the function of P-glycoprotein is important because of implications for cancer chemotherapy. Thus, it is of great interest to study the effect that the P-glycoprotein modulator has on drug distribution and elimination, especially at the level of the brain and bile, where the function of P-glycoprotein may play a role as gatekeeper in BBB and accelerated elimination in biliary excretion.
Methods
Chemicals and reagents
20(S)-Camptothecin was purchased from Aldrich Chem. Co. (Milwaukee, WI, U.S.A.). Berberine, quercetin, naringin, naringenin and chemical reagents were obtained from Sigma (St. Louis, MO, U.S.A.). Cyclosporin A (Sandimmun) was purchased from Novatis Pharma (Basle, Switzerland). Chromatographic solvents were purchased from BDH (Poole, U.K.). Triple de-ionized water from Millipore (Bedford, MA, U.S.A.) was utilized for all preparations.
Liquid chromatography
The liquid chromatographic system comprised of a chromatographic pump (Waters 510, Bedford, MA, U.S.A.), an injector (Rheodyne 7125, Cotati, CA, U.S.A.) equipped with a 20 μl sample loop, and a fluorescence detector (Jasco FP-920, Tokyo, Japan). Dialysates were separated with a reversed phase C18 column (Merck, 250×4.6 mm I.D.; particle size 5 μm, Darmstadt, Germany) preserved at an ambient temperature to perform the ideal chromatographic phase. The mobile phase included of methanol-acetonitrile-1 mM octanesulphonic acid (20 : 30 : 50, v v−1, pH 4.8 adjusted with orthophosphoric acid), with a flow rate 1.0 ml min−1. The mobile phase mixture was filtered through a 0.45 μm Millipore membrane, then degassed before use. The optimal fluorescence response for camptothecin was observed at excitation and emission wavelengths of 360 and 440 nm, respectively. Output data from the detector was integrated utilizing an EZChrom chromatographic data system (Scientific Software, San Ramon, CA, U.S.A.). The chromatographic method was modified from that previously reported (Tsai et al., 1999d; 2000a).
Method validation
All calibration curves of camptothecin (external standards) were established before the experiments, and displayed correlation values of at least 0.995. The intra-day and inter-day variabilities for camptothecin were assayed (six replicates) at concentrations of 5, 10, 20, 50, 100, 200 and 500 ng ml−1 on the same day and on six sequential days, respectively. The accuracy (per cent bias) was calculated from the nominal concentration (Cnom) and the mean value of the observed concentration (Cobs) as follows: Bias (%)=[(Cobs−Cnom)/(Cnom)]×100. The relative standard deviation (RSD) was calculated from the observed concentrations as follows: precision (% RSD)=[standard deviation (s.d.)/Cobs]×100. Accuracy and precision values within±15% covering the actual range of experimental concentrations were considered acceptable.
Animals
The institutional animal experimentation committee of the National Research Institute of Chinese Medicine reviewed and approved all experimental protocols involving animals. Male specific pathogen-free Sprague-Dawley rats were obtained from the Laboratory Animal Center of the National Yang-Ming University, Taipei. The animals had free access to food (Laboratory rodent diet no. 5P14, PMI Feeds Inc., Richmond, IN, U.S.A.) and water until 18 h prior to being supplied for experiments, at which time only food was removed. The rats were initially anaesthetized with urethane 0.8 g ml−1 and α-chloralose 0.08 g ml−1 (1 ml kg−1, i.p.), and remained anaesthetized throughout the experimental period. The femoral vein was exposed for further drug administration. The rats' body temperature was maintained at 37°C with a heating pad during the experiment. The sample treatments of the brain regions and plasma were as previously reported (Tsai et al., 1996).
Blood, brain and bile microdialysis
Blood, brain and bile microdialysis systems comprised of a CMA/100 microinjection pump (CMA, Stockholm, Sweden) and microdialysis probes. The dialysis probes for blood (10 mm in length), brain (3 mm in length) and bile (7 cm in length) were made of silica capillary in a concentric design (Tsai et al., 1999c; 2000a, 2000b). Their tips were covered by dialysis membrane (Spectrum Co., 150 μm outer diameter with a cut-off at nominal molecular weight of 13 000, Laguna Hills, CA, U.S.A.) and all unions were cemented with epoxy. At least 24 h was allowed for the epoxy to dry. The blood microdialysis probe was located within the jugular vein/right atrium and then perfused with anticoagulant dextrose (ACD) solution (mM): citric acid 3.5; sodium citrate 7.5; dextrose 13.6 at a flow rate of 2 μl min−1 employing the microinjection pump (Tsai et al., 1999a). The bile duct microdialysis probes were constructed in our own laboratory based on the design originally described by Scott & Lunte (1993) and Hadwiger et al. (1994). This bile microdialysis method was reported in our previous studies (Tsai et al., 1999b, 1999c). For brain microdialysis, the rat was mounted on a Kopf stereotaxic frame and perfused with Ringer's solution (mM): Na+ 147; Ca2+ 2.2; K+ 4; pH 7.0. After being washed with Ringer's solution at a flow rate of 2 μl min−1, the microdialysis probe was implanted in the right striatum (coordinates: AP 0.2 mm, ML −3.0 mm, DV −7.0 mm) according to the Paxinos & Watson (1986) atlas. The probe positions were confirmed by standard histological procedure after the experiments. This method was modified from our previously reported procedures (Tsai et al., 1999a, 1999d).
Outflows from blood microdialysates were connected to an on-line injector (CMA/160) for automatic injection into the liquid chromatographic system every 10 min. The brain and bile dialysates were collected in a fraction collector (CMA/140) every 12 min and the same chromatographic system was subsequently analysed following blood dialysate. A retrograde calibration technique was utilized during in vivo recovery. The blood, brain and bile microdialysis probes were inserted into the rats' jugular vein, brain striatum and bile duct under anaesthesia with urethane and α-chloralose. Following a stabilization period of 2 h post probe implantation, the perfusate (Cperf) and dialysate (Cdial) concentrations of camptothecin were determined by HPLC. ACD solution (for blood microdialysis) containing camptothecin or Ringer's solution (for brain and bile microdialysis) containing camptothecin was perfused through the probe at a constant flow rate (2 μl min−1) employing the infusion pump (CMA/100). The in vivo relative recovery (Rdial) of camptothecin across the microdialysis probe was calculated by the following equation: Rdial=(Cperf−Cdial)/Cperf. The microdialysate recovery and concentration calculation were performed according to our previous report (Tsai et al., 2000a). Camptothecin microdialysate concentrations (Cm) were converted to unbound concentration (Cu) as follows: Cu=Cm/Rdial.
Drug administration
After a 2 h post-surgical stabilization period of probe implantation, the drug was subsequently administered according to the following study design. Six animals were used in each group. The control group received a 5 mg kg−1 of camptothecin by i.v. bolus injection. For the cyclosporin A treated group, cyclosporin A 20 mg kg−1 was injected via the femoral vein 10 min before camptothecin 5 mg kg−1 injection. For the cyclosporin A pretreated group, cyclosporin A 10 mg kg−1 was injected via intraperitoneally daily for 5 days, then on the sixth day, cyclosporin A 20 mg kg−1 was injected via the femoral vein 10 min prior to camptothecin 5 mg kg−1 injection. The cyclosporin A injection solution was diluted with 5% dextrose-in-water. Berberine (20 mg kg−1), quercetin (0.1 mg kg−1), quercetin (10 mg kg−1), naringin (10 mg kg−1) or naringenin (10 mg kg−1) were injected via femoral vein 10 min prior to camptothecin 5 mg kg−1 injection. The sampling interval was 12 min for each probe. Blood, brain and bile dialysates were measured by HPLC in the same experimental day.
Pharmacokinetics
Pharmacokinetic calculations were performed on each individual animal's data utilizing the pharmacokinetic calculation software WinNonlin Standard Edition Version 1.1 (Scientific Consulting Inc., Apex, NC, U.S.A.) by a noncompartmental method. The area under the concentration-time curve (AUC), the area under the first moment curve (AUMC) and the mean residence time (MRT) were calculated by statistical moments (Gabrielsson & Weiner, 1994). Formation rate constants were calculated from the extrapolated formation slope determined by the residuals method. The AUCs from time zero to time infinity (AUC0-inf) were calculated by the trapezoidal rule and extrapolated to time infinity by the addition of AUCt-inf. An analogous method was employed to calculate the AUMC with the concentration versus time data.
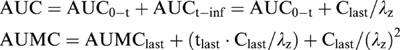 |
Where Clast and tlast are the last observed concentration and time, respectively; and λz is the terminal slope which is estimated by linear regression of the logarithmic value of the last observed data. The clearance (Cl) and MRT were calculated as follows: Cl=dose/AUC and MRT=AUMC/AUC. The blood to tissue distribution was calculated as follows: AUCtissue/AUCblood (Lin et al., 1982).
Statistical analysis
The results are represented as mean±standard error of the mean. The statistical analysis was performed with SPSS version 10.0 (SPSS Inc. Chicago, IL, U.S.A.). One-way ANOVA was followed by a Dunnett's post hoc test comparison between the control (camptothecin treated alone) and P-glycoprotein modulators treated groups. All statistical tests were performed at the two-sided 5% level of significance.
Results
Method validation
The calibration curve of camptothecin was obtained before HPLC analysis of dialysates over concentration ranges of 5 – 500 ng ml−1. This chromatographic system were validated for both inter- and intra-assay accuracy (0.1 – 10%) and the determined limits of this precision (0.1 – 7%) assay were acceptable. The chromatograms of a blank blood, brain and bile dialysate reveal that none of the observed peaks interfered with the analyte. In vivo recovery of camptothecin in blood (100 ng ml−1), brain (100 ng ml−1) and bile (100 ng ml−1) were 26.2±2.9, 8.7±0.6 and 61.0±0.9%, respectively (n=6).
Brain distribution of camptothecin
Utilizing direct sampling of blood and tissue homogenized sample, the biological samples to determine camptothecin were measured with HPLC coupled to fluorescence detection. The brain tissue concentration of camptothecin revealed no significant variation among various regions (cerebral cortex, striatum, hippocampus, cerebellum and brain stem) after 15 min of camptothecin administration (5 mg kg−1, i.v.). The mean concentration of camptothecin in each brain region was approximately one fourth of the camptothecin in plasma (Table 1).
Table 1.
Mean camptothecin concentration in rat brain regions (μg g−1) and plasma (μg ml−1) at 15 min after camptothecin administration (5 mg kg−1, i.v., n=6)

Effect of P-glycoprotein modulators on the pharmacokinetics of camptothecin in blood
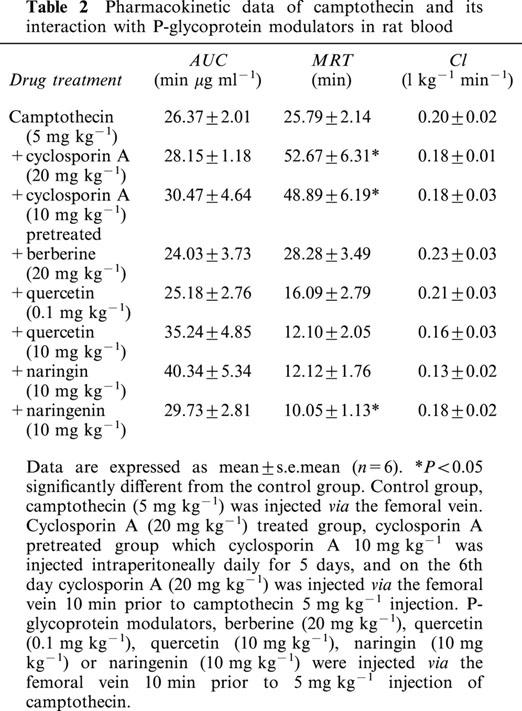
The pharmacokinetic curves of camptothecin in blood were constructed by concentrations versus time curves in each group as Figure 1 reveals. The effect of concomitant administration of P-glycoprotein modulators, cyclosporin A, berberine, quercetin, naringin or naringenin, on the overall disposition of camptothecin was examined by calculating the pharmacokinetic parameters employing non-compartmental analysis. The AUC and Cl demonstrated no significant differences in the blood between control and all the treated groups (Table 2). A prolonged MRT was identified in the treated and pretreated groups of cyclosporin A, and no significant difference was found in the berberine, quercetin and naringin treated groups, although reduced MRT was observed in the group of naringenin treated (Table 2).
Figure 1.
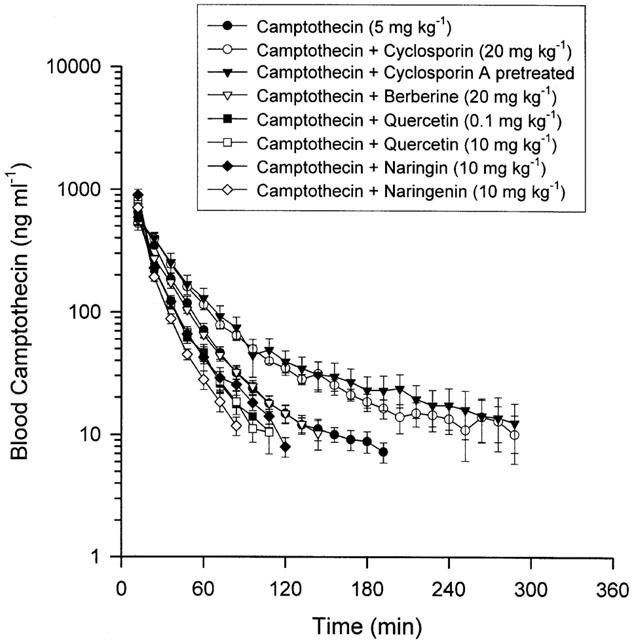
Mean unbound levels of camptothecin in rat blood were divided into the above groups with six individual experimental animals used in each group. Data are presented as mean±s.e.mean.
Table 2.
Pharmacokinetic data of camptothecin and its interaction with P-glycoprotein modulators in rat blood
Effect of P-glycoprotein modulators on the distribution of camptothecin in brain
According to the even distribution of camptothecin in brain regions (Table 1), the microdialysis probe was inserted into the striatum for sampling extracellular tissue. The peak concentrations of camptothecin in the brain were attained in the first 24 min following drug administration (Figure 2). A significant increase in the AUCs of camptothecin in the brain striatum occurred when cyclosporin A was employed for treated and pretreated groups, as Figure 2 reveals. The pharmacokinetic profiles reveal that cyclosporin A treated and pretreated animals display significant increases of camptothecin AUC in the brain, but no significant difference when berberine, quercetin, naringin or naringenin were concomitantly administered (Table 3). Corresponding to P-glycoprotein modulators, the MRT was prolonged in the cyclosporin A pretreated group but other treated groups displayed no significant difference. Following camptothecin administration (5 mg kg−1, i.v.), the distribution ratio of camptothecin from blood to brain (AUCbrain/AUCblood) was 0.27±0.024. Cyclosporin A, the P-glycoprotein inhibitor, in both treated and pretreated groups, elevated the distribution ratio of camptothecin in the rats' brains; however some P-glycoprotein modulators, higher dose of quercetin (10 mg kg−1), naringin and naringenin reduced the distribution ratio (Table 3). Treated groups presented no altered distribution ratio of camptothecin in the rats' brains at a smaller dose of quercetin (0.1 mg kg−1) and berberine (20 mg kg−1). These results suggest that camptothecin penetrates the BBB and the P-glycoprotein might regulate it.
Figure 2.
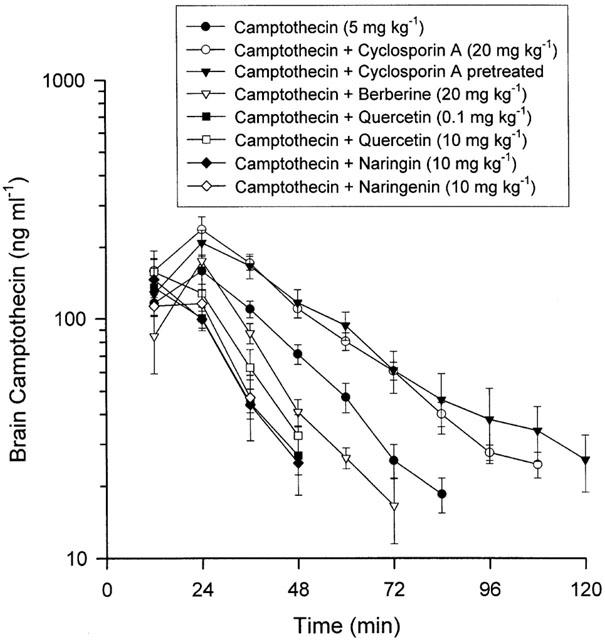
Mean camptothecin levels in rat brain were divided into the above groups with six individual experimental animals used in each group. Data are presented as mean±s.e.mean.
Table 3.
Pharmacokinetic data of camptothecin and its interaction with P-glycoprotein modulators in rat brain
Effect of P-glycoprotein modulators on the disposition of camptothecin in bile
The average concentration of camptothecin in the bile increased during the first 30 min following drug administration. The distribution ratio of camptothecin from blood to bile (AUCbile/AUCblood) in the control group was 4.32±0.51 (Table 4). The amount of camptothecin, as estimated from the AUC, in bile set against the concentration gradient significantly exceeded that in blood, suggesting that camptothecin might be actively excreted into the bile. A decrease in the concentration versus time curve of camptothecin in the bile was observed when other P-glycoprotein modulators were treated, as shown in Figure 3. This result did not apply to the berberine treated group. The camptothecin distribution ratios in bile decreased significantly in the groups of cyclosporin A treated and pretreated, higher dose of quercetin (10 mg kg−1), naringin and naringenin treated groups. However, berberine and a smaller dose of quercetin (0.1 mg kg−1) did not markedly affect the biliary distribution ratio of camptothecin. These results imply that the P-glycoprotein might regulate the hepatobiliary excretion of camptothecin. The variation in biliary distribution ratios suggest that the potency differed among P-glycoprotein modulators.
Table 4.
Pharmacokinetic data of camptothecin and its interaction with P-glycoprotein modulators in rat bile
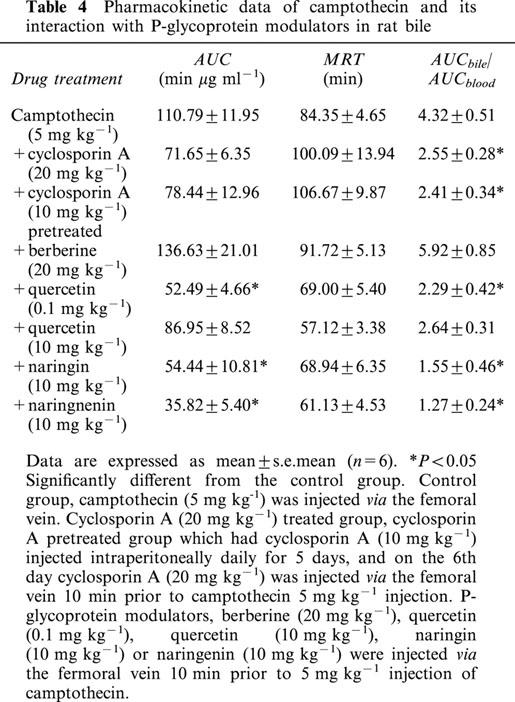
Figure 3.
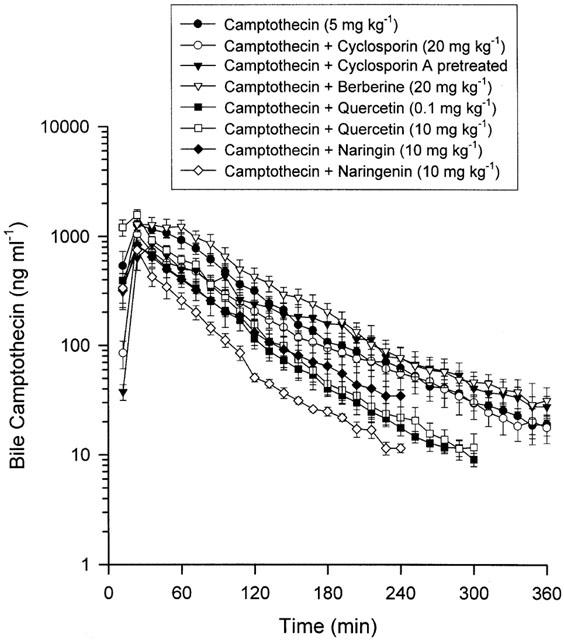
Mean camptothecin levels in rat bile were divided into the above groups with six individual experimental animals used in each group. Data are presented as mean±s.e.mean.
Discussion
In this investigation, the analytical method allowed simultaneous measurement of unbound camptothecin dialysate samples in rat blood, brain, bile as well as total form of camptothecin in plasma and brain tissue. The results indicate that no interference occurred during the analysis of various biological samples. The analysis of brain tissue homogenates provides a measurement of total drug uptake by the brain, without distinguishing between extracellular and intracellular drug concentrations. Table 1 reveals the even distribution of camptothecin in rat brain regions and indicates that the brain concentration to plasma distribution ratio of camptothecin derivative was about 0.3. The cerebrospinal fluid (CSF) concentration to plasma distribution ratio of camptothecin in children ranges from 0.1 to 0.59 after a 24 h i.v. infusion (Baker et al., 1996). It has been reported in a related result that the average CSF concentration to plasma of camptothecin derivative has been found to be 0.3 in nonhuman primates (Blaney et al., 1993). However, different results have been reported in a pharmaceutical modification study, where the brain AUC surpassed the blood AUC at a ratio of 2.94 (Yang et al., 1999).
Microdialysis allows extracellular concentration of drugs in blood and other biological tissues to be measured in vivo (de lange et al., 1997). This sampling technique is extremely useful to investigate the transport of endogenous and exogenous substances across the BBB. This paper demonstrates the pharmacokinetics of unbound camptothecin in blood, brain and bile after intravenous administration to rats. This procedure combined with P-glycoprotein modulators made it possible to estimate the extracellular camptothecin concentration in the brain and the camptothecin's mechanism.
The present microdialysis technique provides protein free samples that can be injected directly into a liquid chromatographic system to monitor continuously in vivo unbound drugs in biological samples such as blood, brain or bile. Furthermore, this sampling method facilitates pharmacokinetic studies by reducing the influence of biological volume changes compared to conventional biological fluid withdrawing assays. The instability of camptothecin (lactone form) has been demonstrated, and the fact that the opening of the ring under basic conditions produces the water soluble carboxylate form. An on-line microdialysis technique was employed in this study to avoid the labile form of camptothecin (Shenderova et al., 1999).
The ratio of the AUC in the brain over that in blood (AUCbrain/AUCblood) was utilized to assess the drug penetration through the BBB (de lange et al., 1997). This steadily increasing ratio further demonstrates that camptothecin penetrated the BBB, which strongly confirms the findings of Blaney et al. (1998). Their findings reveal that the camptothecin analogue, 9-aminocamptothecin could cross the BBB. Our data reveal that the distribution ratio of unbound camptothecin in rat brains was 0.27±0.024 (Table 3). This result indicates that the brain distribution ratio of camptothecin did not differ significantly from the total form of camptothecin concentration (Table 1). A similar phenomenon has been discovered in the pharmacokinetics of theophylline; the pharmacokinetic data demonstrate whole blood sampling does not differ statistically from simultaneous microdialysis sampling (Telting-Diaz et al., 1992).
This investigation has revealed that the groups treated and pretreated with cyclosporin A, a P-glycoprotein inhibitor, exhibited enhanced delivery of camptothecin into the rat brain. However, the P-glycoprotein modulators, berberine, quercetin, naringin and naringenin did not significantly diminish camptothecin AUC in the brain. This finding supports the hypothesis that P-glycoprotein can play an important role in restricting the access of substrate molecules to the brain. Although the herbal alkaloid, berberine, augment the expression of P-glycoprotein in the cell lines (Lin et al., 1999), it did not markedly regulate the camptothecin penetration of BBB. This may be due to the weak potency of berberine in vivo or unsuitable dose. Several flavonoids, dietary compounds, have been reported to strongly up-regulate the apparent activity of P-glycoprotein in cancer cell lines (Chieli et al., 1995). However, these flavonoids did not alter the P-glycoprotein function for camptothecin in BBB penetration in vivo. The contrary report found that the flavonoids also did not affect the action of P-glycoprotein (Choi et al., 1999).
The results of the investigation did not confirm the claim by Mitsunaga et al. (2000), who found a biphasic effect, whereby a smaller dose of quercetin (0.1 mg kg−1) diminished [3H]-vincristine penetration into BBB, but a higher dose of quercetin (1.0 mg kg−1) enhanced this effect in mice. The distribution of mdr 1 gene knockout mouse was possibly the most convincing evidence currently in the literature. Schinkel et al. (1994) demonstrated that the gene knockout mice were over 100 times more sensitive to the CNS toxin, ivermectin, than the mice that were treated with P-glycoprotein modulator. In addition, evidence of ATP-dependent and P-glycoprotein mediated transport of cyclosporin A across the BBB in rats has been reported in vitro and in vivo (Tsuji et al., 1993; Sakata et al., 1994).
As well known, hepatocytes produce bile juice. This process involves the vectorial transport of compounds such as bile salts, phospholipids and cholesterol, as well as endobiotics and xenobiotics. This phenomenon requires P-glycoprotein because it was assumed to be involved in the permeation of compounds across the plasma membrane (Frijters et al., 1997). The biliary excretion of camptothecin was previously reported in an isolated perfused rat liver in vitro system (Platzer et al., 2000). A recent report indicates that the biliary excretion mechanism of camptothecin derivative irinotecan might involve P-glycoprotein (Chu et al., 1999). In this study, we systematically examined the biliary excretion of camptothecin in various P-glycoprotein modulators treated in rats to illuminate the biliary excretion mechanism involved. The P-glycoprotein modulators, cyclosporin A and some flavonoids have been demonstrated to decrease the bile distribution ratio of camptothecin. This phenomenon implies the treatment of P-glycoprotein modulator inhibited the efflux of camptothecin from biliary fluid. These results confirm the recent report which indicates that flavonoid genistein interacts with P-glycoprotein and inhibits P-glycoprotein-mediated drug transport (Castro & Altenberg, 1997).
This investigation convincingly demonstrates the potential for studying the pharmacokinetics of camptothecin in rat blood, brain and bile. Several in vivo techniques have been previously described for brain pharmacokinetic studies, encompassing autoradiography, imaging methods (PET-positron emission tomography and NMR-nuclear magnetic resonance), cerebral fluid sampling, in vivo voltammetry, and intracerebral microdialysis. However, these techniques are too expensive for general laboratory use. Although intracerebral microdialysis has the disadvantage of being invasive, it is much cheaper than PET scanning or NMR, and it can be employed in the general laboratory.
This study has demonstrated that cyclosporin A, plant alkaloid (berberine) and plant flavonoids (quercetin, naringin and naringenin) interact with camptothecin. This result indicates that the P-glycoprotein might regulate the BBB penetration and hepatobiliary elimination of camptothecin. Future studies should incorporate a design avoiding mdr 1a-encoded P-glycoprotein animals.
Acknowledgments
This study was supported in part by research grants from the National Science Council (NSC89-2113-M-077-009; NSC89-2320-B-077-013), Taiwan.
Abbreviations
- AUC
area under the concentration curve
- BBB
blood brain barrier
- CSF
cerebrospinal fluid
- MDR
multidrug resistance
- NMR
nuclear magnetic resonance
- PET
positron emission tomography
- t1/2
elimination half-life
References
- BAKER S.D., HEIDEMAN R.L., CROM W.R., KUTTESCH J.F., GAJJAR A., STEWART C.F. Cerebrospinal fluid pharmacokinetics and penetration of continuous infusion topotecan in children with central nervous system tumors. Cancer Chemother. Pharmacol. 1996;37:195–202. doi: 10.1007/BF00688317. [DOI] [PubMed] [Google Scholar]
- BLANEY S.M., COLE D.E., BALIS F.M., GODWIN K., POPLACK D.G. Plasma and cerebrospinal fluid pharmacokinetic study of topotecan in nonhuman primates. Cancer Res. 1993;53:725–727. [PubMed] [Google Scholar]
- BLANEY S.M., TAKIMOTO C., MURRY D.J., KUTTESCH N., MCCULLY C., COLE D.E., GODWIN K., BALIS F.M. Plasma and cerebrospinal fluid pharmacokinetics of 9-aminocamptothecin (9-AC), irinotecan (CPT-11), and SN-38 in nonhuman primates. Cancer Chemother. Pharmacol. 1998;41:464–468. doi: 10.1007/s002800050768. [DOI] [PubMed] [Google Scholar]
- CASTRO A.F., ALTENBERG G.A. Inhibition of drug transport by genistein in multidrug-resistant cells expressing P-glycoprotein. Biochem. Pharmacol. 1997;53:89–93. doi: 10.1016/s0006-2952(96)00657-0. [DOI] [PubMed] [Google Scholar]
- CHIELI E., ROMITI N., CERVELLI F., TONGIANI R. Effects of flavonols on P-glycoprotein activity in cultured rat hepatocytes. Life Sci. 1995;57:1741–1751. doi: 10.1016/0024-3205(95)02152-9. [DOI] [PubMed] [Google Scholar]
- CHOI S.U., KIM K.H., CHOI E.J., PARK S.H., LEE C.O., JUNG N.P., YOON S.K., RYU S.Y. P-glycoprotein (Pgp) does not affect the cytotoxicity of flavonoids from Sophora flavescens, which also have no effects on Pgp action. Anticancer Res. 1999;19:2035–2040. [PubMed] [Google Scholar]
- CHU X.Y., KATO Y., SUGIYAMA Y. Possible involvement of P-glycoprotein in biliary excretion of CPT-11 in rats. Drug Metab. Dispos. 1999;27:440–441. [PubMed] [Google Scholar]
- CORDON-CARDO C., O'BRIEN J.P., BOCCIA J., CASALS D., BERTINO J.R., MELAMED M.R. Expression of the multidrug resistance gene product (P-glycoprotein) in human normal and tumor tissues. J. Histochem. Cytochem. 1990;38:1277–1287. doi: 10.1177/38.9.1974900. [DOI] [PubMed] [Google Scholar]
- D'ARPA P., LIU L.F. Topoisomerase-targeting antitumor drugs. Biochim. Biophys. Acta. 1989;989:163–177. doi: 10.1016/0304-419x(89)90041-3. [DOI] [PubMed] [Google Scholar]
- DARZYNKIEWICZ Z., BRUNO S., DEL BINO G., TRAGANOS F. The cell cycle effects of camptothecin. Ann. NY. Acad. Sci. 1996;803:93–100. doi: 10.1111/j.1749-6632.1996.tb26379.x. [DOI] [PubMed] [Google Scholar]
- DE LANGE E.C., DANHOF M., DE BOER A.G., BREIMER D.D. Methodological considerations of intracerebral microdialysis in pharmacokinetic studies on drug transport across the blood-brain barrier. Br. Res. Rev. 1997;25:27–49. doi: 10.1016/s0165-0173(97)00014-3. [DOI] [PubMed] [Google Scholar]
- FARDEL O., LECUREUR V., GUILLOUZO A. The P-glycoprotein multidrug transporter. Gen. Pharmacol. 1996;27:1283–1291. doi: 10.1016/s0306-3623(96)00081-x. [DOI] [PubMed] [Google Scholar]
- FRIJTERS C.M.G., GROEN A.K., OUDE ELFERINK R.P.J. MDR2 P-glycoprotein-mediated lipid sectetion and its relevance to biliary drug transport. Adv. Drug Deliv. Rev. 1997;25:201–215. [Google Scholar]
- FROMM M.F. P-glycoprotein: a defense mechanism limiting oral bioavailability and CNS accumulation of drugs. Int. J. Clin. Pharmacol. Ther. 2000;38:69–74. doi: 10.5414/cpp38069. [DOI] [PubMed] [Google Scholar]
- GABRIELSSON J., WEINER D. Pharmacokinetic and Pharmacodynamic Data Analysis Concepts and Applications. Stockholm, Swedish Pharm. Press; 1994. Non-compartmental analysis; pp. 621–626. [Google Scholar]
- GIOVANELLA B.C., STEHLIN J.S., WALL M.E., WANI M.C., NICHOLAS A.W., LIU L.F., SILBER R., POTMESIL M. DNA topoisomerase I-targeted chemotherapy of human colon cancer in xenografts. Sciences. 1989;246:1046–1048. doi: 10.1126/science.2555920. [DOI] [PubMed] [Google Scholar]
- HADWIGER M.E., TELTING-DIAZ M., LUNTE C.E. Liquid chromatographic determination of tacrine and its metabolites in rat bile microdialysates. J. Chromatogr. B. 1994;655:235–241. doi: 10.1016/0378-4347(94)00099-9. [DOI] [PubMed] [Google Scholar]
- HEBERT M.F. Contributions of hepatic and intestinal metabolism and P-glycoprotein to cyclosporine and tacrolimus oral drug delivery. Adv. Drug Deliver. Rev. 1997;27:201–214. doi: 10.1016/s0169-409x(97)00043-4. [DOI] [PubMed] [Google Scholar]
- HSIANG Y.H., HERTZBERG R., HECHT S., LIU L.F. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J. Biol. Chem. 1985;260:14873–14878. [PubMed] [Google Scholar]
- KARTNER N., RIORDAN J.R., LING V. Cell surface P-glycoprotein associated with multidrug resistance in mammalian cell lines. Science. 1983;221:1285–1288. doi: 10.1126/science.6137059. [DOI] [PubMed] [Google Scholar]
- LIN H.L., LIU T.Y., LUI W.Y., CHI C.W. Up-regulation of multidrug resistance transporter expression by berberine in human and murine hepatoma cells. Cancer. 1999;85:1937–1942. [PubMed] [Google Scholar]
- LIN J.H., SUGIYAMA Y., AWAZU S., HANANO M. In vitro and in vivo evaluation of the tissue-to-blood partition coefficient for physiological pharmacokinetic models. J. Pharmacokinet. Biopharm. 1982;10:637–647. doi: 10.1007/BF01062545. [DOI] [PubMed] [Google Scholar]
- LIU L.F., DUANN P., LIN C.T., D'ARPA P., WU J. Mechanism of action of camptothecin. Ann. N. Y. Acad. Sci. 1996;803:44–49. doi: 10.1111/j.1749-6632.1996.tb26375.x. [DOI] [PubMed] [Google Scholar]
- MITSUNAGA Y., TAKANAGA H., MATSUO H., NAITO M., TSURUO T., OHTANI H., SAWADA Y. Effect of bioflavonoids on vincristine transport across blood-brain barrier. Eur. J. Pharmacol. 2000;395:193–201. doi: 10.1016/s0014-2999(00)00180-1. [DOI] [PubMed] [Google Scholar]
- MOERTEL C.G., SCHUTT A.J., REITEMEIER R.J., HAHN R.G. Phase II study of camptothecin (NSC 100880) in the treatment of advanced gastrointestinal cancer. Cancer Chemother. Rep. 1972;56:95–101. [PubMed] [Google Scholar]
- MUGGIA F.M., DIMERY I., ARBUCK S.G. Camptothecin and its analogs. An overview of their potential in cancer therapeutics. Ann. N. Y. Acad. Sci. 1996;803:213–223. doi: 10.1111/j.1749-6632.1996.tb26391.x. [DOI] [PubMed] [Google Scholar]
- O'LEARY J., MUGGIA F.M. Camptothecins: a review of their development and schedules of administration. Eur. J. Cancer. 1998;34:1500–1508. doi: 10.1016/s0959-8049(98)00229-9. [DOI] [PubMed] [Google Scholar]
- PARDRIDGE W.M. CNS drug design based on principles of blood-brain barrier transport. J. Neurochem. 1998;70:1781–1792. doi: 10.1046/j.1471-4159.1998.70051781.x. [DOI] [PubMed] [Google Scholar]
- PAXINOS G., WATSON C. The Rat Brain in Stereotaxic Coordinates. Academic Press, San Diego, California, U.S.A; 1986. [Google Scholar]
- PLATZER P., THALHAMMER T., HAMILTON G., ULSPERGER E., ROSENBERG E., WISSIACK R., JAGER W. Metabolism of camptothecin, a potent topoisomerase I inhibitor, in the isolated perfused rat liver. Cancer Chemother. Pharmacol. 2000;45:50–54. doi: 10.1007/PL00006742. [DOI] [PubMed] [Google Scholar]
- RILEY J., STYLES J., VERSCHOYLE R.D., STANLEY L.A., WHITE I.N., GANT T.W. Association of tamoxifen biliary excretion rate with prior tamoxifen exposure and increased mdr1b expression. Biochem. Pharmacol. 2000;60:233–239. doi: 10.1016/s0006-2952(00)00326-9. [DOI] [PubMed] [Google Scholar]
- RIVORY L.P., ROBERT J. Molecular, cellular, and clinical aspects of the pharmacology of 20(S)camptothecin and its derivatives. Pharmacol. Ther. 1995;68:269–296. doi: 10.1016/0163-7258(95)02009-8. [DOI] [PubMed] [Google Scholar]
- RONINSON I.B. The role of the MDR1 (P-glycoprotein) gene in multidrug resistance in vitro and in vivo. Biochem. Pharmacol. 1992;43:95–102. doi: 10.1016/0006-2952(92)90666-7. [DOI] [PubMed] [Google Scholar]
- SAKATA A., TAMAI I., KAWAZY K., DEGUCHI Y., OHNISHI T., SAHEKI A., TSUJI A. In vivo evidence for ATP-dependent and P-glycoprotein-mediated transport of cyclosporin A at the blood-brain barrier. Biochem. Pharmacol. 1994;48:1989–1992. doi: 10.1016/0006-2952(94)90601-7. [DOI] [PubMed] [Google Scholar]
- SCHINKEL A.H., SMIT J.J., VAN TELLINGEN O., BEIJNEN J.H., WAGENAAR E., VAN DEENTER L., MOL C.A., VAN DER VALK M.A., ROBANUS-MAANDAG E.C., TE RIELE H.P. Disruption of the mouse mdr 1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell. 1994;77:491–502. doi: 10.1016/0092-8674(94)90212-7. [DOI] [PubMed] [Google Scholar]
- SCHULTZ A.G. Camptothecin. Chem. Rev. 1973;73:385–405. doi: 10.1021/cr60284a004. [DOI] [PubMed] [Google Scholar]
- SCOTT D.O., LUNTE C.E. In vivo microdialysis sampling in the bile, blood, and liver of rats to study the disposition of phenol. Pharm. Res. 1993;10:335–342. doi: 10.1023/a:1018971818689. [DOI] [PubMed] [Google Scholar]
- SHAMMA M., STGEORGIEV V. Camptothecin. J. Pharm. Sci. 1974;63:163–183. doi: 10.1002/jps.2600630203. [DOI] [PubMed] [Google Scholar]
- SHENDEROVA A., BURKE T.G., SCHWENDEMAN S.P. The acidic microclimate in poly(lactide-co-glycolide) microspheres stabilizes camptothecins. Pharm. Res. 1999;16:241–248. doi: 10.1023/a:1018876308346. [DOI] [PubMed] [Google Scholar]
- TATSUTA T., NAITO M., OH-HARA T., SUGAWARA I., TSURUO T. Functional involvement of P-glycoprotein in blood-brain barrier. J. Biol. Chem. 1992;267:20383–20391. [PubMed] [Google Scholar]
- TELTING-DIAZ M., SCOTT D.O., LUNTE C.E. Intravenous microdialysis sampling in awake, freely-moving rats. Anal. Chem. 1992;64:806–810. doi: 10.1021/ac00031a019. [DOI] [PubMed] [Google Scholar]
- TSAI T.H., CHEN Y.F., CHEN I.F., CHEN C.F. Measurement of unbound caffeic acid in rat blood by on-line microdialysis coupled with liquid chromatography and its application to pharmacokinetic studies. J. Chromatogr. B. 1999a;729:119–125. doi: 10.1016/s0378-4347(99)00133-4. [DOI] [PubMed] [Google Scholar]
- TSAI T.H., CHOU C.J., CHEN C.F. Pharmacokinetic and brain distribution of magnolol in the rats after intravenous bolus injection. J. Pharm. Pharmacol. 1996;48:57–59. doi: 10.1111/j.2042-7158.1996.tb05877.x. [DOI] [PubMed] [Google Scholar]
- TSAI T.H., HUANG C.T., SHUM A.Y.C., CHEN C.F. Pharmacokinetic and biliary excretion study of esculetin by microdialysis in rat. Life Sci. 1999b;65:1647–1655. doi: 10.1016/s0024-3205(99)00413-0. [DOI] [PubMed] [Google Scholar]
- TSAI T.H., HUNG L.C., CHEN C.F. Biliary excretion of chloramphenicol and chloramphenicol glucuronide in rat by microdialysis. J. Pharm. Pharmacol. 1999c;51:911–915. doi: 10.1211/0022357991773339. [DOI] [PubMed] [Google Scholar]
- TSAI T.H., TSAI T.R., CHEN Y.F., CHOU C.J., CHEN C.F. Determination of unbound 20(S)-camptothecin in rat bile by on-line microdialysis coupled to microbore liquid chromatography with fluorescence detection. J. Chromatogr. B. 1999d;732:221–225. doi: 10.1016/s0378-4347(99)00288-1. [DOI] [PubMed] [Google Scholar]
- TSAI T.H., CHEN Y.F., CHOU C.J., CHEN C.F. Measurement and pharmacokinetics of unbound 20(S)-camptothecin in rat blood and brain by microdialysis coupled to microbore liquid chromatography with fluorescence detection. J. Chromatogr. A. 2000a;870:221–226. doi: 10.1016/s0021-9673(99)00854-7. [DOI] [PubMed] [Google Scholar]
- TSAI T.H., SHUM A.Y.C., CHEN C.F. Enterohepatic circulation of chloramphenicol and chloramphenicol glucuronide in rat. Life Sci. 2000b;66:363–370. doi: 10.1016/s0024-3205(99)00598-6. [DOI] [PubMed] [Google Scholar]
- TSUJI A., TAMAI I., SAKATA A., TENDA Y., TERASAKI T. Restricted transport of cyclosporin A across the blood-brain barrier by a multidrug transporter, P-glycoprotein. Biochem. Pharmacol. 1993;46:1096–1099. doi: 10.1016/0006-2952(93)90677-o. [DOI] [PubMed] [Google Scholar]
- WALL M.E. Camptothecin and taxol: discovery to clinic. Med. Res. Rev. 1998;18:299–314. doi: 10.1002/(sici)1098-1128(199809)18:5<299::aid-med2>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- WALL M.E., WANI M.C. Camptothecin. Discovery to clinic. Ann. N. Y. Acad. Sci. 1996;803:1–12. doi: 10.1111/j.1749-6632.1996.tb26371.x. [DOI] [PubMed] [Google Scholar]
- WALL M.E., WANI M.C., COOK C.E., PALMER K.H., MCPHAIL A.T., SIM G.A. Plant antitumor agents. I. The isolation and structure of camptothecin, a novel alkaloidal leukemia and tumour inhibitor from Camptotheca acuminata. J. Am Chem. Soc. 1966;88:3888–3890. [Google Scholar]
- YANG S.C., LU L.F., CAI Y., ZHU J.B., LIANG B.W., YANG C.Z. Body distribution in mice of intravenously injected camptothecin solid lipid nanoparticles and targeting effect on brain. J. Control. Release. 1999;59:299–307. doi: 10.1016/s0168-3659(99)00007-3. [DOI] [PubMed] [Google Scholar]