

Abstract
Vascular endothelial growth factor (VEGF) is a potent angiogenic and inflammatory mediator. We have recently shown that this latter effect requires the activation of Flk-1 receptor and subsequent endothelial cell (EC) PAF synthesis. However, the intracellular events that regulate EC PAF synthesis upon Flk-1 stimulation by VEGF remain to be elucidated.
Using specific inhibitors and Western blot analysis, we herein report that in bovine aortic endothelial cells (BAEC), VEGF induces the synthesis of PAF through the cascade activation of Flk-1 receptor, phospholipase Cγ (PLCγ), protein kinase C (PKC) and p42/44 mitogen-activated protein kinases (MAPK).
Moreover, we demonstrate that VEGF-mediated PAF synthesis requires the activation of p38 MAPK, likely by directing the conversion of lyso-PAF to PAF.
Interestingly, we observed that VEGF also promoted the activation of the phosphatidyl inositol-3-phosphate kinase (PI3K) pathway, and that its blockade potentiated PAF synthesis following a VEGF treatment. Consequently, it appears that the PI3K pathway acts as a negative regulator of EC PAF synthesis.
Taken together, these results allow a better understanding of the intracellular events activated upon EC stimulation by VEGF, and shed a new light on the mechanisms by which VEGF induces PAF synthesis.
Keywords: VEGF, PAF, PI3K, MAPK, PLC, inflammation, angiogenesis
Introduction
It has been known for several decades that angiogenesis, the formation of new blood vessels from preexisting ones, is a crucial requirement for several physiological conditions, such as wound healing, tissular regeneration, and uterine wall thickening (Folkman & Klagsbrun, 1987; Folkman, 1991). However, numerous reports have recently confirmed that angiogenesis is also involved in the pathogenesis of several disorders such as tumour growth, atherosclerosis, and proliferative retinopathies (Folkman, 1991; Ferrara, 1995; Moulton et al., 1999). During this multistep process, capillary endothelial cells (EC) migrate and proliferate in order to form a three-dimensional structure capable of carrying blood, resulting in part from the concerted action of EC-directed growth factors (Folkman & Klagsbrun, 1987).
By virtue of its multifunctional nature, vascular endothelial growth factor (VEGF) is a likely candidate for the regulation of both physiological and pathological angiogenesis (Ferrara & Davis-Smith, 1997). Initially identified as a tumour-released factor that enhances vascular permeability to circulating macromolecules (Senger et al., 1983), VEGF was subsequently found to promote EC shape changes, migration, proliferation and angiogenesis through interactions with its tyrosine kinase receptors Flt-1 and Flk-1, and also by binding to neuropilin-1 (Ferrara & Davis-Smith, 1997; Connolly et al., 1989; Soker et al., 1998). Although VEGF and other growth factors, such as acidic and basic fibroblast growth factors (aFGF, bFGF) and epidermal growth factor (EGF), can promote EC migration and growth in vitro and angiogenesis in vivo (Unemori et al., 1992), only VEGF is capable of inducing inflammation (Connolly et al., 1989). We have previously shown that the effect of VEGF on vascular permeability is mediated through the induction of platelet-activating factor (PAF) synthesis by EC (Sirois & Edelman, 1997) and that such response is elicited through Flk-1 activation (Bernatchez et al., 1999).
Platelet-activating factor is involved in pathological angiogenesis (Bussolino & Camussi, 1995) and much work has been devoted to determine how the synthesis of this inflammatory lipid is regulated. In EC, PAF synthesis is mediated via a remodelling pathway in which membrane phospholipids are converted by a phospholipase A2 (PLA2) into lyso-PAF, which is then acetylated by the acetyl-CoA:lyso-PAF acetyltransferase (lyso-PAF AT) to form PAF (Bussolino & Camussi, 1995; Snyder et al., 1996). However, there are no data regarding the intracellular events that regulate the activation of the remodelling pathway upon Flk-1 activation.
Previous reports have shown that stimulation of VEGF receptors may lead to: (1) a modest activation of the Ras-dependent pathway through the recruitment of Src homology 2 domain-containing proteins (Takahashi et al., 1999) and subsequent activation of the Ras/Raf cascade, and (2) a much greater activation of a Ras-independent pathway, leading to phospholipase C (PLC)γ activation (Takahashi et al., 1999; He et al., 1999), the subsequent release of diacylglycerol and inositol trisphosphate, which in turn, activate protein kinase C (PKC) (van biesen et al., 1996) and cause a rapid elevation of intracellular Ca2+ (Sa & Fox, 1994). Both Ras-dependent and -independent pathways promote the activation of Raf and the downstream p42/44 mitogen-activated protein kinases (MAPK) (Takahashi et al., 1999; van biesen et al., 1996; Dangelo et al., 1995), also known as ERK1/ERK2, involved in cellular proliferation and PLA2 activation (Sa & Fox, 1994). More recently, others have shown that VEGF also activates the p38 MAPK pathway (Rousseau et al., 1997), whose activity is elevated during inflammatory responses and governed by the mixed lineage kinases (MLK) and the MAPK kinases-3/6 (MKK-3/6) (Tibbles & Woodgett, 1999). Finally, VEGF also activates the phosphatidyl inositol-3-kinase (PI3K) pathway, involved in the effect of VEGF on NO synthesis (Papapetropoulos et al., 1997). However, the relative contribution of these pathways to VEGF-induced PAF synthesis is currently unknown.
Herein we demonstrate that in BAEC, VEGF activates p42/44 MAPK via a Ras-independent pathway, involving PLCγ and PKC, and that this pathway is crucial to PAF synthesis. We also demonstrate that VEGF promotes the cascade activation of MKK-3/6 and p38 MAPK which is as well required to elicit PAF synthesis. Finally, we show that PI3K activation by VEGF acts as an endogenous negative regulator of EC PAF synthesis. These data bring new insights to our understanding of the intracellular events initiated by VEGF on EC and may have broad implications for the regulation of VEGF inflammatory and angiogenic activity.
Methods
Cell culture
Bovine aortic endothelial cells (BAEC) were isolated from freshly harvested bovine aortas, cultured in Dulbecco's modified Eagle's medium (DMEM; Life Technologies, Burlington, Canada) containing 5% foetal bovine serum (Hyclone Lab., Logan, UT, U.S.A.), and antibiotics (Sigma Chem., St-Louis, MO, U.S.A.). BAEC were characterized as described previously (Sirois & Edelman, 1997; Bernatchez et al., 1999). Cells were not passaged for more than nine cycles.
Measurement of PAF synthesis
PAF production by BAEC was measured by the incorporation of 3H-acetate into lyso-PAF as described previously (Sirois & Edelman, 1997; Bernatchez et al., 1999). Briefly, confluent BAEC (100 mm tissue culture plate) were stimulated for 15 min in 1 ml of HBSS-HEPES (10 mM, pH 7.4), CaCl2 (10 mM), 3H-acetate (25 μCi) (New England Nuclear, Boston, MA, U.S.A.) plus VEGF (1 nM) (human VEGF-A165, PeproTech, Rocky Hill, NJ, U.S.A.). Genistein, U73122, U73343, GF109203X, PD98059, SB203580, Wortmannin and LY294002 (Calbiochem, La Jolla, CA, U.S.A.) were dissolved in DMSO and, where indicated, added 10 min prior to the addition of VEGF (1 nM). DMSO was used to dissolve specific inhibitors, and tested alone (as control) at equivalent final concentration used for the inhibitors. The reaction was stopped by addition of acidified methanol (50 mM acetic acid), and lipids were extracted by the Bligh and Dyer method (Bligh & Dyer, 1959). Isolated lipids were dried under a stream of N2 gas, redissolved in elution buffer, and purified by normal-phase HPLC. Fractions corresponding to 3H-PAF were quantified by counting radioactivity with a β-counter. The separation and detection of 3H-PAF was confirmed by comparison with the HPLC elution pattern of a commercial 3H-PAF standard (New England Nuclear), and by the ability of PAF-containing fractions to induce platelet aggregation when compared to standard alkyl-PAF (Avanti Polar Lipids, Alabaster, AL, U.S.A.) (Sirois & Edelman, 1997).
Western blot analysis of PLCγ phosphorylation
Subconfluent BAEC (100 mm tissue culture plate) were serum-starved for 18 h in DMEM and then stimulated with VEGF (1 nM) for various periods of time as described for PAF synthesis±inhibitors. Cells were lysed ((mM): Tris-HCl pH 7.4 20, NaCl 150, Triton X-100 1.2%, EGTA 1, PMSF 1, aprotinin 0.15 Uml−1, leupeptin 10 μg ml−1, NaVO3 1, 0.001 Microcystin LR), the plates scraped, the lysate clarified by centrifugation, and the protein concentration of the supernatant determined by using a protein assay kit (Bio-Rad, Hercules, CA, U.S.A.). Total proteins (1.2 mg) were immunoprecipitated with an anti-PLCγ antibody (Santa Cruz Biotechnologies, Santa Cruz, CA, U.S.A.) coupled to Protein G-coated sepharose beads (Amersham Pharmacia Biotech, Upsala, Sweden), separated on a 7.5% SDS – PAGE and transblotted overnight at 0.07 mA (25 mM Tris-Base, 192 mM Glycine, 5% MeOH) onto a PVDF membrane (Milipore, Bedford, MA, U.S.A.). Membranes were blocked in TTBS (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 150 mM NaCl, 0.1% Triton X-100) containing 3% BSA for 1 h at room temperature, probed with a PY20 anti-phosphotyrosine antiserum (Santa Cruz Biotechnologies, CA, U.S.A.) overnight, and incubated with an anti-mouse antibody coupled to horseradish peroxidase (Santa Cruz Biotechnologies, CA, U.S.A.) which was revealed by chemiluminescence. Kaleidoscope pre-stained proteins were used as standards for SDS – PAGE.
Western blot analysis of p42/44 MAPK, MKK-3/6, p38 MAPK and Akt activation
Confluent BAEC (100 mm tissue culture plate) were serum-starved for 18 h in DMEM and then stimulated with VEGF (1 nM) for various periods of time with or without inhibitors as described above. Cells were lysed and total proteins (100 μg) were separated by a 7.5% SDS – PAGE gel and transblotted for 1 h at 120 V onto an Immunobilon-P PVDF membrane. Membranes were blocked in TTBS buffer containing 5% Blotto (Bio-Rad) for 1 h at room temperature. Membranes were then incubated overnight in blocking buffer containing either an anti-phospho-p42/44 MAPK, MKK-3/6, p38 MAPK, or Akt rabbit polyclonal antisera (dilution 1 : 1,000; New England Biolabs, Beverley, MA, U.S.A.). Membranes were washed with TTBS and incubated with an anti-rabbit antibody coupled to horseradish peroxidase (Santa Cruz Biotechnologies, CA, U.S.A.). Membranes were washed with TTBS and horseradish peroxidase was revealed by chemiluminesence. Kaleidoscope pre-stained standards were used as standards for SDS – PAGE.
Measurement of PLCγ activation by inositol phosphate (IP) production
Confluent BAEC (six well tissue culture plate) were brought to confluence, washed twice with DMEM, supplied with 1 ml of DMEM+1 μCi ml−1 of 3H-myoinositol (Amersham Pharmacia Biotech) and incubated at 37°C for 16 h. The cells were washed twice with DMEM, and supplemented with 0.5 ml of DMEM+0.1% BSA+10 mM CaCl2+10 mM LiCl with or without VEGF (1 nM) for 5 min. The cells were placed on ice and perchloric acid was added to the reaction media (5% v v−1 final concentration). The cells were scraped, isolated in tubes, vortexed and spun at 12,000×g for 5 min. The aqueous phase was extracted with an equal volume (1 : 1 : 1) of 1,1,2-trichlorotrifluoroethane and tri-n-octylamine, vortexed, and spun at 12,000×g for 1 min. The upper phase was isolated and applied to Dowex AG1X8 200 – 400 mesh columns (Bio-Rad) as previously described (Berridge et al., 1983). Two ml fractions were collected and counted by using a β-scintillation counter.
Trypan blue viability assay
Cellular viability was confirmed in BAEC used for PAF synthesis experiments. Cells were treated with inhibitors as described for PAF synthesis experiments. After incubation with VEGF, the media was removed and cells were incubated with a 0.1% Trypan blue-PBS solution (Sigma) for 2 min. Cells were rinsed once with PBS and visualized under a reversed phase microscope to determine the percentage of cells having incorporated the Trypan blue.
Results
Modulation of VEGF-induced PAF synthesis by specific inhibitors of the Ras-independent and MAPK pathways
We recently reported that VEGF dose-dependently induced the synthesis of PAF by BAEC (Sirois & Edelman, 1997; Bernatchez et al., 1999). Hence, in order to determine the intracellular pathways by which VEGF promotes EC PAF synthesis, we pretreated BAEC with a range of selective inhibitors and investigated their effects on VEGF-induced PAF synthesis. This approach allowed the use of native non-transfected EC which possess the intracellular pathways found in normal vascular endothelium. The application of VEGF (1 nM) to confluent BAEC induced a 19.6 fold increase of PAF synthesis as compared to control non-stimulated cells (PBS) (Figure 1). Pretreatment of BAEC with genistein (10 μM; IC50=2.6 μM) (Constantinou & Huberman, 1995), a potent inhibitor of tyrosine kinase receptor autophosphorylation and activation, attenuated VEGF-induced PAF synthesis by 72%. Moreover, specific inhibitors of PLC (U73122; 10 μM; IC50=1 μM) (Thompson et al., 1991) and PKC (GF109203X; 1 μM; IC50=10 nM) (Toullec et al., 1991) attenuated VEGF-induced PAF synthesis by 88 and 71% respectively, whereas an inactive PLC inhibitor analogue (U73343; 10 μM) had no significant effect (data not shown). Pretreatment of BAEC with potent inhibitors of the p42/44 and p38 MAPK pathways (PD98059; 10 μM; IC50=1 μM; and SB203580; 10 μM; IC50=0.6 μM) (Rousse et al., 1994; Cuenda et al., 1995) attenuated by 86 and 85% VEGF-induced PAF synthesis (Figure 1). DMSO alone did not induce PAF synthesis and did not prevent VEGF-mediated effect on PAF synthesis (data not shown). Trypan blue exclusion experiments confirmed that in similar conditions, the inhibitors herein described had no toxicity effect on the BAEC used in these experiments (data not shown).
Figure 1.
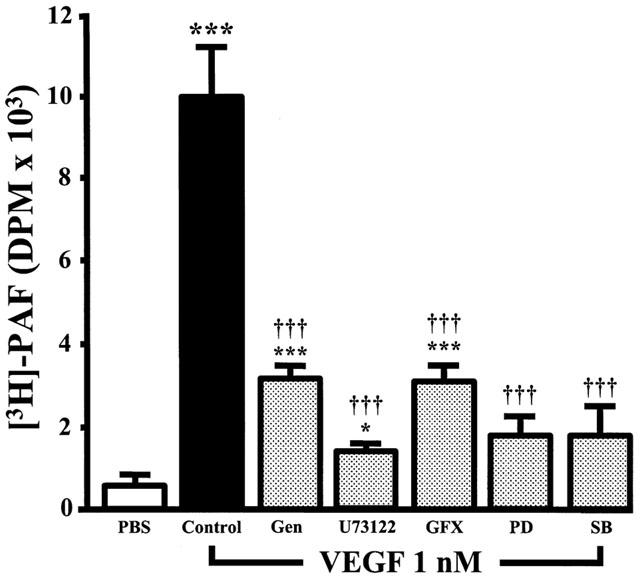
Inhibitors effect on VEGF-induced EC PAF synthesis. BAEC were grown to confluence in six-well tissue culture plate. The media was replaced by 1 ml of HBSS – HEPES (10 mM)+CaCl2 (10 mM), and cells were pretreated with tyrosine kinase inhibitor genistein (Gen, 10 μM), PLC inhibitor U73122 (10 μM), PKC inhibitor GF109203X (GFX, 1 μM), p42/44 MAPK pathway inhibitor PD98059 (PD, 10 μM) or p38 MAPK inhibitor SB203580 (SB, 10 μM). Cells were then incubated with [3H]-acetate, and stimulated with VEGF (1 nM) for 15 min. The values are means of at least eight experiments±s.e.mean. *P<0.05, ***P<0.001 as compared with control buffer (PBS). †††P<0.001 as compared with VEGF (1 nM), as determined by analysis of variance followed by Bonferoni's t-test.
VEGF effect on PLCγ phosphorylation
As the results presented above confirm that PLC activation is involved in VEGF-mediated PAF synthesis, we then sought to investigate if VEGF activates the γ isoform of PLC in BAEC. Treatment of BAEC with VEGF increased PLCγ phosphorylation with a maximal effect observed within 5 min following stimulation (Figure 2A). Pretreatment with U73122 (10 μM) inhibited VEGF effect on PLCγ activation by 87%, whereas U73343, the inactive analogue of U73122, had no effect (Figure 2B). Both U73122 and U73343 had only marginal effects on the basal phosphorylation of PLCγ (Figure 2C).
Figure 2.
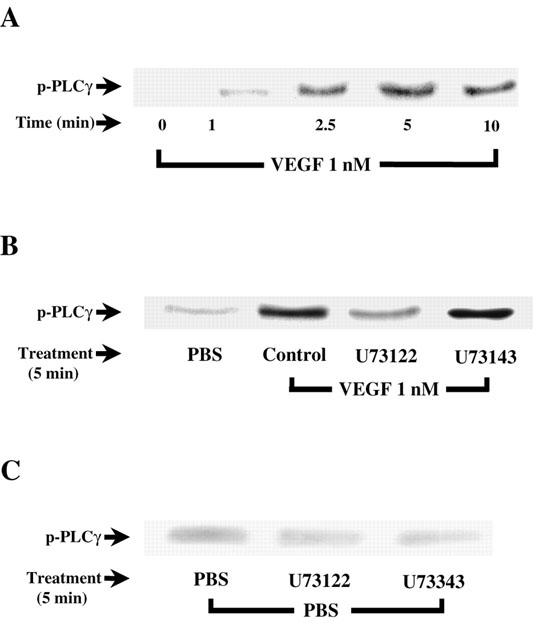
VEGF-induced activation of PLCγ. (A) Western blot analysis of VEGF time-dependent effect on PLCγ activation. Near-confluent BAEC were serum-starved in DMEM for 16 h and stimulated with VEGF (1 nM) for various periods of time. Cells were lysed, proteins were immunoprecipitated with an anti-PLCγ antibody and separated with a 10% SDS – PAGE. Western blot analysis was performed with an anti-PY20 antibody. (B) and (C) effect of U73122 (10 μM) on VEGF- and PBS-induced PLCγ phosphorylation respectively. BAEC were pretreated 10 min with U73122 and U73343 prior to a 5 min VEGF treatment.
VEGF effect on p42/44 MAPK activation
Since p42/44 MAPK appears to play a pivotal role in PAF synthesis induced by VEGF (Figure 1), we sought to determine its activation pattern in BAEC challenged with VEGF for various periods of time by Western blot analysis. The data presented in Figure 3A reveal that VEGF promotes a transient time-dependent activation of p42/44 MAPK which peaked within 10 min following VEGF stimulation. To determine the intracellular events required by VEGF to direct p42/44 MAPK activation, BAEC were pretreated with the previously-described inhibitors, challenged with VEGF (1 nM) for 10 min, which correlates with the peak activation of p42/44 MAPK, and processed as described above. Incubation of BAEC with VEGF (1 nM) for 10 min elicited a 4.2 fold increase in p42/44 MAPK activation as compared to PBS-treated cells (Figure 3B). Pretreatment with genistein (10 μM) completely blocked p42/44 MAPK activation. The application of the PLC and PKC inhibitors U73122 (10 μM) and GF109203X (1 μM) attenuated by as much as 93 and 87% VEGF-induced p42/44 MAPK activation (Figure 3B). Finally, PD98059 (10 μM) completely blocked p42/44 MAPK activation in response to VEGF, whereas the p38 MAPK inhibitor SB203580 (10 μM) had no significant effect. The inactive PLC inhibitor analogue U73343 (10 μM) and DMSO alone had no significant effect (data not shown).
Figure 3.
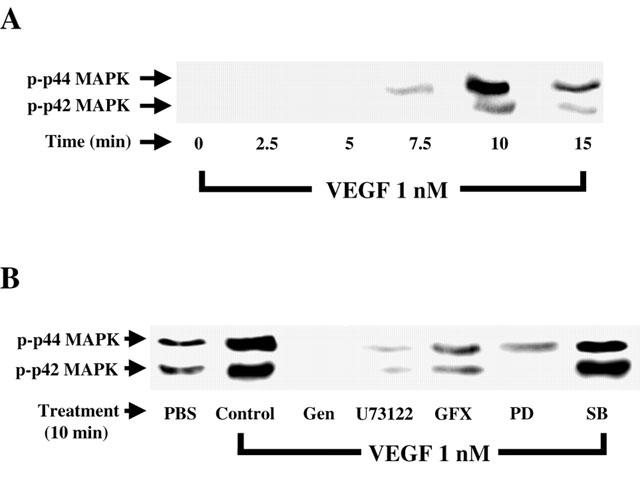
VEGF effect on p42/44 MAPK phosphorylation. (A) Western blot analysis of VEGF time-dependent effect on p42/44 MAPK activation. Confluent BAEC were stimulated with VEGF (1 nM) for various periods of time. Cells were lysed, proteins were separated on a 10% SDS – PAGE. Western blot analysis was performed with an anti-phospho-p42/44 MAPK antibody. (B) BAEC were pretreated with genistein (Gen, 10 μM), U73122 (10 μM), GF109203X (GFX, 1 μM), PD98059 (PD, 10 μM), SB203580 (SB, 10 μM), then the cells were treated as in (A).
VEGF effect on p38 MAPK activation
Knowing that p38 MAPK activation is directly involved in the synthesis of PAF following the stimulation of BAEC with VEGF (Figure 1), we first sought to confirm that VEGF can elicit a sustained elevation in p38 MAPK phosphorylation. Treatment of BAEC with VEGF (1 nM) elicited a time dependent increase in p38 MAPK phosphorylation as determined by Western blot analysis, with a peak phosphorylation observed at 7.5 min (Figure 4A). We then sought to establish if the intracellular mechanisms investigated so far were involved in the activation of p38 MAPK by VEGF. First, a 7.5 min treatment of BAEC with VEGF (1 nM) caused a 4.5 fold increase in p38 MAPK activation as compared to a PBS treatment (Figure 4B). The application of the tyrosine kinase receptor inhibitor (genistein; 10 μM) completely blocked this VEGF effect. However, pretreatment with inhibitors of PLC (U73122, 10 μM), PKC (GF109203X, 1 μM) or p42/44 MAPK (PD98059, 10 μM) had no effect on p38 MAPK activation, whereas the inhibitor of p38 MAPK (SB203580, 10 μM) completely blocked the phosphorylation p38 MAPK mediated by a VEGF treatment (Figure 4B).
Figure 4.
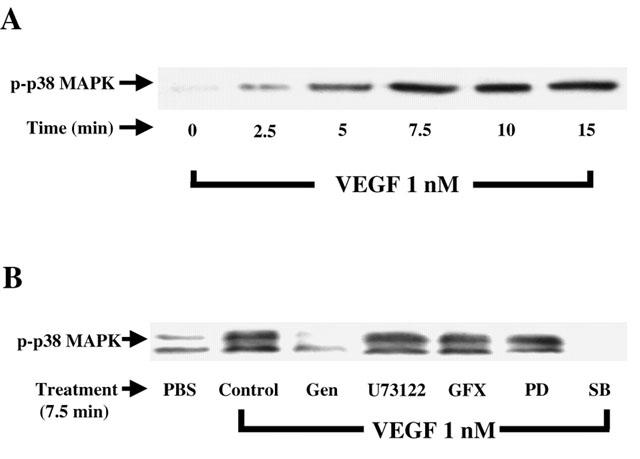
Effect of VEGF on p38 MAPK phoshorylation. (A) Western blot analysis of the phosphorylated form of p38 MAPK was conducted as described in Figure 3A with the use of an anti-phospho-p38 MAPK antibody. (B) Cells were pretreated as described in Figure 3B, and processed as described in A.
VEGF effect on MKK-3/6 activation
Considering that the activation of p38 MAPK appears to be dissociated from the PLCγ, PKC and p42/44 MAPK pathway (Figure 4B), and that MKK-3/6 were previously shown to induce the activation of p38 MAPK, we investigated if VEGF could indeed promote the phosphorylation of MKK-3/6. Treatment of BAEC with VEGF (1 nM) induced a time-dependent phosphorylation of the MKK-3/6 with a peak phosphorylation observed within 5 min following VEGF stimulation (Figure 5)
Figure 5.
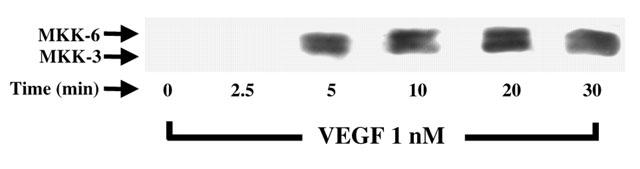
Effect of VEGF on MKK-3/6 phoshorylation. Western blot analysis of the phosphorylated form of MKK-3/6 was conducted as described in Figure 3A with the use of an anti-phospho-MKK-3/6 antibody.
Effect of selective PI3K inhibitors on VEGF-induced PAF synthesis
Since previous reports have confirmed that PI3K is activated by VEGF in EC, we aimed to determine its role in regulating PAF synthesis in response to VEGF stimulation. Pretreatment of EC with Wortmannin (10 and 100 nM; IC50=5 nM) (Bonser et al., 1991) elicited a concentration-dependent increase in PAF synthesis induced by VEGF (1 nM) by 39 and 102%, whereas LY294002 (1 and 10 μM; IC50=1.4 μM) (Vlahos et al., 1995) was less potent, elevating PAF synthesis by 28 and 65%, thereby suggesting that the PI3K pathway acts as a negative regulator of VEGF-induced PAF synthesis (Figure 6). In contrast, pretreatment with these inhibitors did not significantly affect basal PAF synthesis (data not shown).
Figure 6.
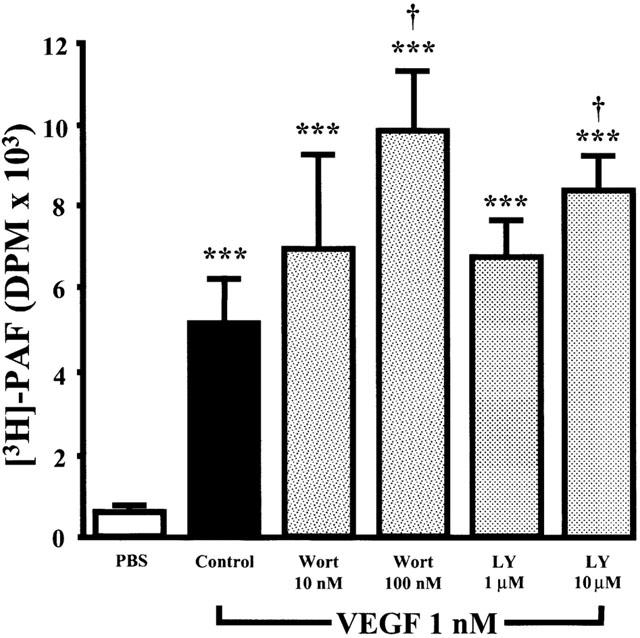
Effect of Wortmannin and LY294002 on VEGF-induced EC PAF synthesis. Experiments were conducted as described in Figure 1, with the exception that BAEC were pretreated for 10 min without or with the PI3K pathway inhibitors Wortmannin (Wort, 10 – 100 nM) or LY294002 (1 – 10 μM). The values are means of at least eight experiments. ***P<0.001 as compared with control buffer (PBS). †P<0.05 as compared with VEGF (1 nM) as determined by analysis of variance followed by Bonferoni's t-test.
Effect of VEGF on PI3K pathway activation
Several studies have shown that activation of PI3K directly leads to Akt phosphorylation. Hence, Western blot analysis of Akt phosphorylation is a useful approach to investigate PI3K activity. Stimulation of BAEC with VEGF (1 nM) elicited a time-dependent increase of Akt phosphorylation with a peak activity observed within 7.5 min following VEGF stimulation. These results support a possible role of the PI3K in regulating PAF synthesis in BAEC (Figure 7).
Figure 7.
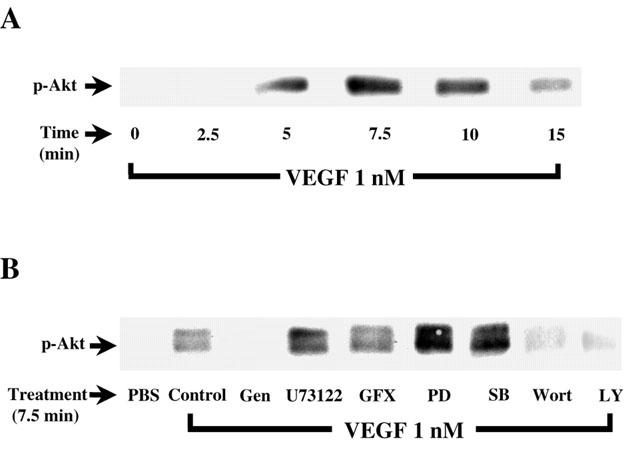
Effect of VEGF on PI3K pathway activation. (A) Confluent BAEC were stimulated with VEGF (1 nM) for various periods of time. Cells were lysed and separated on a 10% SDS – PAGE. Western blot analysis was performed with an anti-phospho-Akt antibody. (B) Western blot determination of the phosphorylated form of Akt reveals the effect of VEGF-induced Akt phosphorylation. Experiments were carried out as in Figure 6A, except that BAEC were pretreated 5 min with genistein (Gen, 10 μM), U73122 (10 μM), GF109203X (GFX, 1 μM), PD98059 (PD, 10 μM), SB203580 (SB, 10 μM), Wortmannin (Wort, 100 nM) or LY294002 (10 μM) prior to a 7.5 min treatment with VEGF (1 nM).
Effect of selective inhibitors on VEGF-induced PI3K pathway activation
To determine if the intracellular mechanisms described herein are involved in the activation of the PI3K by VEGF, confluent BAEC were pretreated with various inhibitors, stimulated with VEGF, and Akt phosphorylation was revealed by Western blot analysis. Figure 7B shows that VEGF induced an increase in PI3K activation as compared to PBS-treated cells. As expected, the tyrosine kinase inhibitor (genistein; 10 μM), the PI3K inhibitors (Wortmannin; 100 nM, and LY294002; 10 μM) attenuated by 98, 76 and 78% the effect of VEGF on PI3K activation, respectively. In contrast, inhibition of PLC (U73122; 10 μM), PKC (GF109203X, 1 μM), p42/44 MAPK (PD98059; 10 μM) and p38 MAPK (SB203580; 10 μM) did not attenuate VEGF-induced phosphorylation of Akt (Figure 7B).
Effect of two selective PI3K inhibitors on VEGF-induced p42/44 and p38 MAPK activation
We investigated the ability of Wortmannin and LY294002 to modulate the activation of the p42/44 and p38 MAPK pathways in response to VEGF treatment. The results presented in Figure 8A,B show that a pretreatment of BAEC with Wortmannin (100 nM) or LY294002 (10 μM) did not modulate the effect of VEGF on these two intracellular pathways.
Figure 8.
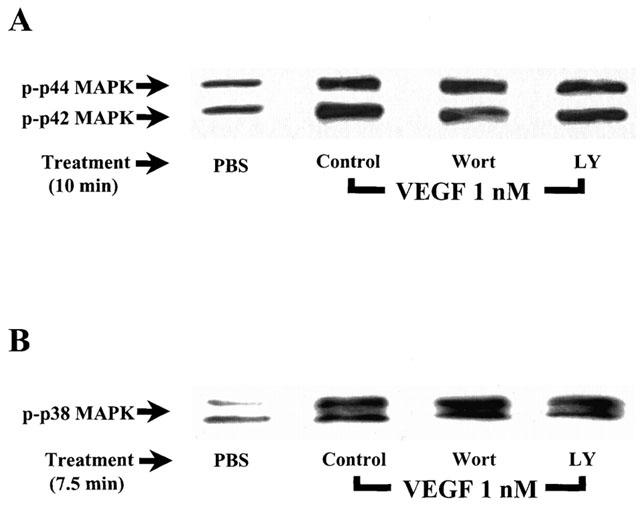
Effect of Wortmannin and LY294002 on VEGF-induced activation of the p42/44 and p38 MAPK. (A) Confluent BAEC were pretreated with or without Wortmannin (Wort; 100 nM) or LY294002 (LY; 10 μM), and were stimulated and processed as described in Figure 3A. Western blot analysis was performed with an anti-phospho-p42/44 MAPK antibody. (B) Confluent BAEC were pretreated with or without Wortmannin (Wort; 100 nM) or LY294002 (LY; 10 μM), and were stimulated and processed as described in Figure 3B. Western blot analysis was performed with an anti-phospho-p38 MAPK antibody.
Effect of PI3K inhibitors on IP production induced by VEGF in BAEC
As the results presented above suggest that VEGF activates PLCγ and that PI3K inhibition does not affect p42/44 and p38 MAPK phosphorylation but does potentiate PAF synthesis, we investigated the possibility that the inhibition of PI3K activity might potentiate VEGF effect on PAF synthesis by increasing PLCγ activation. First, we confirmed that PLCγ was activated by VEGF in BAEC by measuring the production of inositol phosphates (IP). Treatment of BAEC with VEGF elicited a time-dependent increase in total IP production as compared to PBS-treated cells, and maximal effect was observed within 5 min following a stimulation with VEGF (Figure 9A). Pretreatment with U73122 (10 μM) completely abrogated the synthesis of IP following a 5 min stimulation with VEGF (1 nM), whereas its inactive analogue U73343 (10 μM) had no effect. Pretreatment with the two PI3K inhibitors Wortmannin (100 nM) or LY294002 (10 μM) had no significant effect on IP synthesis mediated by VEGF (Figure 9B).
Figure 9.
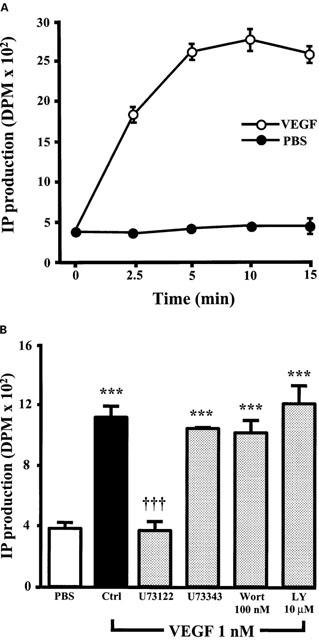
VEGF effect on IP production. (A) Confluent BAEC were labelled with [3H]-myoinositol, and stimulated with or without VEGF (1 nM) for various periods of time. The cells were scraped, total IP were isolated by extraction with 1,1,2-trichlorotrifluoroethane and tri-n-octylamine, purified by chromatography with a Dowex AG1X8 column (200 – 400 mesh) and quantified by scintillation counting. The values are means of at least four experiments±s.e.mean. (B) Confluent BAEC were pretreated either with U73122 (10 μM), U73343 (10 μM), Wortmannin (Wort; 100 nM) or LY294002 (LY; 10 μM), and stimulated with VEGF (1 nM) for 5 min. IP production was determined as in A. ***P<0.001 as compared with control buffer (PBS). †††P<0.001 as compared with VEGF (1 nM) as determined by analysis of variance followed by a Bonferoni t-test.
Discussion
Signal transduction following Flk-1 activation is mediated mostly through a Ras-independent pathway and leads to PAF synthesis
We have shown recently that VEGF could induce PAF synthesis and that such a response was Flk-1 dependent (Sirois & Edelman, 1997; Bernatchez et al., 1999). These results were the first to show that autophosphorylation of a tyrosine kinase receptor could promote the activation of the phospholipid remodelling pathway in a similar fashion to G protein-coupled receptors (Bussolino & Camussi, 1995). To confirm these results, we used a potent inhibitor of tyrosine kinase receptors. Pretreatment with Genistein reduced the ability of VEGF to induce PAF synthesis. The incomplete suppression of PAF synthesis by Genistein may result from its lack of selectivity since Genistein has previously been shown to block partially the intracellular phosphorylation cascade elicited by a VEGF treatment (Xia et al., 1996).
Autophosphorylation of Flk-1 receptor is known to induce phosphorylation of PLCγ and downstream activation of PKC, Raf and the p42/44 MAPK pathway (van biesen et al., 1996). As PLCγ and PKC are activated within minutes by VEGF in BAEC (Takahashi et al., 1999; Guo et al., 1995; He et al., 1999), we investigated their involvement in VEGF-induced PAF synthesis. As shown in Figure 1, the PLC inhibitor (U73122) almost completely blocked PAF synthesis whereas PKC inhibition (GFX109203) was slightly less effective. These results confirm a crucial role for the PLCγ – PKC pathway in mediating cellular responses following Flk-1 activation. This statement is supported by the fact that PKC activation by other inflammatory mediators has been shown to be involved in EC PAF production (Bussolino & Camussi, 1995; Whatley et al., 1989). One may hypothesize that the incapacity of GF109203X to completely abrogate PAF synthesis may be attributable to its inability to block completely the PKC isoforms involved in PAF synthesis. Pretreatment of BAEC with a potent inhibitor of the Ras-dependent pathway (BMS191563) (Muthalif et al., 2000) had only a minor effect on VEGF activity (data not shown), which supports our current hypothesis that VEGF induces the synthesis of PAF through a Ras-independent pathway.
Role of the p42/44 and p38 MAPK pathway in EC PAF synthesis
Activation of a Ras-independent pathway (PLCγ – PKC) by VEGF may lead to the downstream phosphorylation of p42/44 MAPK in BAEC (Takahashi et al., 1999), and promote the activation of PLA2 (Geijsen et al., 2000; Hazan-Halevy et al., 2000; Hiller & Sundler, 1999). Pretreatment of BAEC with PD98059 completely blocked EC PAF synthesis, illustrating the crucial role played by the p42/44 MAPK pathway. p38 MAPK is known to be activated by cellular stresses, inflammatory cytokines (Rouse et al., 1994) as well as by VEGF (Rousseau et al., 1997). Since p38 MAPK was shown to directly activate the lyso-PAF AT (Nixon et al., 1999), an enzyme required for VEGF-induced PAF synthesis (Bernatchez et al., 2001) we hypothesized that p38 MAPK may be involved in the induction of PAF synthesis by VEGF. Pretreatment of BAEC with an inhibitor of p38 MAPK (SB203580) completely blocked EC PAF synthesis, also illustrating the crucial role played by the p38 MAPK pathway.
VEGF activates PLCγ
To support our initial hypothesis that PLCγ is involved in EC PAF synthesis initiated by VEGF, we tested the ability of VEGF to promote tyrosine phosphorylation of PLCγ. The data presented in Figure 2 confirm that VEGF promoted a rapid time-dependent phosphorylation of PLCγ. This effect was nearly abolished when the cells were pretreated with a PLC inhibitor. VEGF effect on other PLC isozymes (β and δ) (Tobin, 1997) was not investigated herein since they contain neither SH2 nor SH3 domains.
VEGF activates p42/44 and p38 MAPK
Next, we sought to determine if p42/44 and p38 MAPK are activated by VEGF in the BAEC used for PAF synthesis experiments. Figures 3A and 4A reveal that VEGF does induce a transient activation of both p42/44 and p38 MAPK. To investigate further the intracellular mechanisms required for VEGF to trigger EC PAF synthesis, we pretreated BAEC with the above-described inhibitors and monitored their effects on p42/44 and p38 MAPK activation by VEGF. The data presented in Figures 3B and 4B reveal that VEGF induces p42/44 MAPK phosphorylation through the cascade activation of receptor tyrosine autophosphorylation, PLCγ, and PKC. In contrast, the phosphorylation of p38 MAPK by VEGF appears to be dependent of receptor tyrosine autophosphorylation but independent of the PLCγ, PKC or p42/44 pathways.
MKK-3/6 are two MAPK kinases that are capable of directly phosphorylating p38 MAPK, and whose activities are governed by the mixed lineage kinases (MLK) (Tibbles & Woodgett, 1999). Hence, the MLK – MKK-3/6 pathway is a potential candidate for mediating the activation of p38 MAPK. Western blot analysis revealed that VEGF causes a rapid elevation in MKK-3/6 phosphorylation (Figure 5), thereby suggesting that VEGF activates two parallel pathways which are both crucial to EC PAF synthesis: firstly, the PLCγ – PKC pathway which activates p42/44 MAPK, and secondly, the MLK – MKK-3/6 pathway which activates p38 MAPK.
p42/44 MAPK, p38 MAPK and the phospholipid remodelling pathway
We have recently shown that PAF synthesis induced by VEGF requires the activation of PLA2 leading to the release of the inactive precursor form of PAF, lyso-PAF, from membrane phospholipids (Prescott et al., 1990; Bernatchez et al., 2001). Interestingly, several other studies have shown that p42/44 MAPK phosphorylation by growth factors and other agonists directly induces the activation of PLA2 (Sa & Fox, 1994; Hiller & Sundler, 1999; Geijsen et al., 2000; Hazan-Halevy et al., 2000). Hence, the capacity of the p42/44 MAPK pathway inhibitor to completely block the synthesis of PAF mediated by VEGF may reside in its ability to prevent the PLA2 activation involved in EC PAF synthesis.
Once synthesized, lyso-PAF is then converted into PAF by the addition of an acetate group by lyso-PAF AT (Wykle et al., 1980). Recently, it has been reported that p38 MAPK, and not p42/44 MAPK, can directly activate the lyso-PAF AT and promote a 3 fold increase in its activity (Nixon et al., 1999). Hence, the capacity of the p38 MAPK inhibitor (SB203580) to regulate the synthesis of PAF elicited by a VEGF treatment is likely attributable to its capacity to prevent the activation of lyso-PAF-AT. However, these authors did not investigate the role of p38 MAPK in the synthesis of the final product (PAF) in living cells. To the best of our knowledge, the data herein presented are the first to show that p38 MAPK is involved in regulating PAF synthesis in intact cells.
bFGF is a growth factor capable of activating both p42/44 MAPK and PLA2 isozymes (Sa & Fox, 1994). In contrast to VEGF, bFGF is unable to induce either EC PAF synthesis or inflammation (Connolly et al., 1989; Sirois & Edelman, 1997). Interestingly, bFGF fails to activate p38 MAPK in EC (Rousseau et al., 1997). Hence, these observations link the phosphorylation of p42/44 MAPK by VEGF with PLA2 activation, and the phosphorylation of p38 MAPK with the activation of the lyso-PAF-AT.
The PI3K pathway is a negative regulator of EC PAF synthesis and acts independently of PLC, p42/44 and p38 MAPK pathways
PI3K is a SH2 domain-containing enzyme known to participate in the production of nitric oxide (NO) mediated by VEGF (Papapetropoulos et al., 1997). Since both NO and PAF are synthesized by EC within minutes when challenged with VEGF (Murohara et al., 1998), we investigated if the PI3K pathway was involved in EC PAF synthesis mediated by VEGF. As shown in Figure 6, pretreatment of EC with Wortmannin and LY294002 potentiated the synthesis of PAF mediated by VEGF. Hence, PI3K acts as a negative regulator of EC PAF synthesis. Western blot analysis confirmed that the PI3K pathway is indeed rapidly activated by VEGF in BAEC (Figure 7A). Moreover, pretreatment with inhibitors of the Ras-independent, p42/44 and p38 MAPK pathways did not prevent Akt phosphorylation induced by VEGF, which demonstrates that Flk-1 activation mediates PI3K activation without requiring the PLCγ-PKC, p42/44 and p38 MAPK pathways (Figure 7B). Since the inhibition of PI3K pathway potentiated the effect of VEGF on EC PAF synthesis, we investigated the effect of Wortmannin and LY294002 on the activation of PLCγ, p42/44 and p38 MAPK. PLCγ activation was determined by measuring the production of IP by BAEC. We observed that VEGF induced a time-dependent increase in IP production (Figure 9A). Pretreatment of EC with U73122 greatly abrogated IP production by EC (Figure 9B). Interestingly, pretreatment of EC with Wortmannin or LY294002 did not alter the ability of VEGF to promote the synthesis of IP (Figure 9B). In addition, Wortmannin and LY294002 did not inhibit the activation of p42/44 and p38 MAPK by VEGF (Figure 8A, B). These results suggest that PI3K regulates PAF synthesis downstream from the PLCγ, p42/44 and p38 MAPK pathway, by directly interacting with the phospholipid remodeling pathway.
Since VEGF promotes the activation of several intracellular pathways that regulate PAF synthesis, we propose an intracellular model by which VEGF induces PAF synthesis and we also report that such activity is in fact tightly regulated since VEGF-induced PAF synthesis can be schematized as an intracellular balance between pro-PAF (PLCγ, PKC, p42/44 and p38 MAPK) and anti-PAF (PI3K) signaling pathways (Figure 10).
Figure 10.
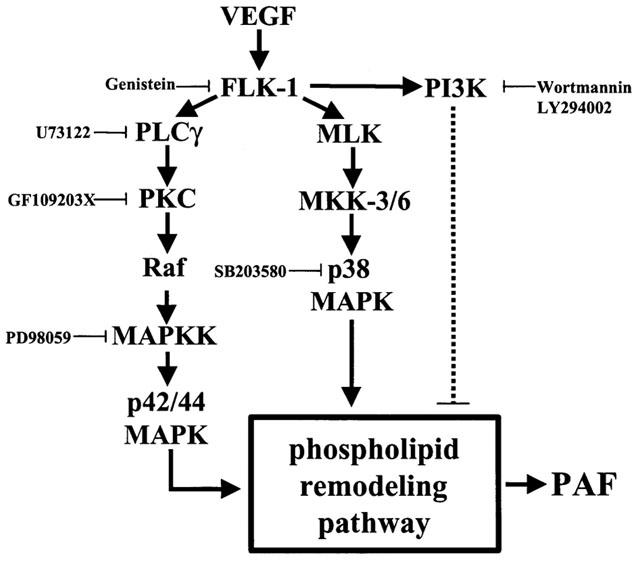
Proposed pathway for the induction of EC PAF synthesis by VEGF. The synthesis of PAF upon Flk-1 autophosphorylation by VEGF is predominantly mediated through the activation of a Ras-independent pathway (PLCγ and PKC) (solid line), promoting the downstream activation of the Raf and p42/44 MAPK pathway. In addition, VEGF activates the MKK-3/6 which induces the activation of p38 MAPK. Both p42/44 and p38 MAPK are known to activate the phospholipid remodelling pathway which leads to PAF synthesis. Meanwhile, Flk-1 autophosphorylation also activates PI3K, which appears to act as a negative regulator of PAF synthesis in BAEC (dashed line). Since VEGF-induced p42/44 and p38 MAPK phosphorylation were not affected by Wortmannin or LY294002, PI3K likely exerts its inhibitory effect downstream of p42/44 and p38 MAPK. Hence, the PI3K pathway might act on PAF synthesis in BAEC by downregulating the activity of the phospholipid remodelling pathway.
In conclusion, our results illustrate the potency of VEGF to activate, via a Ras-independent mechanism, both p42/44 and p38 MAPK, which are leading to the synthesis of PAF in BAEC. Moreover, our results show that VEGF is also capable of activating the PI3K pathway which acts as an endogenous negative regulator of EC PAF synthesis. Future studies are aimed at determining how and which MAPK are involved in the activation of the phospholipid remodelling pathway including the type of enzymes involved in such mechanism.
Acknowledgments
This study was supported by grants from the Canadian Institutes of Health Research (CIHR) (M.G. Sirois; MT-14378 and MOP-43919, and B.G. Allen; MT-14725), and the Heart and Stroke Foundation of Québec to Dr Sirois. Drs Sirois and Allen are recipients of scholarships from the Heart and Stroke Foundation of Canada. P.N. Bernatchez is a recipient of a studentship from the CIHR.
Abbreviations
- BAEC
bovine aortic endothelial cells
- bFGF
basic fibroblast growth factor
- DMEM
Dulbecco's modified Eagle's medium
- EC
endothelial cells
- lyso-PAF AT
acetyl-CoA:lyso-PAF acetyltransferase
- MAPK
mitogen-activated protein kinase
- MKK
MAPK kinase
- PI3K
phosphatidyl inositol-3 kinase
- PKC
protein kinase C
- PLA2
phospholipase A2
- PLC
phospholipase C
- VEGF
vascular endothelial growth factor
References
- BERNATCHEZ P.N., SOKER S., SIROIS M.G. VEGF effect on endothelial cell proliferation migration and PAF synthesis is mediated through the activation of Flk-1 receptor. J. Biol. Chem. 1999;274:31047–31054. doi: 10.1074/jbc.274.43.31047. [DOI] [PubMed] [Google Scholar]
- BERNATCHEZ P.N., WINSTEAD M.V., DENNIS E.A., SIROIS M.G. VEGF activation of endothelial cell PAF synthesis mediated by secretory phospholipase A2. Br. J. Pharmacol. 2001;134:197–205. doi: 10.1038/sj.bjp.0704215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERRIDGE M.J., DAWSON R.M., DOWNES C.P., HESLOP J.P., IRVINE R.F. Changes in the levels of inositol phosphates after agonist-dependent hydrolysis of membrane phosphoinositides. Biochem. J. 1983;212:473–482. doi: 10.1042/bj2120473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BLIGH E.G., DYER W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- BONSER R.W., THOMPSON N.T., RANDALL R.W., TATESON J.E., SPACEY G.D., HODSON H.F., GARLAND L.G. Demethoxyviridin and Wortmannin block phospholipase C and D activation in the human neutrophil. Br. J. Pharmacol. 1991;103:1237–1241. doi: 10.1111/j.1476-5381.1991.tb12330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUSSOLINO F., CAMUSSI G. Platelet-activating factor produced by endothelial cells: a molecule with autocrine and paracrine properties. Eur. J. Biochem. 1995;229:327–337. [PubMed] [Google Scholar]
- CONNOLLY D.T., HEUVELMAN D.M., NELSON R., OLANDER J.V., EPPLEY B.L., DELFINO J.J., SIEGEL N.R., LIMGRUBER R.M., FEDER J. Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J. Clin. Invest. 1989;84:1470–1478. doi: 10.1172/JCI114322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONSTANTINOU A., HUBERMAN E. Genistein as an inducer of tumor cell differentiation: possible mechanisms of action. Exp. Biol. Med. 1995;208:109–115. doi: 10.3181/00379727-208-43841. [DOI] [PubMed] [Google Scholar]
- CUENDA A., ROUSE J., DOZA Y.N., MEIER R., COHEN P., GALLAGHER T.F., YOUNG P.R., LEE J.C. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Letters. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- DANGELO G., STRUMAN I., MARTIAL J., WEINER R.J. Activation mitogen-activated protein kinases by vascular endothelial growth factor and basic fibroblast growth factor in capillary endothelial cells is inhibited by the antiangiogenic factor 16-kDa N-terminal fragment of prolactin. Proc. Natl. Acad. Sci. U.S.A. 1995;92:6374–6378. doi: 10.1073/pnas.92.14.6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FERRARA N. Vascular endothelial growth factor. The trigger for neovascularization in the eye. Lab. Invest. 1995;72:615–618. [PubMed] [Google Scholar]
- FERRARA N., DAVIS-SMITH T. The biology of vascular endothelial growth factor. Endo. Rev. 1997;18:4–25. doi: 10.1210/edrv.18.1.0287. [DOI] [PubMed] [Google Scholar]
- FOLKMAN J. What is the evidence that tumors are angiogenesis-dependent. J. Natl. Cancer Inst. 1991;82:4–6. doi: 10.1093/jnci/82.1.4. [DOI] [PubMed] [Google Scholar]
- FOLKMAN J., KLAGSBRUN M. Angiogenic Factors. Science. 1987;235:442–447. doi: 10.1126/science.2432664. [DOI] [PubMed] [Google Scholar]
- GEIJSEN N., DIJKERS P.F., LAMMERS J.J., KOENDERMAN L., COFFER P.J. Cytokine-mediated cPLA2 phosphorylation is regulated by multiple MAPK family members. FEBS Letters. 2000;471:83–88. doi: 10.1016/s0014-5793(00)01373-9. [DOI] [PubMed] [Google Scholar]
- GUO D., JIA Q., SONG H.Y., WARREN R.S., DONNER D.B. Vascular endothelial cell growth factor promotes tyrosine phosphorylation of mediators of signal transduction that contain SH2 domains. Association with endothelial cell proliferation. J. Biol. Chem. 1995;270:6729–6733. doi: 10.1074/jbc.270.12.6729. [DOI] [PubMed] [Google Scholar]
- HAZAN-HALEVY I., SEGER R., LEVY R. The requirement of both extracellular regulated kinase and p38 mitogen-activated protein kinase for stimulation of cytosolic phospholipase A2 activity by either FcγRIIA or FcγRIIIB in human neutrophils. A possible role for Pyk2 but not for the Grb2-Sos-Shc complex. J. Biol. Chem. 2000;275:12416–12423. doi: 10.1074/jbc.275.17.12416. [DOI] [PubMed] [Google Scholar]
- HE H., VENEMA V.J., GU X., VENEMA R.C., MARRERO M.B., CALDWELL R.B. Vascular endothelial growth factor signals endothelial cell production of nitric oxide and prostacyclin through flk-1/KDR activation of c-Src. J. Biol. Chem. 1999;274:25130–25135. doi: 10.1074/jbc.274.35.25130. [DOI] [PubMed] [Google Scholar]
- HILLER G., SUNDLER R. Activation of arachidonate release and cytosolic phospholipase A2 via extracellular signal-regulated kinase and p38 mitogen-activated protein kinase in macrophages stimulated by bacteria or zymosan. Cell. Signal. 1999;11:863–869. doi: 10.1016/s0898-6568(99)00058-3. [DOI] [PubMed] [Google Scholar]
- MOULTON K.S., HELLER E., KONERDING M.A., FLYNN E., PALINSKI W., FOLKMAN J. Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation. 1999;99:1726–1732. doi: 10.1161/01.cir.99.13.1726. [DOI] [PubMed] [Google Scholar]
- MUROHARA T., HOROWITZ J.R., SILVER M., TSURUMI Y., CHEN D., SULLIVAN A., ISNER J.M. Vascular endothelial growth factor/vascular permeability factor enhances vascular permeability via nitric oxide and prostacyclin. Circulation. 1998;97:99–107. doi: 10.1161/01.cir.97.1.99. [DOI] [PubMed] [Google Scholar]
- MUTHALIF M.M., BENTER I.F., KHANDEKAR Z., GABER L., ESTES A., MALIK S., PARMENTIER J.H., MANNE V., MALIK K.U. Contribution of Ras GTPase/MAP kinase and cytochrome P450 metabolites to deoxycorticosterone-salt-induced hypertension. Hypertension. 2000;35:457–463. doi: 10.1161/01.hyp.35.1.457. [DOI] [PubMed] [Google Scholar]
- NIXON A.B., O'FLAHERTY J.T., SALYER J.K., WYKLE R.L. Acetyl-CoA:1-O-alkyl-2-lyso-sn-glycero-3-phosphocholine acetyltransferase is directly activated by p38 kinase. J. Biol. Chem. 1999;274:5469–5473. doi: 10.1074/jbc.274.9.5469. [DOI] [PubMed] [Google Scholar]
- PAPAPETROPOULOS A., GARCIA-CARDENA G., MADRI J.A., SESSA W.C. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J. Clin. Invest. 1997;100:3131–3139. doi: 10.1172/JCI119868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRESCOTT S.M., ZIMMERMAN G.A., MCINTYRE T.M. Platelet-activating factor. J. Biol. Chem. 1990;265:17381–17384. [PubMed] [Google Scholar]
- ROUSSEAU S., HOULE F., LANDRY J., HUOT J. p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene. 1997;15:2169–2177. doi: 10.1038/sj.onc.1201380. [DOI] [PubMed] [Google Scholar]
- SA G., FOX P.L. Activation of cytosolic phospholipase A2 by basic fibroblast growth factor via a p42 mitogen-activated protein kinase-dependent phosphorylation pathway in endothelial cells. J. Biol. Chem. 1994;269:3219–3225. doi: 10.1074/jbc.270.5.2360. [DOI] [PubMed] [Google Scholar]
- SENGER D.R., GALLI S.J., DVORAK A.M., PERRUZI C.A., HARVEY V.S., DVORAK H.F. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983–985. doi: 10.1126/science.6823562. [DOI] [PubMed] [Google Scholar]
- SIROIS M.G., EDELMAN E.R. VEGF effect on vascular permeability is mediated by synthesis of platelet-activating factor. Am. J. Physiol. 1997;272:H2746–H2756. doi: 10.1152/ajpheart.1997.272.6.H2746. [DOI] [PubMed] [Google Scholar]
- SNYDER F., FITZGERALD V., BLANK M.L. Biosynthesis of Platelet-activating factor and enzyme inhibitors. Adv. Exp. Med. Biol. 1996;416:5–10. doi: 10.1007/978-1-4899-0179-8_2. [DOI] [PubMed] [Google Scholar]
- SOKER S., TAKASHIMA S., MIAO H.Q., NEUFELD G., KLAGSBRUN M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell. 1998;92:735–745. doi: 10.1016/s0092-8674(00)81402-6. [DOI] [PubMed] [Google Scholar]
- TAKAHASHI T., UENO H., SHIBUYA M. VEGF activates protein kinase C-dependent, but Ras-independent Raf-MEK-MAP kinase pathway for DNA synthesis in primary endothelial cells. Oncogene. 1999;18:2221–2230. doi: 10.1038/sj.onc.1202527. [DOI] [PubMed] [Google Scholar]
- THOMPSON N.T., BONSER R.W., TATESON J.E., SPACEY G.D., RANDALL R.W., HODSON H.F., GARLAND L.G. Demethoxyviridin and wortmannin block phospholipase C and D activation in the human neutrophil. Br. J. Pharmacol. 1991;103:1237–1241. doi: 10.1111/j.1476-5381.1991.tb12330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TIBBLES L.A., WOODGETT J.R. The stress-activated protein kinase pathways. Cell. Mol. Life Sci. 1999;55:1230–1254. doi: 10.1007/s000180050369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOBIN A.B. Phosphorylation of phospholipase C-coupled receptors. Pharmacol. Therap. 1997;75:135–151. doi: 10.1016/s0163-7258(97)00053-3. [DOI] [PubMed] [Google Scholar]
- TOULLEC D., PIANETTI P., COSTE H., BELLEVERGUE P., GRAND-PARRET T., AJAKANE M., BAUDET V., BOISSIN P., BOURSIER E., LORIOLLE F. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J. Biol. Chem. 1991;266:15771–15781. [PubMed] [Google Scholar]
- UNEMORI E.N., FERRARA N., BAUER E.A., AMENTO E.P. Vascular endothelial growth factor induces interstitial collagenase expression in human endothelial cells. J. Cell. Physiol. 1992;153:557–562. doi: 10.1002/jcp.1041530317. [DOI] [PubMed] [Google Scholar]
- VAN BIESEN T., LUTTRELL L.M., HAWES B.E., LEFKOWITZ R.J. Mitogenic signaling via G protein-coupled receptors. Endoc. Rev. 1996;17:698–711. doi: 10.1210/edrv-17-6-698. [DOI] [PubMed] [Google Scholar]
- VLAHOS C.J., MATTER W.F., BROWN R.F., TRAYNOR-KAPLAN A.E., HEYWOTH P.G., PROSSNITZ E.R., YE R.D., MARDER P., SCHELM J.A., ROTHFUSS K.J. Investigation of neutrophil signal transduction using a specific inhibitor of phosphatidylinositol 3-kinase. J. Imuunol. 1995;154:2413–2422. [PubMed] [Google Scholar]
- WHATLEY R.E., NELSON P., ZIMMERMAN G.A., STEVENS D.L., PARKER C.J., MCINTYRE T.M., PRESCOTT S.M. The regulation of platelet-activating factor production in endothelial cells. The role of calcium and protein kinase C. J. Biol. Chem. 1989;264:6325–6333. [PubMed] [Google Scholar]
- WYKLE R.L., MALONE B., SNYDER F. Enzymatic synthesis of 1-alkyl-2-acetyl-sn-glycero-3-phosphocholine, a hypotensive and platelet-aggregating lipid. J. Biol. Chem. 1980;255:10256–10260. [PubMed] [Google Scholar]
- XIA P., AIELLO L.P., ISHII H., JIANG Z.Y., PARK D.J., ROBINSON G.S., TAKAGI H., NEWSOME W.P., JIROUSEK M.R., KING G.L. Characterization of vascular endothelial growth factor's effect on the activation of protein kinase C, its isoforms, and endothelial cell growth. J. Clin. Invest. 1996;98:2018–2026. doi: 10.1172/JCI119006. [DOI] [PMC free article] [PubMed] [Google Scholar]