

Introduction
The proposition that cyclo-oxygenase-2 (COX-2) is causally linked to cancer offers a new approach to extending our knowledge of neoplasia and of improving treatment of the human disease. The identification of an enzyme catalysing fatty acid oxidation as a rate limiting step in the progress from normal cell growth through hyperplasia on to neoplasia has opened up a whole new field of cancer research, far from the usual approaches based on nucleic acid metabolism. The precise interactions and links between lipid metabolism and DNA replication have still to be elucidated and defined but they are already having clinical consequences. In practical terms, this surprising proposition offers a sound scientific basis for the successful prevention of cancer and a real prospect of drug treatment of diagnosed disease without the serious side effects usually associated with cancer chemotherapy. Chemoprevention based on COX-2 has already been achieved in a restricted group of patients (Steinbach et al., 2000) and clinical enthusiasm has already been expressed for COX-2 based treatment to be extended as chemoprevention in a wider range of patients or as adjuvant therapy of established disease (Vainio & Morgan, 1998; Lord et al., 1999; Cuendet & Pezzuto, 2000; Gately, 2000).
This review will summarize the evidence for the COX-2 and cancer proposition in general. However because the majority of the work, both experimental and clinical, has focused on the relevance of COX-2 to colorectal cancer (CRC), this form of cancer will also be the focus of this review. At the end of the review some selected references to COX-2 in other forms of cancer are given as points of entry into those areas, but with no attempt at a comprehensive coverage.
Biology of COX-2
First, a brief description of the enzyme, COX-2, concentrating on those of its characteristics particularly relevant to its participation in the processes of carcinogenesis. Further details can be found in a number of reviews (Bakhle & Botting, 1996; Vane et al., 1998; Bakhle, 1999; Smith et al., 2000a).
Cyclo-oxygenase-2 (COX-2) is also formally called prostaglandin H2 synthase-2 (PGHS-2), mostly in the American literature. COX-2 and COX-1 are isoforms of an enzyme which catalyses the first stage in the oxidation of arachidonic acid to the prostanoids (Figure 1). Most of the biochemical cascade outlined in this Figure was well characterized for what is now known as COX-1, before a new protein with COX like activity was described in the early 1990s (Xie et al., 1991; Kujubu et al., 1991). This new protein proved to be a true isoform of COX-1, i.e., it accepts the same substrate and yields the same product, by the same reactions. In addition, the linear sequence and three-dimensional structure of the two isoforms are very similar. Even the active site of the isoforms differs minimally (valine/isoluecine substitutions) at only two positions. The crystal structure of COX-2 (Luong et al., 1996; Kurumbail et al., 1996) was solved by superimposition on that already described for COX-1 (Picot et al., 1994). Nevertheless, inhibitors with high and clinically demonstrable selectivity for the isoforms are available (Smith et al., 1998; Chan et al., 1999; Talley et al., 2000; Riendeau et al., 2001).
Figure 1.
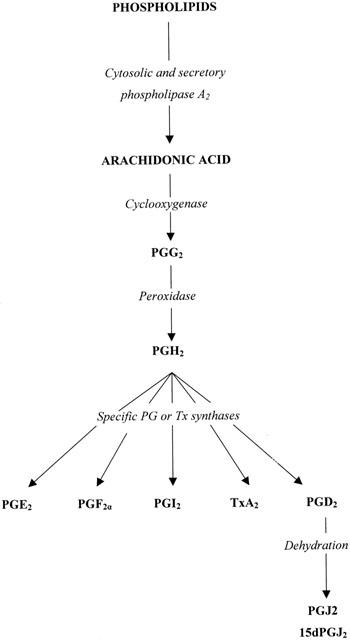
Biosynthesis of prostanoids. The resting levels of free arachidonic acid (AA) in cells are low and PG synthesis is dependent on the provision of AA from phospholipid. The PLA2 enzymes are the major providers but free AA can be generated indirectly via PLC or PLD activities. The COX protein— in either isoform —exhibits two separate enzymic activities, as shown. Inhibition of the COX activity does not inhibit peroxidase activity. All the final prostanoid products have a common precursor, PGH2, and separate synthase enzymes. So far only PGE2 synthase has been found to be inducible, by the same stimuli as induce COX-2. The cyclopentenone PG ligands for the nuclear receptor PPARγ, PGJ2 and 15dPGJ2, are derived only from PGD2 by ill-defined pathways in vivo.
Although the development of such inhibitors (Vane et al., 1998; Bakhle, 1999) will not be discussed here, it is necessary for the rest of this review to note that the clinically used, non-steroid anti-inflammatory drugs (NSAIDs), such as aspirin, indomethacin, ibuprofen or diclofenac, are inhibitors of COX activity. These are either non-selective, acting on both isoforms, or are more effective on COX-1. The term NSAID will be used to denote this type of inhibitor in contrast to the selective inhibitors of COX-2, which will be referred to as C2Is.
The important biological difference between the isoforms is that COX-1 is normally present in most types of cells and is a constitutive, housekeeping enzyme. The latter characteristic is derived from its DNA and RNA structures and in practical terms implies that amounts of COX-1 protein remain virtually constant (about 2 – 5 fold variation) under either physiological or pathological conditions. By contrast, COX-2 protein is normally absent from most cells – with some notable exceptions – but appears rapidly (2 – 4 h) in large amounts in a range of pathological, often inflammatory, situations and in many cell types.
In the present context, it is important that COX-2 was first described as being induced by a viral oncogene (Xie et al., 1991) or by a tumour promoter (Kujubu et al., 1991). Subsequent work has shown it to be inducible by a variety of growth factors and mitogens (Bakhle & Botting, 1996; Smith et al., 2000a), making this isoform particularly relevant to the processes of cell growth and carcinogenesis.
One further aspect of COX biochemistry must be emphasised. Both COX-1 and COX-2 form the same product, PGH2. This PG is the common precursor for the biosynthesis of thromboxane A2 (TxA2), prostacylin (PGI2) and the other prostaglandins PGD2, PGE2, PGF2α (see Figure 1). These ‘post COX' transformations are catalysed by quite separate enzymes. It is these prostanoids that determine the final biological response to the action of COX. For instance, it is the same isoform of COX that in platelets leads to the formation of the vasoconstrictor and pro-aggregatory TxA2 but in endothelial cells provides the vasodilator and anti-aggregatory PGI2. This biological selectivity reflects the absence of PGI2 synthase in platelets and of TxA2 synthase in endothelial cells. The importance of the ‘post COX' enzymes has recently been underlined by the identification of an inducible isoform of PGE2 synthase. This isoform is induced, like COX-2, by bacterial lipopolysaccharide (LPS) or interleukin-1 (IL-1), it is located on the perinuclear membrane and it appears to be functionally coupled to COX-2 (Naraba et al., 1998; Jakobsson et al., 1999; Murakami et al., 2000). Thus the final biological effect of COX-2 activity may be predominantly expressed by PGE2 and susceptible to control by inhibitors selective for the inducible PGE2 synthase.
Natural history of CRC
CRC provides a significant proportion of cancer deaths in the Western world and is second only to lung cancer in the U.S. Its incidence is age related – hardly any below the age of 40 and rising to about 300 per 100,000 over 65 years. Mortality is high, about 150 per 100,000. It might be reasonable to predict an increase in deaths from CRC, purely on an age-related basis, as other causes of death in the over 50s (mainly cardiovascular at present) are reduced, by the relative success of modern treatment of cardiovascular disease.
Although the great majority of CRC cases are sporadic, i.e. have no clear cause, in a small proportion (5 – 13%; Emery et al., 2001) there is a family history suggesting a heritable susceptibility to the disease. In two very small groups there is clear evidence for an autosomal dominant genetic mutation that leads to CRC. The most clearly defined is the group with familial adenomatous polyposis (FAP) who comprise about 0.5% of all CRC cases. In patients with FAP, hundreds of small polyps occur spontaneously in the colon and rectum by about 20 years of age. These growths are initially benign but within the next 20 years as they grow in size and number, some will become malignant. If untreated, most of the subjects will develop colorectal cancer. The genetic fault is in the APC (adenomatous polyposis coli) gene and several mutations are known, giving rise to defective APC proteins (Dubois et al., 1996; Syngal et al., 2000). The penetrance of this mutation is almost 100%, i.e., all who carry the defective gene will develop CRC (Emery et al., 2001). Because of this high penetrance, this small group of subjects have been very important in the development of potential chemopreventive agents and C2Is are already used in this condition (Steinbach et al., 2000).
The other group of heritable CRC is hereditary non-polyposis colon cancer (HNPCC) and is less clearly defined than FAP although it is larger in size (about 2% of all CRC) (Syngal et al., 2000, Emery et al., 2001). The genetic defect here is also less specific, being in a number of genes related to DNA repair including MLH1, MSH2, PMS1 & 2. The corresponding proteins identify and repair errors in DNA arising from mismatching during replication (MMR enzymes) and their dysfunction allows errors in DNA to persist into the next cycle of cell division. The penetrance here is significantly less than in FAP and all the genetic mutations associated with HNPCC have still to be identified.
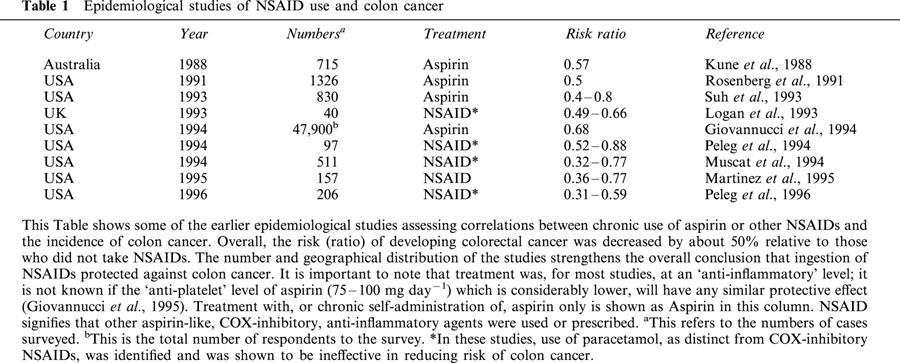
However, it must be emphasised that the large majority of CRC cases (over 80%) are sporadic and exhibit no obvious heritable tendency. Therefore the epidemiological finding that chronic NSAID use decreased CRC incidence (summarized in Thun, 1996; Table 1) applies mainly to sporadic CRC and is not restricted to any genetically determined sub-group of the disease. If, as suggested from experimental work, COX-2 may be a rate-limiting component at several stages in the development of neoplasia (Reddy et al., 2000) and in the growth of the fully transformed cell, inhibition of this enzyme would be effective in the genesis of CRC and in its subsequent growth, irrespective of the initiating mechanisms.
Table 1.
Epidemiological studies of NSAID use and colon cancer
Carcinogenesis in CRC
Carcinogenesis is a multi-stage process, in which a succession of mutations is needed to progress from the normal cell to the fully neoplastic cell (Kinzler & Vogelstein, 1996; Marks & Furstenberger, 2000; Syngal et al., 2000; Emery et al., 2001). Such a progress is well exemplified in human CRC and in related experimental systems. The first mutation in colonic epithelial cells is in the gene for APC, a protein that is a significant component of the apoptotic pathway in these cells (Kinzler & Vogelstein, 1996; Marks & Furstenberger, 2000; Fosslien, 2000). This mutation is followed by others and one scheme of the progress via hyperplasia to neoplasia in epithelium is summarized in Figure 2. The need for many different gene mutations to accumulate within a cell before transformation to a neoplastic form is compatible with the relatively slow development of CRC from the precursor adenoma and of the adenoma from normal epithelium (CRC is uncommon before 50 years of age). It must be emphasised that although COX-2 induction appears in this scheme there is no suggestion or evidence that the COX-2 gene or the protein itself is mutated in colonic epithelial cells during carcinogenesis.
Figure 2.
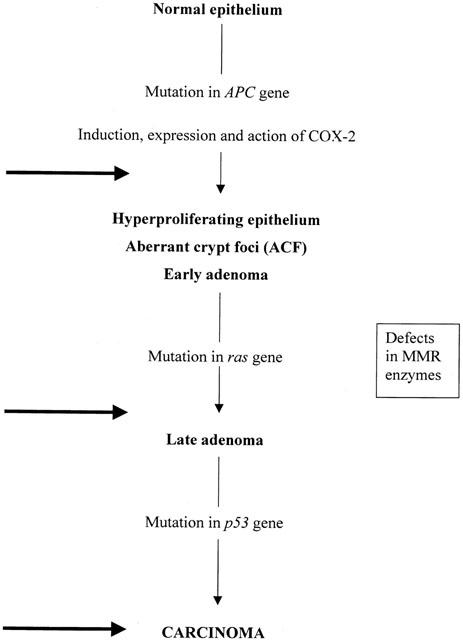
Carcinogenesis in CRC and action of NSAIDs or selective COX-2 inhibitors (C2Is). The progression from normal epithelium to carcinoma has several identifiable stages, some associated with particular genetic mutations with the loss of the related protein and its function. Loss of the APC gene product is accepted as the initiator of the whole process. Expression and consequent action of COX-2 in the APC-deficient cells is required for carcinogenesis to progress. The nature of the link between loss of APC and induction of COX-2 is not clear, e.g., is it direct or coincidental. Further progression from the early adenoma onwards is associated with mutations of RAS and of p53 but these mutations alone, i.e., in the absence of the APC mutation do not lead to carcinoma. At these stages, the defects in the DNA repair enzymes (MMR) appear to be expressed. The heavy arrows show the points at which inhibition of COX-2 will block progression. COX-2 activity is also needed for growth of transformed cells i.e. after carcinogenesis is complete.
Genetic models of CRC
There are many animal models of genetic faults relating to cancer (Alexander, 2000) and the most important for CRC is a model with mutations in the gene for the APC protein, with a phenotype closely resembling that of FAP (Shoemaker et al., 1997; Fodde & Smits, 2001). Such Min (multiple intestinal neoplasia) mice spontaneously developed intestinal polyposis which was prevented by NSAIDs (Boolbol et al., 1996; Jacoby et al., 1996; Nakatsugi et al., 1997). A more direct involvement of COX-2 with this phenotype was found when a strain of the Min mice was cross-bred with mice lacking the gene for COX-2 (Oshima et al., 1996). The number and size of the polyps was successively diminished in the Min mice lacking one or both genes for COX-2. The effect of COX-2 deletion on polyposis in the Min mice was comparable to that of treating Min mice with an experimental C2I. Subsequently both the C2Is in present clinical use were shown to decrease polyposis in APC mutant Min mice (Jacoby et al., 2001; Oshima et al., 2001). A new variation on the theme of polyp prevention in the Min mouse is combination chemoprevention (Torrance et al., 2000). Here the NSAID sulindac was used together with inhibitors of the epidermal growth factor (EGF) receptor kinase to give better polyp suppression than either agent used alone. It would be interesting to combine a C2I with the EGF kinase inhibitors in this model.
The close correlation between responses of the Min mouse models and human FAP to NSAIDs and C2Is has strengthened confidence in results obtained in this model, in spite of some differences between the phenotypes of the Min mouse and FAP (Alexander, 2000; Shoemaker et al., 1997).
Development of the COX-2 and cancer concept
The correlation between COX-2 and CRC emerged at a uniquely appropriate time, with several factors coming together from quite disparate sources. A number of case study reports showing a correlation of chronic NSAID use and a lower incidence of CRC appeared in the early '90s (Table 1). The epidemiological case for chronic NSAID use protecting against CRC was strong but there was no clear existing scientific explanation of these observations.
At the same time, COX-2 had been characterized as one of the proteins induced during the transformation of cells by the viral oncogene, v-src, and as a mitogen inducible protein (Xie et al., 1991; 1992; Kujubu et al., 1991).
Another highly significant contributory event was the small clinical trial of the NSAID sulindac in patients with FAP (Giardiello et al., 1993). Although this trial was short term (only 12 months), the marked reduction in polyp number and size while sulindac was taken together with the increase after the treatment was stopped, showed very clearly that inhibition of either COX-1 or COX-2 or both did have real benefit in this condition.
These independent findings were then synthesized into a testable hypothesis that a COX activity associated with growth and oncogene action was causally related to the protection offered by inhibitors of this enzymic activity against CRC. An early result crucial to the hypothesis was provided by Eberhart et al. (1994) who showed the presence of COX-2 in neoplastic tissue from CRC patients and its absence from adjacent histologically normal intestinal tissue. COX-1 was present in both normal and neoplastic tissue equally. In the many laboratory and clinical studies since then the presence and action of COX-2 has been causally related to carcinogenesis.
The strength of the correlation between COX-2 and CRC does not deny the efficacy of many other agents lacking COX-inhibitory actions (Laird et al., 1995; Levy, 1997), nor even the actions of NSAIDs and C2Is unrelated to inhibition of PG biosynthesis (Ahnen, 1998; Shiff & Rigas, 1999). The other isoform, COX-1, was also crucial in development of polyposis in Min mice (Chulada et al., 2000). Clearly with a multi-stage development of CRC, a similar multiplicity of inhibitory mechanisms could be expected. Nevertheless because the ‘COX-2 and cancer' hypothesis provides a new and clearly defined mechanism for, and identifies another stage in, the development of CRC, this hypothesis has become the basis for an important new therapeutic target in tumorigenesis in general.
Mechanisms and pathways involved in modulation of cancer by COX-2
PG-dependent and COX-2 dependent mechanisms appear to influence both the progression from normality to neoplasia, i.e., carcinogenesis and also the replication of neoplastic cells after transformation, in the epithelial cell. Here I shall discuss first the mechanisms that may influence the neoplastic transformation and then those that may control the growth of the tumour.
Apoptosis
Decreased apoptosis of epithelial cells appears to play a crucial role in the genesis of CRC. The ‘gate keeping' mutation required for the development of CRC is in the APC gene, which codes for a pro-apoptotic protein (Kinzler & Vogelstein, 1996; Shoemaker et al., 1997; Marks & Furstenberger, 2000; Fodde & Smits, 2001). Modulation of apoptosis by APC involves other proteins including β-catenin and E-cadherin (Fosslien, 2000; Marks & Furstenberger, 2000) and the Wnt signalling pathway (Bienz & Clevers, 2000; Polakis, 2000).
In Min mice, intestinal epithelium showed a decreased apoptotic rate, which was reversible by the NSAID sulindac (Mahmoud et al., 1997). A more direct connection between COX-2 and apoptosis was shown in rat intestinal epithelial cells (Tsujii & Dubois, 1995). In cultures of these rat cells, apoptosis was decreased after transfection with the COX-2 gene. This was accompanied by increased spontaneous output of PGE2 and increased levels of Bcl-2, another anti-apoptotic protein, suggesting a possible molecular mechanism for the pro-neoplastic effects of COX-2 action. Inhibition of PGE2 biosynthesis with sulindac restored the apoptotic rate. There was a direct effect of PGE2 on Bcl-2 and apoptosis in cell lines derived from human colon cancer samples (Sheng et al., 1998). In HCA-7 and HT 29 cell lines, NSAIDs and C2Is inhibited growth and induced apoptosis (Smith et al., 2000b).
Another colon cancer cell line, HCT-29, expresses a truncated APC protein and high levels of COX-2 (Hsi et al., 1999). After transfection with a normal APC gene, the cells exhibited increased apoptosis, decreased COX-2 and changes in β-catenin signalling pathways. These findings are compatible with the proposed ‘self promotion' interaction between APC and COX-2 in which decreased activity of the APC protein increased COX-2 biosynthesis (Prescott & White, 1996).
Peroxisome proliferator-activated receptors (PPARs)
There are three different PPARs – α, β (or δ), and γ – and each acts as a transcription factor controlling gene expression as a heterodimer with another nuclear protein, the retinoic acid receptor, (RXR; Figure 3). Each PPAR affects a different range of genes, most of which are generally involved in lipid metabolism (Lowell, 1999; Clarke et al., 1999; Kersten et al., 2000; Willson et al., 2000). In the present context, although PPAR β/δ may be involved (Kersten et al., 2000; Gupta et al., 2000), most attention has been paid to PPARγ (Debril et al., 2001). This PPAR is particularly involved with PG action because of the high potency of the cyclopentenone PGs derived from PGD2 – PGJ2 and its further derivatives, particularly 15-deoxy-delta 12,14-PGJ2 (15dPGJ2) – as agonist ligands for this nuclear receptor (Kliewer et al., 1995; Forman et al., 1995).
Figure 3.
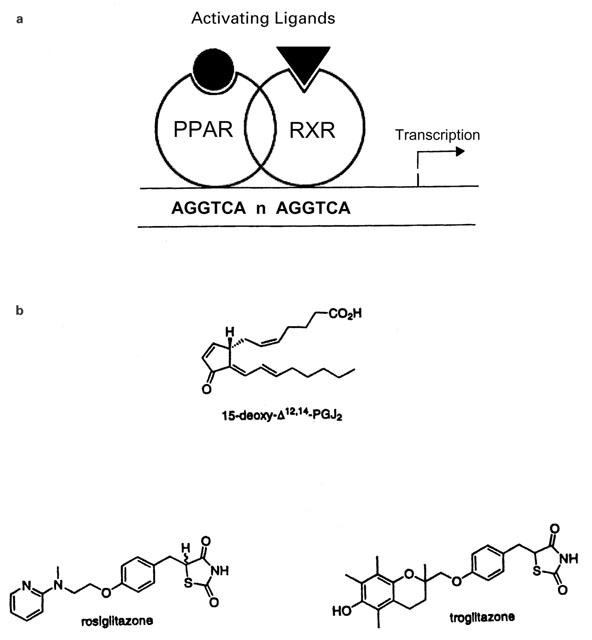
The PPAR nuclear receptor is one part of a transcription factor complex. In (A) the heterodimer of a PPAR and a retinoic acid receptor, RXR, is shown binding to the DNA sequence that functions as a response element for these proteins. Both receptors require activating ligands to bind other co-activating proteins necessary for the transcription of the downstream gene. In (B) two types of activating ligand for the PPARγ are shown. The PGJ2 derivative is the most potent endogenous agonist known so far; long-chain fatty acids (arachidonate, linoleinate, eicosapentenoate) are also agonists. Several synthetic thiazolidinediones (TZDs) are agonists with sub-micromolar potency and two of those at present used as anti-diabetic therapy are also shown.
PPARγ
The originally described function of the cyclopentenone PG ligands for PPARγ was to drive the differentiation of adipocytes from precursor cells (Kliewer et al., 1995; Forman et al., 1995). Many other fatty acids are less potent ligands for PPARγ, including another oxidized metabolite of AA, 15-HETE (Kersten et al., 2000). Another group of highly active agonist ligands are the thiazolidinediones (TZDs) such as troglitazone and rosiglitazone, but the link between activation of PPARγ and anti-diabetic action is not clear (Murphy & Holder, 2000).
A pro-carcinogenic role for activation of PPARs and cancer was originally proposed on the basis of the correlation of CRC with dietary fat (Hill, 1999; Reddy, 2000), the latter providing the agonist ligands for the PPARs. Later, work using Min mice showed that two TZD agonists of PPARγ, troglitazone and rosiglitazone, increased the numbers of polyps in the colon (Saez et al., 1998; Lefebvre et al., 1998). This coupled with the agonist potency of PGs and the anti-carcinogenic effect of C2Is in Min mice suggested that a PG agonist for PPARs activates pro-carcinogenic mechanisms and that the PG concerned is derived from COX-2. A potential positive feedback loop is suggested by the induction of COX-2 by PPARγ agonists and identification of a PPAR-response element in the promoter region of the COX-2 gene in epithelial cells (Meade et al., 1999).
An opposing hypothesis that stimulation of PPARγ is anti-carcinogenic also has experimental support. Early reports established the anti-neoplastic and anti-proliferative effects of PGD2 and PGJ2 (Fukushima, 1992; Negishi et al., 1995) before these PGs were recognized as ligands for PPARγ (Kliewer et al., 1995; Forman et al., 1995). The TZD troglitazone inhibited growth of several CRC cell lines in culture and of implants of these cells in mice (Sarraf et al., 1998). In mammary cancer cell lines, both troglitazone and 15dPGJ2 inhibited proliferation and induced apoptosis (Clay et al., 1999). This action was independent of the oestrogen responsiveness of the cells. Recently, a synthetic, highly selective and potent non-TZD PPARγ agonist, GW7845, decreased tumour incidence, weight, and number in chemically induced mammary cancer in rats (Suh et al., 1999).
These conflicting results must be resolved if action at PPARγ is to provide viable explanations for the anti-carcinogenic effects of the C2Is. Further the procarcinogenic effects of TZDs in Min mice raises questions about their long-term effects in the elderly for the treatment of Type 2 diabetes. It is also tempting to speculate that the recent correlation between cancer and obesity (WHO, 2001) could be attributed to stimulation of PPARγ driving both adipocyte formation and cancer growth.
Angiogenesis
Most solid tumours require new blood vessels to provide the nutrients necessary to ensure growth and survival (Holmgren, 1996). The provision of this new blood supply – angiogenesis – is also a crucial determinant of metastasis. Angiogenic factors could be secreted either by the tumour cells themselves or by the adjacent host cells and would elicit the new outgrowth of the host blood vessels (Augustin, 1998). Such pro-angiogenic actions of colon cancer cells were shown with HCA-7 or Caco-2 cells over expressing COX-2 [Caco-2+COX-2], co-cultured with endothelial cells (Tsujii et al., 1998). Formation of endothelial cell tubules (an angiogenic response) was increased with [Caco-2+COX-2] cells and these cells also secreted 4 fold more angiogenic factors than the parent Caco-2 cells. Both tubule formation and secretion of angiogenic factors was inhibited by the addition of C2Is or aspirin. The crucial contribution of the host cells was clearly demonstrated by the marked inhibition of growth of Lewis lung carcinoma cells in mice with COX-2 deletions, whereas COX-1 deletions had no effect relative to the wild type strain (Williams et al., 2000). Vascularity of the tumours (Factor VIII staining) in the COX-2 null mice was also reduced by about 30% compared with those in the wild type strain.
Histological analysis of metastases derived from HT29 or Lewis lung carcinoma cells showed COX-2 in the adjacent normal host blood vessels and in the tumour neovasculature but not in the tumour cells themselves (Masferrer et al., 2000). Incidence of metastasis from either HT29 or Lewis lung carcinomas was markedly reduced by treatment with a C2I, celecoxib. There is thus strong evidence to support the positive correlation between COX-2 activity, angiogenesis and primary or metastatic tumour growth.
Nevertheless, it is important to remember that angiogenesis in the adult is a normal physiological response, as, for instance, in wound healing or endometrial development during the menstrual cycle, and is driven by the same endogenous angiogenic signal molecules which stimulate tumour angiogenesis. In models of non-tumour angiogenesis, COX-2 has also been implicated. For instance, neovascularisation in rat cornea stimulated by exogenous bFGF was strongly inhibited by a C2I, celecoxib but not by a selective inhibitor of COX-1, SC 560 (Masferrer et al., 2000). Healing of gastric ulcers is also delayed by C2Is (Schmassman et al., 1998; Jones et al., 1999). Inhibition of physiological angiogenesis would then be a possible side effect of COX-2 inhibition in treatment of tumour angiogenesis in CRC or other cancers.
Effects of COX-2 on immunological responses
Many cancers are associated with ‘immunosuppressed' states, often expressed by changes in secretion of immuno-active cytokines. Thus IL-10 is increased and IL-12, TNF and IL-1 decreased in experimental models of cancer and these changes have been linked to increased synthesis of PGE2 and COX-2 activity (Kambayashi et al., 1995; Shattuck-Brandt et al., 2000; Stolina et al., 2000). In CRC patients, a PGE2-mediated immunosuppression has been observed (Balch et al., 1984). In this ‘immuno-suppressive' mode, PGE2 or other products of COX-2 may act via PPARγ as activation of this nuclear receptor by 15dPGJ2 and other non-PG agonists reduced output of the inflammatory cytokines, IL-1, TNFα and IL-6 (Ricote et al., 1999). Another mode of action would be by interference with the expression of immunological receptors. Expression of HLA antigens was decreased in samples of human CRC and adenomas (Tsioulias et al., 1992; 1993). Exposure of SW 116 cells to PGE2 downregulated the HLA-DR (class II) antigen and aspirin induced expression of this antigen on HT 29 cells (Arvind et al., 1995).
Effects of COX-2 not mediated by PGs
Although most of the biological consequences of COX-catalysed oxidation of arachidonate are attributable to the formation of PGs, this may not always be so in carcinogenesis. For instance the oxidation of arachidonate by COX generates other oxidative species and thus raises the overall oxidative state of the cell. One consequence of this is that COX-2 will co-oxidise compounds such as benzo [a] pyrene (Eling et al., 1990) to highly carcinogenic derivatives. Direct oxidative damage to DNA is a well recognized mutagenic event and such damage is increased following induction of COX-2 (Nikolic & van Breeman, 2001). Another cellular oxidation-response is the activation of NFkB known to be a transcription factor for COX-2 (Lim et al., 2001; Hardwick et al., 2001), setting up a possible positive feedback loop to maintain high oxidative levels in the cell. Accumulation of free arachidonate may also be involved since this appeared to be the trigger for apoptosis in HCT 116 cells, experimental mammary tumours or clinical samples of CRC (Chan et al., 1998; Trimboli et al., 1999; Cao et al., 2001). However, all these possible alternative mechanisms would still be susceptible to inhibition of COX-2 activity by selective inhibitors.
Prevention and treatment of CRC
Existing programmes
Prevention schemes for CRC are at present based on early detection of polyps or tumours. Typically, they comprise a combination of genetic analysis to identify ‘at risk' groups followed by physical screening – faecal blood tests (FOBT), sigmoidoscopy or, if justified, colonoscopy (Midgley & Kerr, 1999; Podolsky, 2000, Dove-Edwin & Thomas, 2001).
The most at risk group consists of the families with FAP; these would be the most intensively screened. A lesser risk group are the first-degree relatives of those with diagnosed HNPCC. A much larger third group (maybe as high as 5% of the population in the U.S.) comprises those with a family history of CRC, but which do not fall into either of the first two categories.
The treatment of diagnosed CRC is still resection of the affected bowel but although macroscopic clearance of tumours is good (80% in colon cancer), recurrence rates are high. Recurrence in the bowel may be treated by further resection in a small proportion of cases (3 – 5%) but usually the treatment is radiotherapy and chemotherapy. Chemotherapy is also given as an adjuvant therapy immediately after resection to minimise the development of metastases already disseminated (Leen et al., 2000). The main chemotherapeutic agent is 5-fluorouracil (5-FU) which interferes with thymidine incorporation into DNA. This and other cytotoxic agents (ralitrexed, oxaplatin and the newer topoisomerase inhibitors such as irinotecan) exert characteristic and unpleasant side effects such as diarrhoea, mucositis, alopecia and neutropaenia (Midgley & Kerr, 1999).
What is the role of COX-2 inhibition in prevention and treatment of CRC?
Chemoprevention programmes for CRC have hardly started and any programme based on COX-2 inhibition would reflect the ‘at risk' assessments already described above (Steinbach et al., 2000; Lynch, 2001). Indeed, the value of COX-2 inhibition has already been recognized for FAP (Steinbach et al., 2000) and such inhibitors may be used to decrease polyp number before colectomy or post-operatively to suppress growth of the polyps remaining. Although colectomy will remain the treatment of choice in FAP, it may be possible to delay that intervention, safely, by early ‘prophylactic' use of C2Is in those who have the characteristic genetic profile but no physical signs.
Extension of the use of C2Is in the close relatives of those with HNPCC who are symptom-free is certainly worth a clinical trial, bearing in mind the inherently low side effect profile of these compounds. Furthermore, since HNPCC is also associated with tumours elsewhere in the body (Midgley & Kerr, 1999; Emery et al., 2001) and COX-2 expression is elevated in cervical and endometrial tumour tissue (Kulkarni et al., 2001; Munir et al., 2000), as well from many other sites (Table 2), extra-colonic action of C2Is could be beneficial.
Table 2.
Association of COX-2 with cancer in other tissues

Wider, more general chemoprevention of CRC with C2Is would ideally take the form of a prophylactic daily dose in, say, all those over 50 years old. The theoretical justification of this much wider use comes from the clear results from epidemiological analysis of NSAID use and CRC incidence (Table 1). These by themselves would have been enough to initiate clinical trials but for the low tolerability of the NSAIDs in subjects in the appropriate age range but otherwise symptom-free. The much lower potential of the C2Is for gastro-intestinal side effects have already been demonstrated (Simon et al., 1999; Bombardier et al., 2000). Direct support for a general chemopreventive application should be available from an analysis of those using C2Is for chronic inflammatory disease (rheumatoid or osteoarthritis), by analogy with the analyses already carried out for the NSAIDs (Thun, 1996). Refinement of this ‘shotgun', age-based, approach to CRC prevention may also be possible with recent advances in gene analysis, using, for instance, screening for germline mutations in MMR enzymes or other new data from the Human Genome Project.
Treatment
For most forms of CRC, resection will remain the preferred treatment. However, there is enough evidence from experimental models to propose a role for C2Is in treatment of diagnosed CRC. At present, the most logical place for C2Is is in adjuvant therapy mixtures given after resection. Several advantages would accrue. First, the low side effect profile of the C2Is would create little or no additional side effect to those already associated with the cytotoxic agents, routinely used. The anti-inflammatory effects of the C2Is may actually alleviate some of these side effects, which have inflammatory aspects (diarrhoea, mucositis). The anti-metastatic effects of C2Is in experimental systems (Masferrer et al., 2000) would be valuable in suppressing metastases which are frequently the cause of recurrence after potentially curative resections (Leen et al., 2000; Midgley & Kerr, 1999). Finally, there is experimental evidence for radio-sensitization by C2Is (Milas et al., 1999; Kishi et al., 2000; Gallo, 2000) and such an effect would be beneficial in those recurrences that are treated with radiotherapy. All these would be in addition to any inhibition of adenoma or tumour growth exerted directly by the C2Is.
The first trials of C2Is in treatment of CRC would have to be as an addition to the usual mixture of agents. It may however be possible to reduce the level of cytotoxic agents by combining them with the C2Is to achieve equal or better survival. This may need a careful selection of patients, perhaps by genetic screening to maximise benefit. The consequent reduction in side effect intensity due to the cytotoxic agents would have marked benefits for the patient and improve compliance and acceptance of the therapy.
COX-2 in other forms of cancer
Most of the work, experimental or clinical, relating COX-2 to cancer has involved CRC and, to a lesser extent, gastric and oesophageal cancer. However this correlation has been tested in a wider range of neoplastic cells, generally those derived from epithelial cells in the organs involved, typically lung, prostate or breast (see Table 2). Although there is less work with the cancers in these organs and the conclusions are correspondingly less secure, overall, there is general support for the correlation. Thus, in clinical samples, there is more COX-2 in the tumour tissue than in normal tissue (Soslow et al., 2000). In cultures of cells derived from these human tumours, growth was inhibited by inhibitors of COX activity (Hsu et al., 2000). Finally, in models of chemically induced carcinogenesis, development of tumours was inhibited by NSAIDs or by C2Is (Alshafie et al., 2000).
There is one important practical difference between CRC and other cancers – the epidemiological evidence for protection with NSAID use, which is very clear for colon, rectal, gastric and oesophageal cancers, is much less strong for breast or lung cancer (Thun et al., 1993; Egan et al., 1996; Harris et al., 1996; Thun, 1996; Schreinermachers & Everson, 1999). Nonetheless there are positive findings, particularly in breast cancer, which deserve further attention.
The success of tamoxifen in chemoprevention of breast cancer (Decensi & Costa, 2000) and the links to the BRCA1 and BRCA2 genes (Brown & Lippman, 2000) should encourage further genetic correlations using the large library of samples already typed for BRCA which could now be assessed for COX-2. Further analyses could look for other signs of defects (such as microsatellite instability) in the genes for the MMR enzymes in breast cancer, bearing in mind that the phenotype of HNPCC also includes breast, uterine and ovarian tumours (Emery et al., 2001). Another very important feature of breast cancer is the hormonal responsiveness of the tumour which is frequently lost in the more invasive forms. A particular advantage of the C2Is is that they are equally effective in oestrogen-sensitive or -resistant cell lines (Trimboli et al., 1999). If this result were to be substantiated clinically, it would give the C2Is a very valuable feature. Further, on the basis of experimental results, the C2Is are likely to inhibit endometrial cancer, and this is one of the major side effects of tamoxifen treatment. These benefits coupled with the inherently low side effect profile of the C2Is will strengthen the case for clinical trial of C2Is as a component of the chemopreventive mixture in breast cancer.
Adjuvant therapy in breast cancer is well established and a number of regimens have been validated (Hortobagyi, 2000). However the agents used have serious, though reversible, side effects and even now recurrence is a real problem. Another useful modality is pre-operative chemotherapy to reduce tumour size and induce a degree of regression. This may be combined with radiotherapy (Kishi et al., 2000). In all of these regimens C2Is could contribute positively at best and, at least, add little to the side effect burden of the existing therapies (Vainio & Morgan, 1998; Howe et al., 2001).
Concluding remarks
There is now enough strong and unequivocal evidence for a casual link between COX-2 and CRC, which could be extended to a significant proportion of other epithelial derived cancers. The use of C2Is in FAP patients (Steinbach et al., 2000) is proof of concept in a small sub-group of the susceptible population and the extension of C2I use into the adjuvant chemotherapy mixture for CRC should follow soon. The low level of side effects associated with C2Is relative either to the old NSAIDs or to the usual cytotoxic agents (5-FU, irinotecan, etc) will be a key component of the tolerability of C2I therapy – at least do no harm. More and further analysis of mammary and lung cancers, two major and intractable forms of human disease, by study both of human samples and of experimental models can only enlarge the therapeutic possibilities of the C2Is, even if they turn out to be effective in a small proportion of the cases.
Finally, the experimental approaches and models involving COX-2 that are now available have expanded our knowledge and understanding of neoplasia and their potential has not yet been exhausted nor even fully realized. The roles of COX-2, PGE2 synthase, 5- and 15-lipoxygenase and other enzymes of fatty acid oxidation, when fully elucidated, will add significantly to our understanding of carcinogenesis. Even the demonstration of non-PG mechanisms exerted by the NSAIDs or C2Is and their congeners will open new possibilities and suggest new approaches to the central question of growth control in neoplasia.
Not all CRC or all epithelial cell-derived cancers will be dependent on COX-2 in their development. The variable phenotypes of the same genotype seen in Min mice (Shoemaker et al., 1997; Hong et al., 2001) will undoubtedly be reproduced in the genetically much more diverse human population and there may indeed be even more variation (Spirio et al., 1996; Tomlinson et al., 1996). In this context it would be valuable to screen CRC patients for genetic variants in phospholipases, as well as mutations in the genes for APC and MMR enzymes. This programme would be analogous to the pharmacogenomic analysis of receptors that is presently being undertaken to improve treatment of hypertension and mental illness. The techniques for such genetic screening are available and becoming increasingly routine and the enhancement of outcome resulting from better targeting would clearly justify the effort.
Inhibition of COX-2 will not be a panacea for cancer but it will at least provide a very significant therapy for a very significant proportion of the patient population. At best, that population may actually decrease, as chemoprevention by C2Is becomes a clinical reality.
Acknowledgments
It is a pleasure to acknowledge the efforts of many of my colleagues in encouraging me to write this review and then in criticising what I had written, thereby greatly increasing its clarity and intelligibility. Nonetheless, the facts, interpretations and mistakes are solely my own responsibility.
Abbreviations
- APC
adenomatous polyposis coli
- bFGF
basic fibroblast growth factor
- COX
cyclooxygenase
- CRC
colorectal cancer
- C2I
selective inhibitor of COX-2
- EGF
epidermal growth factor
- FAP
familial adenomatous polyposis
- FOBT
faecal occult blood tests
- 5-FU
5-fluorouracil
- HNPCC
hereditary non-polyposis colon cancer
- IL
interleukin
- LPS
bacterial lipopolysaccharide
- Min
multiple intestinal neoplasia
- MMR
mismatch repair
- NFκB
nuclear factor kappa B
- NSAID
non-steroid anti-inflammatory drug
- PG
prostaglandin
- PGI2
prostacyclin
- 15dPGJ2
15-deoxy-delta 12,14-PGJ2;
- PPAR
peroxisome proliferator-activated receptor
- RXR
retinoic acid receptor
- TNFα
tumour necrosis factor alpha
- TxA2
thromboxane A2
- TZD
thiazolidinedione
References
- AHNEN D.J. Colon cancer prevention by NSAIDs: what is the mechanism of action. Eur. J. Surg. 1998;164 Suppl. 582:111–114. doi: 10.1080/11024159850191544. [DOI] [PubMed] [Google Scholar]
- ALEXANDER J. Use of transgenic mice in identifying chemopreventive agents. Toxicol. Lett. 2000;112–113:507–512. doi: 10.1016/s0378-4274(99)00213-1. [DOI] [PubMed] [Google Scholar]
- ALSHAFIE G., ABOU-ISSA H.M., SEIBERT K., HARRIS R.E. Chemotherapeutic evaluation of Celecoxib, a cyclo-oxygenase-2 inhibitor in a rat mammary tumor model. Oncol. Rep. 2000;7:1377–1381. doi: 10.3892/or.7.6.1377. [DOI] [PubMed] [Google Scholar]
- ARVIND P., PAPAVASSILIOU E.D., TSIOULIAS G.T., QIAO L., LOVELACE C.I.P., DUCEMAN B., RIGAS B. Prostaglandin E2 down-regulates the expression of HLA-DR antigen in human colon adenocarcinoma cell lines. Biochemistry. 1995;34:5604–5609. doi: 10.1021/bi00016a035. [DOI] [PubMed] [Google Scholar]
- ATTIGA F.A., FERNANDEZ P.M., WEERATNA A.T., MANYAK M.J., PATIERNO S.R. Inhibitors of prostaglandin synthesis inhibit human prostate tumor cell invasiveness and reduce the release of matrix metalloproteinases. Cancer Res. 2000;60:4629–4637. [PubMed] [Google Scholar]
- AUGUSTIN H.G. Antiangiogenic tumour therapy: will it work. Trends in Pharmacological Sciences. 1998;19:216–222. doi: 10.1016/s0165-6147(98)01211-5. [DOI] [PubMed] [Google Scholar]
- BAKHLE Y.S. Structure of COX-1 and COX-2 enzymes and their interaction with inhibitors. Drugs of Today. 1999;35:237–250. doi: 10.1358/dot.1999.35.4-5.552200. [DOI] [PubMed] [Google Scholar]
- BAKHLE Y.S., BOTTING R.M. Cyclooxygenase-2 and its regulation in inflammation. Mediat. Inflamm. 1996;5:305–323. doi: 10.1155/S0962935196000452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BALCH C.M., DOUGHERTY P.A., CLOUD G.A., TILDEN A.B. Prostaglandin E2-mediated suppression of cellular immunity in colon cancer patients. Surgery. 1984;95:71–77. [PubMed] [Google Scholar]
- BIENZ M., CLEVERS H. Linking colorectal cancer to Wnt signalling. Cell. 2000;103:311–320. doi: 10.1016/s0092-8674(00)00122-7. [DOI] [PubMed] [Google Scholar]
- BOMBARDIER C., LAINE L., REICIN A., SHAPIRO D., BURGOS-VARGAS R., DAVIS B., DAY R., FERRAZ M.B., HAWKEY C.J., HOCHBERG M.C., KVIEN T.K., SCHNITZER T.J. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis; VIGOR Study Group. N. Engl. J. Med. 2000;343:1520–1528. doi: 10.1056/NEJM200011233432103. [DOI] [PubMed] [Google Scholar]
- BOOLBOL S.K., DANNENBERG A.J., CHADBURN A., MARTUCCI C., GUO X.J., RAMONETTI J.T., ABREU-GORIS M., NEWMARK H.L., LIPKIN M.L., DECOSSE J.J., BERTAGNOLLI M.M. Cyclooxygenase-2 overexpression and tumor formation are blocked by sulindac in a murine model of familial adenomatous polyposis. Cancer Res. 1996;56:2556–2560. [PubMed] [Google Scholar]
- BROWN P.H., LIPPMAN S.M. Chemoprevention of breast cancer. Breast Cancer Res. Treat. 2000;62:1–17. doi: 10.1023/a:1006484604454. [DOI] [PubMed] [Google Scholar]
- CAO Y., PEARMAN A.T., ZIMMERMAN G.A., MCINTYRE T.M., PRESCOTT S.M. Intracellular unesterified arachidonic acid signals apoptosis. Proc. Natl. Acad. Sci. U.S.A. 2001;97:11280–11285. doi: 10.1073/pnas.200367597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAN C.C., BOYCE S., BRIDEAU C. Rofecoxib (Vioxx, MK-0966; 4-(4′-methylsulfonylphenyl)-3-phenyl-2(5H)-furanone: a potent and orally active cyclooxygenase inhibitor. Pharmacological and biochemical profiles. J. Pharm. Exp. Ther. 1999;290:551–560. [PubMed] [Google Scholar]
- CHAN T.A., MORIN P.J., VOGELSTEIN B., KINZLER K.W. Mechanisms underlying nonsteroidal antiinflammatory drug-mediated apoptosis. Proc. Natl. Acad. Sci. U.S.A. 1998;95:681–686. doi: 10.1073/pnas.95.2.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHULADA P.C., THOMPSON M.B., MAHLER J.F., DOYLE C.M., GAUL B.W., LEE C., TIANO H.F., MORHAM S.G., SMITHIES O., LANGENBACH R. Genetic disruption of Ptgs-1, as well as Ptgs-2, reduces intestinal tumorigenesis in Min mice. Cancer Res. 2000;60:4705–4708. [PubMed] [Google Scholar]
- CLARKE S.D., THUILLIER P., BAILIE R.A., SHA X. Peroxisome proliferator-activated receptors: a family of lipid-activated transcription factors. Am. J. Clin. Nutr. 1999;70:566–571. doi: 10.1093/ajcn/70.4.566. [DOI] [PubMed] [Google Scholar]
- CLAY C.E., NAMEN A.M., ATSUMI G., WILLINGHAM M.C., HIGH K.P., KUTE T.E., TRIMBOLI A.J., FONTEH A.N., DAWSON P.A., CHILTON F.H. Influence of J series prostaglandins on apoptosis and tumorigenesis of breast cancer cells. Carcinogenesis. 1999;20:1905–1911. doi: 10.1093/carcin/20.10.1905. [DOI] [PubMed] [Google Scholar]
- CUENDET M., PEZZUTO J.M. The role of cyclooxygenase and lipoxygenase in cancer chemoprevention. Drug Metabol. Drug Interact. 2000;17:109–157. doi: 10.1515/dmdi.2000.17.1-4.109. [DOI] [PubMed] [Google Scholar]
- DEBRIL M.B., RENAUD J.P., FAJAS L., AUWERX J. The pleiotropic functions of peroxisome proliferator-activated receptor gamma. J. Mol. Med. 2001;79:30–47. doi: 10.1007/s001090000145. [DOI] [PubMed] [Google Scholar]
- DECENSI A., COSTA A. Recent advances in cancer chemoprevention, with emphasis on breast and colorectal cancer. Eur. J. Cancer. 2000;36:694–709. doi: 10.1016/s0959-8049(00)00040-x. [DOI] [PubMed] [Google Scholar]
- DOHADWALA M., LUO J., ZHU L., LIN Y., DOUGHERTY G.J., SHARMA S., HUANG M., POLD M., BATRA R.K., DUBINETT S.M. Non-small cell lung cancer cyclooxygenase-2 -dependent invasion is mediated by CD44. J. Biol. Chem. 2001;276:20809–20812. doi: 10.1074/jbc.C100140200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOVE-EDWIN I., THOMAS H.J. The prevention of colorectal cancer. Aliment. Pharmacol. Ther. 2001;15:323–336. doi: 10.1046/j.1365-2036.2001.00934.x. [DOI] [PubMed] [Google Scholar]
- DUBOIS R.N., GIARDIELLO F.M., SMALLEY W.E. Nonsteroidal anti-inflammatory drugs, eicosanoids, and colorectal cancer prevention. Gastroenterol. Clin. North. Am. 1996;25:773–791. doi: 10.1016/s0889-8553(05)70274-0. [DOI] [PubMed] [Google Scholar]
- EBERHART C.E., COFFEY R.J., RADHIKA A., GIARDIELLO F.M., FERRENBACH S., DUBOIS R.N. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107:1183–1188. doi: 10.1016/0016-5085(94)90246-1. [DOI] [PubMed] [Google Scholar]
- EGAN K.M., STAMPFER M.J., GIOVANNUCCI E., ROSNER B.A., COLDITZ G.A. Prospective study of regular aspirin use and the risk of breast cancer. J. Natl. Cancer. Inst. 1996;88:988–993. doi: 10.1093/jnci/88.14.988. [DOI] [PubMed] [Google Scholar]
- ELI Y., PRZEDECKI F., LEVIN G., KARIV N., RAZ A. Comparative effects of indomethacin on cell proliferation and cell cycle progression in tumor cells grown in vitro and in vivo. Biochem. Pharmacol. 2001;61:565–571. doi: 10.1016/s0006-2952(00)00578-5. [DOI] [PubMed] [Google Scholar]
- ELING T.E., THOMPSON D.C., FOUREMAN G.L., CURTIS J.F., HUGHES M.F. Prostaglandin H synthase and xenobiotic oxidation. Annu. Rev. Pharmacol. Toxicol. 1990;30:1–45. doi: 10.1146/annurev.pa.30.040190.000245. [DOI] [PubMed] [Google Scholar]
- EMERY J., LUCASSEN A., MURPHY M. Common hereditary cancers and implications for primary care. Lancet. 2001;358:56–63. doi: 10.1016/S0140-6736(00)05257-0. [DOI] [PubMed] [Google Scholar]
- FODDE R., SMITS R. Disease model: familial adenomatous polyposis. Trends in Molecular Medicine. 2001;7:369–373. doi: 10.1016/s1471-4914(01)02050-0. [DOI] [PubMed] [Google Scholar]
- FORMAN B.M., TONTONOZ P., CHEN J., BRUN R.P., SPIEGELMAN B.M., EVANS R.M. 15-Deoxy-delta-12,14-prostaglandin J2 is a ligand for the adipocyte determination factor PPARgamma. Cell. 1995;83:803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- FOSSLIEN E. Molecular pathology of cyclooxygenase-2 in neoplasia. Ann. Clin. Lab. Sci. 2000;30:3–21. [PubMed] [Google Scholar]
- FUKUSHIMA M. Biological activities and mechanisms of action of PGJ2 and related compounds; an update. Prostaglandins Leukot. Essent. Fatty Acids. 1992;47:1–12. doi: 10.1016/0952-3278(92)90178-l. [DOI] [PubMed] [Google Scholar]
- GAFFNEY D.K., HOLDEN S., DAVIS M., ZEMPOLICH K., MURPHY K.J., DODSON M. Elevated cyclo-oxoygenase-2 expression correlates with diminished survival in carcinoma of the cervix treated with radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2001;49:1213–1217. doi: 10.1016/s0360-3016(00)01583-2. [DOI] [PubMed] [Google Scholar]
- GALLO O. Re: Enhancement of tumor response to gamma-radiation by an inhibitor of cyclooxygenase-2 enzyme. J. Natl. Cancer. Inst. 2000;92:346–347. doi: 10.1093/jnci/92.4.346. [DOI] [PubMed] [Google Scholar]
- GATELY S. The contributions of cyclooxygenase-2 to tumor angiogenesis. Cancer Metastasis Rev. 2000;19:19–27. doi: 10.1023/a:1026575610124. [DOI] [PubMed] [Google Scholar]
- GIARDIELLO F.M., HAMILTON S.R., KRUSH A.J., PIANTADOSI S., HYLIND L.M., CELANO P., BOOKER S.V., ROBINSON C.R., OFFERHAUS G.J.A. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N. Engl. J. Med. 1993;328:1313–1316. doi: 10.1056/NEJM199305063281805. [DOI] [PubMed] [Google Scholar]
- GIOVANNUCCI E., EGAN K.M., HUNTER D.J., STAMPFER M.J., COLDITZ G.A., WILLETT W.C., SPEIZER F.E. Aspirin and the risk of colorectal cancer in women. N. Engl. J. Med. 1995;333:609–614. doi: 10.1056/NEJM199509073331001. [DOI] [PubMed] [Google Scholar]
- GIOVANNUCCI E., RIMM E.B., STAMPFER M.J., COLDITZ G.A., ASCHERIO A., WILLETT W.C. Aspirin use and the risk for colorectal cancer and adenoma in male health professionals. Ann. Intern. Med. 1994;121:241–246. doi: 10.7326/0003-4819-121-4-199408150-00001. [DOI] [PubMed] [Google Scholar]
- GRUBBS C.J., LUBET R.A., KOKI A.T., LEAHY K.M., MASFERRER J.L., STEELE V.E., KELLOFF G.J., HILL D.L., SEIBERT K. Celecoxib inhibits N-butyl-N-(4-hydroxybutyl)-nitrosamine-induced urinary bladder cancers in male B6D2F1 mice and female Fischer-344 rats. Cancer Res. 2000;60:5599–5602. [PubMed] [Google Scholar]
- GUPTA R.A., TAN J., KRAUSE W.F., GERACI M.W., WILLSON T.M., DEY S.K., DUBOIS R.N. Prostacyclin-mediated activation of peroxisome proliferator-activated receptor δ in colorectal cancer. Proc. Natl. Acad. Sci. U.S.A. 2000;97:13275–13280. doi: 10.1073/pnas.97.24.13275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARDWICK J.C., VAN DEN BRINK G.R., OFFERHAUS G.J.A., VAN DEVENTER S.J., PEPPELENBOSCH M.P. NF-kappaB, p38 MAPK and JNK are highly expressed and active in the stroma of human colonic adenomatous polyps. Oncogene. 2001;20:819–827. doi: 10.1038/sj.onc.1204162. [DOI] [PubMed] [Google Scholar]
- HARRIS R.E., NAMBOODIRI M.M., FARRAR W.D. Non-steroidal anti-inflammatory drugs and breast cancer. Epidemiology. 1996;7:203–205. doi: 10.1097/00001648-199603000-00017. [DOI] [PubMed] [Google Scholar]
- HIDA T., KOZAKI K., MURAMATSU H., MASUDA A., SHIMIZU S., MITSUDOMI T., SUGIURA T., OGAWA M., TAKAHASHI T. Cyclooxygenase-2 inhibitor induces apoptosis and enhances cytotoxicity of various anticancer agents in non-small cell lung cancer cell lines. Clin. Cancer Res. 2000;6:2006–2011. [PubMed] [Google Scholar]
- HIGASHI Y., KANEKURA T., KANZAKI T. Enhanced expression of cyclo-oxygenase (COX)-2 in human skin epidermal cancer cells: evidence for growth suppression by inhibiting COX-2 expression. Int. J. Cancer. 2000;86:667–671. doi: 10.1002/(sici)1097-0215(20000601)86:5<667::aid-ijc10>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- HILL J. Mechanisms of diet and colon carcinogenesis. Eur. J. Cancer Prev. 1999;8:S95–S98. [PubMed] [Google Scholar]
- HOLMGREN L. Antiangiogenesis restricted tumor dormancy. Cancer Metastasis Rev. 1996;15:241–245. doi: 10.1007/BF00437478. [DOI] [PubMed] [Google Scholar]
- HONG K.H., BONVENTRE J.C., O'LEARY E., BONVENTRE J.V., LANDER E.S. Deletion of cytosolic phospholipase A(2) suppresses Apc(Min)-induced tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 2001;98:3935–3939. doi: 10.1073/pnas.051635898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HORTOBAGYI G. Adjuvant therapy for breast cancer. Annu. Rev. Med. 2000;51:377–392. doi: 10.1146/annurev.med.51.1.377. [DOI] [PubMed] [Google Scholar]
- HOSOMI Y., YOKOSE T., HIROSE Y., NAKAJIMA R., NAGAI K., NISHIWAKI Y., OCHIAI A. Increased cyclo-oxygenase-2 (COX-2) expression occurs frequently in precursor lesions of human adenocarcinoma of the lung. Lung Cancer. 2000;30:73–81. doi: 10.1016/s0169-5002(00)00132-x. [DOI] [PubMed] [Google Scholar]
- HOWE L.R., SUBBARAMAIAH K., BROWN A.M., DANNENBERG A.J. Cyclo-oxygenase-2: a target for the prevention and treatment of breast cancer. Endocr. Relat. Cancer. 2001;8:97–114. doi: 10.1677/erc.0.0080097. [DOI] [PubMed] [Google Scholar]
- HSI L.C., ANGERMAN-STEWART J., ELING T.E. Introduction of full-length APC modulates cyclo-oxygenase-2 expression in HT-29 human colorectal carcinoma cells at the translational level. Carcinogenesis. 1999;20:2045–2049. doi: 10.1093/carcin/20.11.2045. [DOI] [PubMed] [Google Scholar]
- HSU A.L., CHING T.T., WANG D.S., SONG X., RANGNEKAR V.M., CHEN C.S. The cyclo-oxygenase-2 inhibitor celecoxib induces apoptosis by blocking Akt activation in human prostate cancer cells independently of Bcl-2. J. Biol. Chem. 2000;275:11397–11403. doi: 10.1074/jbc.275.15.11397. [DOI] [PubMed] [Google Scholar]
- HUNG W.C., CHANG H.C., PAN M.R., LEE T.H., CHUANG L.Y. Induction of p21 (KIP1) as a mechanism underlying NS-398-induced growth inhibition in human lung cancer cells. Mol. Pharmacol. 2000;58:1398–1403. doi: 10.1124/mol.58.6.1398. [DOI] [PubMed] [Google Scholar]
- JAKOBSSON P.-J., THOREN S., MORGENSTERN R., SAMUELSSON B. Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc. Natl. Acad. Sci. U.S.A. 1999;96:7220–7225. doi: 10.1073/pnas.96.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JACOBY R.F., MARSHALL D.J., NEWTON M.A., NOVAKOVIC K., TUTSCH K., COLE C.E., LUBET R., KELLOFF G.J., VERMA A., MOSER A.R., DOVE W.F. Chemoprevention of spontaneous intestinal adenomas in the ApcMin mouse model by the nonsteroidal anti-inflammatory drug piroxicam. Cancer Res. 1996;56:710–714. [PubMed] [Google Scholar]
- JACOBY R.F., SEIBERT K., COLE C.E., KELLOFF G., LUBET R.A. The cyclo-oxygenase-2 inhibitor celecoxib is a potent preventive and therapeutic agent in the Min mouse model of adenomatous polyposis. Cancer Res. 2001;60:5040–5044. [PubMed] [Google Scholar]
- JONES M.K., WANG H., PESKAR B.M., LEVIN E., ITANI R.M., SARFEH I.J., TARNAWSKI A.S. Inhibition of angiogenesis by nonsteroidal anti-inflammatory drugs: insight into mechanisms and implications for cancer growth and ulcer healing. Nat. Med. 1999;5:1418–1423. doi: 10.1038/70995. [DOI] [PubMed] [Google Scholar]
- KAMBAYASHI T., ALEXANDER H.R., FONG M., STRASSMAN G. Potential involvement of IL-10 in suppressing tumor-associated macrophages; colon-26-derived prostaglandin E2 inhibits TNF-α release via a mechanism involving IL-10. J. Immunol. 1995;154:3383–3390. [PubMed] [Google Scholar]
- KANEKURA T., HIGASHI Y., KANZAKI T. Inhibitory effects of 9-cis-retinoic acid and pyrrolidinedithiocarbamate on cyclo-oxygenase(COX)-2 expression and cell growth in human skin squamous carcinoma cells. Cancer. Lett. 2000;161:177–183. doi: 10.1016/s0304-3835(00)00604-2. [DOI] [PubMed] [Google Scholar]
- KERSTEN S., DESVERGNE B., WAHLI W. Roles of PPARs in health and disease. Nature. 2000;405:421–424. doi: 10.1038/35013000. [DOI] [PubMed] [Google Scholar]
- KHURI F.R., WU H., KEMP B.L., LOTAN R., LIPPMAN S.M., FENG L., HONG W.K., XU X.C. Cyclo-oxygenase-2 overexpression is a marker of poor prognosis in Stage I non-small cell lung cancer. Clin. Cancer. Res. 2001;7:861–867. [PubMed] [Google Scholar]
- KINZLER K.W., VOGELSTEIN B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- KIRSCHENBAUM A., KLAUSNER A.P., LEE R., UNGER P., YAO S., LIU X.H., LEVINE A.C. Expression of cyclo-oxygenase-1 and cyclooxygenase-2 in the human prostate. Urology. 2000;56:671–676. doi: 10.1016/s0090-4295(00)00674-9. [DOI] [PubMed] [Google Scholar]
- KISHI K., PETERSEN S., PETERSEN C., HUNTER N., MASON K., MASFERRER J.L., TOFILON P.J., MILAS L. Preferential enhancement of tumor radioresponse by a cyclo-oxygenase-2 inhibitor. Cancer Res. 2000;60:1326–1331. [PubMed] [Google Scholar]
- KLIEWER S.A., LENHARD J.M., WILLSON T.M., PATEL I., MORRIS D.C., LEHMANN J.M. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell. 1995;83:813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- KUJUBU D.A., FLETCHER B.S., VARNUM B.C., LIM R.W., HERSCHMAN H.R. TIS10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclo-oxygenase homologue. J. Biol. Chem. 1991;266:12866–12872. [PubMed] [Google Scholar]
- KULKARNI S., RADER J.S., ZHANG F., LIAPIS H., KOKI A.T., MASFERRER J., SUBBARAMAIAH K., DANNENBERG A.J. Cyclo-oxygenase-2 is overexpressed in human cervical cancer. Clin. Cancer Res. 2001;7:429–434. [PubMed] [Google Scholar]
- KUNE G.A., WATSON A.J., WATSON L.F. Colorectal cancer risk, chronic illnesses, operations and medications: case control results from the Melbourne Colorectal Cancer Study. Cancer Res. 1988;48:4399–4404. [PubMed] [Google Scholar]
- KURUMBAIL R.G., STEVENS A.M., GIERSE J.K., MCDONALD J.J., STEGEMAN R.A., PAK J.Y., GILDEHAUS D., MIYASHIRO J.M., PENNING T.D., SEIBERT K., ISAKSON P.C., STALLINGS W.C. Structural basis for selective inhibition of cyclo-oxygenase-2 by anti-inflammatory agents. Nature. 1996;384:644–648. doi: 10.1038/384644a0. [DOI] [PubMed] [Google Scholar]
- LAIRD P.W., JACKSON-GRUSBY L., FAZELI A., DICKINSON S.L., JUNG W.E., LI E., WEINBERG R.A., JAENISCH R. Suppression of intestinal neoplasia by DNA hypomethylation. Cell. 1995;81:197–205. doi: 10.1016/0092-8674(95)90329-1. [DOI] [PubMed] [Google Scholar]
- LEE L.M., PAN C.C., CHENG C.J., CHI C.W., LIU T.Y. Expression of cyclo-oxygenase-2 in prostate adenocarcinoma and benign prostatic hyperplasia. Anticancer Res. 2001;21:1291–1294. [PubMed] [Google Scholar]
- LEEN E., GOLDBERG J.A., ANGERSON W.J., MCARDLE C.S. Potential role of Doppler perfusion index in selection of patients with colorectal cancer for adjuvant chemotherapy. Lancet. 2000;355:34–37. doi: 10.1016/S0140-6736(99)06322-9. [DOI] [PubMed] [Google Scholar]
- LEFEBVRE A.M., CHEN I., DESREUMAUX P., NAJIB J., FRUCHART J.C., GEBOES K., BRIGGS M., HEYMAN R., AUWERX J. Activation of the peroxisome proliferator-activated receptor gamma promotes the development of colon tumors in C57BL/6J-APCMin/+mice. Nat. Med. 1998;4:1053–1057. doi: 10.1038/2036. [DOI] [PubMed] [Google Scholar]
- LEVY G.N. Prostaglandin H synthases, non-steroidal anti-inflammatory drugs and colon cancer. FASEB, J. 1997;11:234–247. [PubMed] [Google Scholar]
- LIM J.W., KIM H., KIM K.H. Nuclear factor-kappa B regulates cyclooxygenase-2 expression and cell proliferation in human gastric cancer cells. Lab. Invest. 2001;81:349–360. doi: 10.1038/labinvest.3780243. [DOI] [PubMed] [Google Scholar]
- LIU X.H., KIRSCHENBAUM A., YAO S., LEE R., HOLLAND J.F., LEVINE A.C. Inhibition of cyclo-oxygenase-2 suppresses angiogenesis and the growth of prostate cancer in vivo. J. Urol. 2000;164:820–825. doi: 10.1097/00005392-200009010-00056. [DOI] [PubMed] [Google Scholar]
- LOGAN R.F., LITTLE J., HAWTIN P.G., HARDCASTLE J.D. Effect of aspirin and non-steroidal anti-inflammatory drugs on colorectal adenomas: case-control study of subjects participating in the Nottingham faecal occult blood screening programme. Brit. Med. J. 1993;307:285–289. doi: 10.1136/bmj.307.6899.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LORD R.V., DANENBERG K.D., DANENBERG P.V. Cyclo-oxygenase-2 in Barrett's esophagus, Barrett's adenocarcinomas, and esophageal SCC: ready for clinical trials. Am. J. Gastroenterol. 1999;94:2313–2315. doi: 10.1111/j.1572-0241.1999.02313.x. [DOI] [PubMed] [Google Scholar]
- LOWELL B.B. PPARγ: an essential regulator of adipogenesis and modulator of fat cell function. Cell. 1999;99:239–242. doi: 10.1016/s0092-8674(00)81654-2. [DOI] [PubMed] [Google Scholar]
- LUONG C., MILLER A., BARNETT J., CHOW J., RAMESHA C., BROWNER M.F. Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2. Nat Struc Biol. 1996;3:927–933. doi: 10.1038/nsb1196-927. [DOI] [PubMed] [Google Scholar]
- LYNCH P.M. COX-2 inhibition in clinical cancer prevention. Oncology, (Huntingt). 2001;15:21–26. [PubMed] [Google Scholar]
- MADAAN S., ABEL P.D., CHAUDHARY K.S., HEWITT R., STOTT M.A., STAMP G.W., LALANI E.N. Cytoplasmic induction and over-expression of cyclooxygenase-2 in human prostate cancer; implications for prevention and treatment. BJU. Int. 2000;86:736–741. doi: 10.1046/j.1464-410x.2000.00867.x. [DOI] [PubMed] [Google Scholar]
- MAHMOUD N.N., BOOLBOL S.K., BILINSKI R.T., MARTUCCI C., CHADBURN A., BERTAGNOLLI M.M. Apc gene mutation is associated with a dominant-negative effect upon intestinal cell migration. Cancer Res. 1997;57:5045–5050. [PubMed] [Google Scholar]
- MARKS F., FURSTENBERGER G. Cancer chemoprevention through interruption of multistage carcinogenesis. The lessons learnt by comparing mouse skin carcinogenesis and human large bowel cancer. Eur. J Cancer. 2000;36:314–329. doi: 10.1016/s0959-8049(99)00318-4. [DOI] [PubMed] [Google Scholar]
- MARROGI A.J., TRAVIS W.D., WELSH J.A., KHAN M.A., RAHIM H., TAZELAAR H., PAIROLERO P., TRASTEK V., JETT J., CAPAORASO N.E., LIOTTA L.A., HARRIS C.C. Nitric oxide synthase, cyclo-oxygenase 2 and vascular endothelial growth factor in the angiogenesis of non-small cell lung carcinoma. Clin. Cancer Res. 2000;6:4739–4744. [PubMed] [Google Scholar]
- MARTINEZ M.E., MCPHERSON R.S., LEVIN B., ANNEGERS J.F. Aspirin and other non-steroidal anti-inflammatory drugs and the risk of colorectal adenomatous polyps among endoscoped individuals. Cancer Epidemiol. Biomarkers Prev. 1995;4:703–707. [PubMed] [Google Scholar]
- MASFERRER J.L., LEAHY K.M., KOKI A.T., ZWEIFEL B.S., SETTLE S.L., WOERNER B.M., EDWARDS D.A., FLICKINGER A.G., MOORE R.J., SEIBERT K. Antiangiogenic and antitumor activities of cyclo-oxygenase-2 inhibitors. Cancer Res. 2000;60:1306–1311. [PubMed] [Google Scholar]
- MEADE E.A., MCINTYRE T.M., ZIMMERMAN G.A., PRESCOTT S.M. Peroxisome proliferators enhance cyclo-oxygenase-2 expression in epithelial cells. J. Biol. Chem. 1999;274:8328–8334. doi: 10.1074/jbc.274.12.8328. [DOI] [PubMed] [Google Scholar]
- MIDGLEY R., KERR D. Colorectal cancer. Lancet. 1999;353:391–399. doi: 10.1016/S0140-6736(98)07127-X. [DOI] [PubMed] [Google Scholar]
- MILAS L., KISHI K., HUNTER N., MASON K., MASFERRER J.L., TOFILON P.J. Enhancement of tumor response to gamma-radiation by an inhibitor of cyclooxygenase-2 enzyme. J. Natl. Cancer Inst. 1999;91:1501–1504. doi: 10.1093/jnci/91.17.1501. [DOI] [PubMed] [Google Scholar]
- MUNIR I., FUKUNAGA K., KANASAKI H., MIYAZAKI K., OHBA T., OKAMURA H., MIYAMOTO E. Expression of cyclo-oxygenase-2 by prostaglandin E2 in human endometrial adenocarcinoma cell line HEC-1B. Biol. Reprod. 2000;63:933–941. doi: 10.1095/biolreprod63.3.933. [DOI] [PubMed] [Google Scholar]
- MURAKAMI M., NARABA H., TANIOKA T., SEMMYO N., NAKATANI Y., KOJIMA F., IKEDA T., FUEKI M., UENO A., OH-ISHI S., KUDO I. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclo-oxygenase-2. J. Biol. Chem. 2000;275:32783–32792. doi: 10.1074/jbc.M003505200. [DOI] [PubMed] [Google Scholar]
- MURPHY G.J., HOLDER J.C. PPAR-γ agonists:therapeutic role in diabetes, inflammation and cancer. Trends in Pharmacological Sciences. 2000;21:469–474. doi: 10.1016/s0165-6147(00)01559-5. [DOI] [PubMed] [Google Scholar]
- MUSCAT J.E., STELLMAN S.D., WYNDER E.L. Nonsteroidal antiinflammatory agents and colorectal cancer. Cancer. 1994;74:1847–1854. doi: 10.1002/1097-0142(19941001)74:7<1847::aid-cncr2820740704>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- NAKATSUGI S., FUKUTAKE M., TAKAHASHI M., FUKUDA K., ISOI T., TANIGUCHI Y., SUGIMURA T., WAKABAYASHI K. Suppression of intestinal polyp development by nimesulide, a selective cyclo-oxygenase-2 inhibitor, in Min mice. Jpn. J. Cancer Res. 1997;88:1117–1120. doi: 10.1111/j.1349-7006.1997.tb00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAKATSUGI S., OHTA T., KAWAMORI T., MUTOH M., TANIGAWA T., WATANBE K., SUGIE S., SUGIMURA T., WAKABAYASHI K. Chemoprevention by nimesulide, a selective cyclo-oxygenase inhibitor, of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP)-induced mammary gland carcinogenesis in rats. Jpn. J. Cancer Res. 2000;91:886–892. doi: 10.1111/j.1349-7006.2000.tb01030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NARABA H., MURAKAMI M., MATSUMOTO H., SHIMBARA S., UENO A., KUDO I., OH-ISHI S. Segregated coupling of phospholipases A2, cyclo-oxygenases and terminal prostanoid synthases in different phases of prostanoid biosynthesis in rat peritoneal macrophages. J. Immunol. 1998;160:2974–2982. [PubMed] [Google Scholar]
- NEGISHI M., KOIZUMI T., ICHIKAWA A. Biological actions of delta-12-prostaglandin J2. J. Lipid Mediat. Cell Signal. 1995;12:443–448. doi: 10.1016/0929-7855(95)00029-p. [DOI] [PubMed] [Google Scholar]
- NIKOLIC D., VAN BREEMAN R.B. DNA oxidation induced by cyclo-oxygenase-2. Chem. Res. Toxicol. 2001;14:351–354. doi: 10.1021/tx010004x. [DOI] [PubMed] [Google Scholar]
- OSHIMA M., DINCHUK J.E., KARGMAN S.L., OSHIMA H., HANCOCK B., KWONG E., TRZASKOS J.M., EVANS J.F., TAKETO M.M. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclo-oxygenase 2 (COX-2) Cell. 1996;87:803–809. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- OSHIMA M., MURAI N., KARGMAN S., ARGUELLO M., LUK P., KWONG E., TAKETO M.M., EVANS J.F. Chemoprevention of intestinal polyposis in the Apcdelta716 mouse by rofecoxib, a specific cyclo-oxygenase-2 inhibitor. Cancer Res. 2001;61:1733–1740. [PubMed] [Google Scholar]
- PELEG I.I., LUBIN M.F., COTSONIS G.A., CLARK W.S., WILCOX C.M. Long-term use of nonsteroidal antiinflammatory drugs and other chemopreventors and risk of subsequent colorectal neoplasia. Dig. Dis. Sci. 1996;41:1319–1326. doi: 10.1007/BF02088554. [DOI] [PubMed] [Google Scholar]
- PELEG I.I., MAIBACH H.T., BROWN S.H., WILCOX C.M. Aspirin and nonsteroidal anti-inflammatory drug use and the risk of subsequent colorectal cancer. Arch. Intern. Med. 1994;154:394–399. [PubMed] [Google Scholar]
- PICOT D., LOLL P.J., GARAVITO R.M. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature. 1994;367:243–249. doi: 10.1038/367243a0. [DOI] [PubMed] [Google Scholar]
- PODOLSKY D.K. Going the distance - the case for true colorectal-cancer screening. N. Engl. J. Med. 2000;343:207–208. doi: 10.1056/NEJM200007203430309. [DOI] [PubMed] [Google Scholar]
- POLAKIS P. Wnt signalling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- PRESCOTT S.M., WHITE R.L. Self-promotion? Intimate connections between APC and prostaglandin H synthase. Cell. 1996;87:783–786. doi: 10.1016/s0092-8674(00)81983-2. [DOI] [PubMed] [Google Scholar]
- REDDY B.S. Novel approaches to the prevention of colon cancer by nutritional manipulation and chemoprevention. Cancer Epidemiol. Biomarkers. Prev. 2000;9:239–247. [PubMed] [Google Scholar]
- REDDY B.S., HIROSE Y., LUBET R., STEELE V., KELLOFF G., PAULSON S., SEIBERT K., RAO C.V. Chemoprevention of colon cancer by specific cyclooxygenase-2 inhibitor, celecoxib, administered during different stages of carcinogenesis. Cancer Res. 2000;60:293–297. [PubMed] [Google Scholar]
- RICOTE M., HUANG J.T., WELCH J.S., GLASS C.K. The peroxisome proliferator-activated receptorγ (PPARγ) as a regulator of monocyte/macrophage function. J. Leuk. Biol. 1999;66:733–739. doi: 10.1002/jlb.66.5.733. [DOI] [PubMed] [Google Scholar]
- RIENDEAU D., PERCIVAL M.D., BRIDEAU C., CHARLESON S., DUBE D., ETHIER D., FALGUEYRET J.-P., FRIESEN R.W., GORDON R., GREIG G., GUAY J., MANCINI J.A., OUELLET M., WONG E., XU L., BOYCE S., VISCO D., GIRARD Y., PRASIT P., RODGER I.W., GRESSIER M., FORD-HUTCHINSON A.W., YOUNG R.N., CHAN C.C. Etoricoxib (MK-0663): preclinical profile and comparison with other agents that selectively inhibit cyclooxygenase-2. J. Pharm. Exp. Ther. 2001;296:558–566. [PubMed] [Google Scholar]
- RISTIMAKI A., NIEMINEN O., SAUKKONEN K., HOTAKAINEN K., NORDLING S., HAGLUND C. Expression of cyclo-oxygenase-2 in human transitional cell carcinoma of the urinary bladder. Am. J. Pathol. 2001;158:849–853. doi: 10.1016/S0002-9440(10)64033-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROSENBERG L., PALMER J., ZAUBER A., WARSHAUER M., STOLLEY P., SHAPIRO S. A hypothesis: non-steroidal anti-inflammatory drugs reduce the incidence of large bowel cancer. J. Natl. Cancer Inst. 1991;83:355–358. doi: 10.1093/jnci/83.5.355. [DOI] [PubMed] [Google Scholar]
- RYU H.S., CHANG K.H., YANG H.W., KIM M.S., KWON H.C., OH K.S. High cyclo-oxygenase-2 expression in stage IB cervical cancer with lymph node metastasis or parametrial invasion. Gynecol. Oncol. 2000;76:320–325. doi: 10.1006/gyno.1999.5690. [DOI] [PubMed] [Google Scholar]
- SAEZ E., TONTONOZ P., NELSON M.C., ALVAREZ J.G.A., U T.M., BAIRD S.M., THOMASZY V.A., EVANS R.M. Activators of the nuclear receptor PPARγ enhance colon polyp formation. Nat. Med. 1998;4:1058–1061. doi: 10.1038/2042. [DOI] [PubMed] [Google Scholar]
- SARRAF P., MUELLER E., JONES D., KING F.J., DEANGELO D.J., PARTRIDGE J.B., HOLDEN S.A., CHEN L.B., SINGER S., FLETCHER C., SPIEGELMAN B.M. Differentiation and reversal of malignant changes in colon cancer through PPARγ. Nat. Med. 1998;4:1046–1052. doi: 10.1038/2030. [DOI] [PubMed] [Google Scholar]
- SCHMASSMAN A., PESKAR B.M., STETTLER C. Effects of inhibition of prostaglandin endoperoxide synthase-2 in chronic gastro-intestinal ulcer models in rats. Br. J. Pharmacol. 1998;123:795–804. doi: 10.1038/sj.bjp.0701672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHREINERMACHERS D.M., EVERSON R.B. Aspirin use and lung, colon and breast cancer incidence in a prospective study. Epidemiology. 1999;5:138–146. doi: 10.1097/00001648-199403000-00003. [DOI] [PubMed] [Google Scholar]
- SHATTUCK-BRANDT R.L., VARILEK G.W., RADHIKA A., YANG F., WASHINGTON M.K., DUBOIS R.N. Cyclooxygenase 2 expression is increased in the stroma of colon carcinomas from IL-10(−/−) mice. Gastroenterology. 2000;118:337–345. doi: 10.1016/s0016-5085(00)70216-2. [DOI] [PubMed] [Google Scholar]
- SHENG H., SHAO J., MORROW J.D., BEAUCHAMP R.D., DUBOIS R.N. Modulation of apoptosis and Bcl-2 expression by prostaglandin E2 in human colon cancer cells. Cancer Res. 1998;58:362–366. [PubMed] [Google Scholar]
- SHIFF S.J., RIGAS B. The role of cyclo-oxygenase inhibition in the antineoplastic effects of nonsteroidal anti-inflammatory drugs (NSAIDs) J. Exp. Med. 1999;190:445–450. doi: 10.1084/jem.190.4.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIOTA G., OKUBO M., NOUMI T., NOGUCHI N., OYAMA K., TAKANO Y., YASHIMA K., KISHIMOTO Y., KAWASAKI H. Cyclo-oxygenase-2 expression in hepatocellular carcinoma. Hepatogastroenterology. 1999;46:407–412. [PubMed] [Google Scholar]
- SHOEMAKER A.R., GOULD K.A., LUONGO C., MOSER A.R., DOVE W.F. Studies of neoplasia in the Min mouse. Biochim. Biophys. Acta. 1997;1332:F25–F48. doi: 10.1016/s0304-419x(96)00041-8. [DOI] [PubMed] [Google Scholar]
- SIMON L.S., WEAVER A.L., GRAHAM D.Y., KIVITZ A.J., LIPSKY P.E., HUBBARD R.C., ISAKSON P., VERBURG K.M., YU S.S., ZHAO W.W., GEIS G.S. Anti-inflammatory and upper gastrointestinal effects of celecoxib in rheumatoid arthritis; a randomised controlled trial. JAMA. 1999;282:1921–1928. doi: 10.1001/jama.282.20.1921. [DOI] [PubMed] [Google Scholar]
- SMITH J.M., ZHANG Y., KOBOLDT C.M., MUHAMMAD J., ZWEIFEL B.S., SHAFFER A.F., TALLEY J.J., MASFERRER J., SEIBERT K., ISAKSON P. Pharmacological analysis of cyclo-oxygenase-1 in inflammation. Proc. Natl. Acad. Sci. U.S.A. 1998;95:13313–13318. doi: 10.1073/pnas.95.22.13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMITH M.L., HAWCROFT G., HULL M.A. The effect of non-steroidal anti-inflammatory drugs on human colorectal cancer cells: evidence of different mechanisms of action. Eur. J. Cancer. 2000b;36:664–674. doi: 10.1016/s0959-8049(99)00333-0. [DOI] [PubMed] [Google Scholar]
- SMITH W.L., DEWITT D.L., GARAVITO R.M. Cyclo-oxygenases: structural, cellular and molecular biology. Annu. Rev. Biochem. 2000a;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- SOSLOW R.A., DANNENBERG A.J., RUSH D., WOERNER B.M., KHAN K.N., MASFERRER J., KOKI A.T. Cox-2 is expressed in human pulmonary, colonic and mammary tumors. Cancer. 2000;89:2637–2645. doi: 10.1002/1097-0142(20001215)89:12<2637::aid-cncr17>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- SPIRIO L.N., KUTCHERA W., WINSTEAD M.V., PEARSON B., KAPLAN C., ROBERTSON M., LAWRENCE E., BURT R.W., TISCHFIELD J.A., LEPPERT M.F., PRESCOTT S.M., WHITE R. Three secretory phospholipase A2 genes that map to human chromosome 1P35-36 are not mutated in individuals with attenuated adenomatous polyposis coli. Cancer Res. 1996;56:955–958. [PubMed] [Google Scholar]
- STEINBACH G., LYNCH P.M., PHILLIPS R.K., WALLACE M.H., HAWK E., GORDON G.B., WAKABAYASHI N., SAUNDERS B., SHEN Y., FUJIMURA T., SU L.-K., LEVIN B. The effect of Celecoxib, a cyclo-oxygenase inhibitor, in Familial Adenomatous Polyposis. N. Engl. J. Med. 2000;342:1946–1952. doi: 10.1056/NEJM200006293422603. [DOI] [PubMed] [Google Scholar]
- STOLINA M., SHARMA S., LIN Y., DOHADWALA M., GARDNER B., LUO J., ZHU L., KRONENBERG M., MILLER P.W., PORTANOVA J., LEE J.C., DUBINETT S.M. Specific inhibition of cyclo-oxygenase 2 restores antitumor reactivity by altering the balance of IL-10 and IL-12 synthesis. J. Immunol. 2000;164:361–370. doi: 10.4049/jimmunol.164.1.361. [DOI] [PubMed] [Google Scholar]
- SUH N., WANG Y., WILLIAMS C.R., RISINGSONG R., GILMER T., WILLSON T.M., SPORN M.B. A new ligand for the peroxisome proliferator-activated receptor-γ (PPAR-γ) GW7845, inhibits rat mammary carcinogenesis. Cancer Res. 1999;59:5671–5673. [PubMed] [Google Scholar]
- SUH O., METTLIN C., PETRELLI N.J. Aspirin use, cancer and polyps of the large bowel. Cancer. 1993;72:1171–1177. doi: 10.1002/1097-0142(19930815)72:4<1171::aid-cncr2820720407>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- SYNGAL S., CLARKE G., BANDIPALLIAM P. Potential roles of genetic biomarkers in colorectal cancer chemoprevention. J. Cell Biochem. 2000;34:S28–S34. doi: 10.1002/(sici)1097-4644(2000)77:34+<28::aid-jcb7>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- TALLEY J.J., BROWN D.L., CARTER J.S., GRANETO M.J., KOBOLDT C.M., MASFERRER J., PERKINS W.E., ROGERS R.S., SHAFFER A.F., ZHANG Y.Y., ZWEIFEL B.S., SEIBERT K. 4-[5-Methyl-3-phenylisoxazol-4-yl]-benzenesulfonamide, Valdecoxib: a potent and selective inhibitor of COX-2. J. Med. Chem. 2000;43:775–777. doi: 10.1021/jm990577v. [DOI] [PubMed] [Google Scholar]
- THUN M.J. NSAID use and decreased risk of gastrointestinal cancers. Gastroenterol. Clin. North Am. 1996;25:333–348. doi: 10.1016/s0889-8553(05)70250-8. [DOI] [PubMed] [Google Scholar]
- THUN M.J., NAMBOODIRI M.M., CALLLE E.E., FLANDERS D.W., HEATH C.W.J. Aspirin use and the risk of fatal cancer. Cancer Res. 1993;53:1322–1327. [PubMed] [Google Scholar]
- TOMLINSON I.P., NEALE K., TALBOT I.C., SPIGELMAN A.D., WILLIAMS C.B., PHILLIPS R.K., BODMER W.F. A modifying locus for familial adenomatous polyposis may be present on chromosome 1p35–36. J. Med. Genet. 1996;33:268–273. doi: 10.1136/jmg.33.4.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TORRANCE C.J., JACKSON P.E., MONTGOMERY E., KINZLER K.W., VOGELSTEIN B., WISSNER A., NUNES M., FROST P., DISCAFANI C.M. Combinatorial chemoprevention of intestinal neoplasia. Nat. Med. 2000;6:1024–1028. doi: 10.1038/79534. [DOI] [PubMed] [Google Scholar]
- TRIMBOLI A.J., WAITE B.M., ATSUMI G., FONTEH A.N., NAMEN A.M., HIGH K.P., WILLINGHAM M.C., CHILTON F.H. Influence of coenzyme A-independent transacylase and cyclo-oxygenase inhibitors on the proliferation of breast cancer cells. Cancer Res. 1999;59:6171–6177. [PubMed] [Google Scholar]
- TSIOULIAS G.T., GODWIN T.A., GOLDSTEIN M.F., MCDOUGALL C.J., DECOSSE J.J., RIGAS B. Loss of colonic HLA antigens in Familial Adenomatous Polyposis. Cancer Res. 1992;52:3449–3452. [PubMed] [Google Scholar]
- TSIOULIAS G.T., TRIADAFILOPOULOS G., GOLDIN E., PAPAVASSILIOU E.D., RIZOS S., BASSIOKAS P., RIGAS B. Expression of HLA class I antigens in sporadic adenomas and histologically normal mucosa of the colon. Cancer Res. 1993;53:2374–2378. [PubMed] [Google Scholar]
- TSUBOUCHI Y., MUKAI S., KAWAHITO Y., YAMADA R., KOHNO M., INOUE K., SANO H. Meloxicam inhibits the growth of non-small cell lung cancer. Anticancer Res. 2000;20:2867–2872. [PubMed] [Google Scholar]
- TSUJII M., DUBOIS R.N. Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell. 1995;83:493–501. doi: 10.1016/0092-8674(95)90127-2. [DOI] [PubMed] [Google Scholar]
- TSUJII M., KAWANO S., TSUJI S., SAWAOKA H., HORI M., DUBOIS R.N. Cyclo-oxygenase regulates angiogenesis induced by colon cancer cells. Cell. 1998;93:705–716. doi: 10.1016/s0092-8674(00)81433-6. [DOI] [PubMed] [Google Scholar]
- UOTILA P., VALVE E., MARTIKAINEN P., NEVALAINEN M., NURMI M., HARKONEN P. Increased expression of cyclooxygenase-2 and nitric oxide synthase-2 in human prostate cancer. Urol. Res. 2001;29:23–28. doi: 10.1007/s002400000148. [DOI] [PubMed] [Google Scholar]
- VAINIO H., MORGAN G. Cyclo-oxygenase 2 and breast cancer prevention. Non-steroidal anti-inflammatory agents are worth testing in breast cancer. Brit. Med. J. 1998;317:828–828. doi: 10.1136/bmj.317.7162.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VANE J.R., BAKHLE Y.S., BOTTING R.M. Cyclo-oxygenases 1 and 2. Annu. Rev. Pharmacol. Toxicol. 1998;38:97–120. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- WHO The WHO report is at www.iarc.fr/pageroot/UNITS/Chemoprevention2.html. 2001.
- WILLIAMS C.S., TSUJII M., REESE J., DEY S.K., DUBOIS R.N. Host cyclooxygenase-2 modulates carcinoma growth. J. Clin. Invest. 2000;105:1589–1594. doi: 10.1172/JCI9621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILLSON T.M., BROWN P.J., STERNBACH D.D., HENKE B.R. The PPARs: from orphan receptors to drug discovery. J. Med. Chem. 2000;43:527–550. doi: 10.1021/jm990554g. [DOI] [PubMed] [Google Scholar]
- XIE W., CHIPMAN J.G., ROBERTSON D.L., ERIKSON R.L., SIMMONS D.L. Expression of a mitogen-responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. Proc. Natl. Acad. Sci. U.S.A. 1991;88:2692–2696. doi: 10.1073/pnas.88.7.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XIE W., ROBERTSON D.L., SIMMONS D.L. Mitogen-inducible prostaglandin G/H synthase: a new target for nonsteroidal anti-inflammatory drugs. Drug Devel. Res. 1992;25:249–265. [Google Scholar]
- YAO S., RIOUX N., CASTONGUAY A., YOU M. Inhibition of COX-2 and induction of apoptosis: two determinants of non-steroidal anti-inflammatory drugs' chemopreventive efficacies in mouse lung tumorigenesis. Exp. Lung. Res. 2000;26:731–742. doi: 10.1080/01902140150216783. [DOI] [PubMed] [Google Scholar]
- YOSHIMURA R., SANO H., MASUDA C., KAWAMURA M., TSUBOUCHI Y., CHARGUI J., YOSHIMURA N., HLA T., WADA S. Expression of cyclo-oxygenase-2 in prostate carcinoma. Cancer. 2000;89:589–596. [PubMed] [Google Scholar]