

Abstract
Previous data demonstrate that the tricyclic antidepressant, desipramine, induces glucocorticoid receptor (GR) translocation from the cytoplasm to the nucleus in L929 cells and increases dexamethasone-induced GR-mediated gene transcription in L929 cells stably transfected with the mouse mammary tumour virus-chloramphenicol acetyltransferase (MMTV-CAT) reporter gene (LMCAT cells) (Pariante et al., 1997).
To extend these findings, the present study has investigated the effects of 24 h coincubation of LMCAT cells with dexamethasone and amitriptyline, clomipramine, paroxetine, citalopram or fluoxetine.
All antidepressants, except fluoxetine, enhanced GR-mediated gene transcription, with clomipramine having the greatest effect (10 fold increase). Twenty-four hours coincubation of cells with desipramine, clomipramine or paroxetine, also enhanced GR function in the presence of cortisol, but not of corticosterone.
It is proposed that these effects are due to the antidepressants inhibiting the L929 membrane steroid transporter, which actively extrudes dexamethasone and cortisol from the cell, but not corticosterone. This is further confirmed by the fact that clomipramine failed to enhance GR-mediated gene transcription in the presence of dexamethasone when the membrane steroid transporter was blocked by verapamil.
The membrane steroid transporters that regulate access of glucocorticoids to the brain in vivo, like the multiple drug resistance p-glycoprotein, could be a fundamental target for antidepressant action.
Keywords: Cortisol, corticosterone, dexamethasone, glucocorticoid receptor, H-89, hypothalamic-pituitary-adrenal (HPA) axis, L929 cells, multiple drug resistance (MDR) p-glycoprotein, tricyclic antidepressants, selective serotonin reuptake inhibitor (SSRI), verapamil
Introduction
The hypothesis that antidepressants exert their clinical effects through direct modulation of the glucocorticoid receptor (GR) is one of the most striking and innovative models of the mechanism of action of this class of drugs (Barden, 1999; Holsboer, 2000; McQuade & Young, 2000; Pariante & Miller, 2001).
The GR is a ligand-induced transcription factor that belongs to the steroid/thyroid receptor superfamily. The unactivated receptor resides primarily in the cytoplasm, and, when bound by steroid, undergoes a conformational change and exposes the nuclear localization signal region, that allows translocation from the cytoplasm to the nucleus. In the nucleus, the activated GR influences gene transcription either by binding to glucocorticoid response elements (GREs) or interacting with other transcription factors (de kloet et al., 1998).
Together with the mineralocorticoid receptor, the GR mediates negative feedback regulation of the hypothalamic-pituitary-adrenal (HPA) axis by endogenous glucocorticoids, and is believed to be more important in the regulation of HPA when endogenous levels of glucocorticoids are high (de kloet et al., 1998). Consistent with the fact that patients with major depression exhibit impaired HPA negative feedback in the context of elevated circulating levels of cortisol (Nemeroff, 1996), a number of studies have described that the function of GR is reduced in depressed patients (GR resistance) and that antidepressants act by reversing these putative GR changes (Pariante & Miller, 2001). Specifically, studies in depressed patients, animals, and cellular models, have demonstrated that antidepressants increase GR expression, enhance GR function and promote GR nuclear translocation; this, in turn, is associated with enhanced GR-mediated negative feedback by endogenous glucocorticoids, and thus with reduced resting and stimulated HPA axis activity (Pariante & Miller, 2001).
Work by our group has described in L929 cells (mouse fibroblasts) that incubation with the tricyclic antidepressant, desipramine, induces GR translocation from the cytoplasm to the nucleus in the absence of steroids (Pariante et al., 1997). Moreover, we have found that coincubation of desipramine and dexamethasone (Dex) leads to enhanced GR-mediated gene transcription, while preincubation of desipramine followed by Dex leads to reduced GR-mediated gene transcription (Pariante et al., 1997). This latter finding has been recently replicated by Budziszewska et al. (2000), who also found that preincubation of L929 mouse fibroblast cells with various antidepressants (including desipramine) reduce GR-mediated gene transcription induced by a subsequent treatment with corticosterone or Dex. However, the molecular mechanism underlying the effects of antidepressants on GR function remains unclear.
Recently, the role of membrane steroid transporters in regulating the function of the GR, by modulating the intracellular access of steroid hormones, has been increasingly recognized. Some GR ligands like cortisol (the endogenous glucocorticoid in humans) and Dex – but not corticosterone and progesterone – are actively excreted from cells by membrane transporters belonging to the ATP-binding cassette family of transporters, like the multiple drug resistance (MDR) p-glycoprotein, (Kralli & Yamamoto, 1996; Krishna & Mayer, 2000; Medh et al., 1998; Pariante et al., 2001; Ueda et al., 1992). Moreover, the MDR p-glycoprotein localized on the apical membrane of the endothelial cells of the blood-brain barrier has been found, in mice and humans, to limit the access of Dex and cortisol (but not corticosterone) to the brain (Karssen et al., 2001; Meijer et al., 1998).
Because antidepressants have been shown to inhibit the MDR p-glycoprotein in tumor cells (Merry et al., 1991; Szabo et al., 1999; Varga et al., 1996) we hypothesized that one mechanism by which antidepressants regulate GR function in vitro is by regulating the function of membrane steroid transporters and therefore the intracellular concentration of glucocorticoids. In the present study we explore this hypothesis by examining the effects of a range of antidepressants on GR function (GR-mediated gene transcription) in the presence of steroids that are differentially affected by the L929 membrane steroid transporter. Moreover, we assessed the ability of the inhibitor of the membrane steroid transporter, verapamil, to reverse the effects of antidepressants on GR function. Since glucocorticoids are reliably available in vivo, the conditions under which antidepressants act clinically are best modelled by coincubation of antidepressants and hormone (Pariante & Miller, 2001), and this is the experimental design we have used in the present study.
Methods
Materials
Amitriptyline, clomipramine, desipramine and fluoxetine were obtained from SIGMA. Citalopram was a kind gift from Lundbeck (Denmark). Paroxetine was a kind gift from SmithKline Beecham (U.K.). The antidepressants were tested at 1 or 10 μM concentrations. This range of concentrations resembles the therapeutic plasma and brain levels of antidepressants (Burke & Preskorn, 1995; Glotzbach & Preskorn, 1982; Hrdina & Dubas, 1981). Moreover, micromolar concentrations of antidepressants have been previously used by others and us in studies that have investigated the in vitro effects of antidepressants on the GR (Budziszewska et al., 2000; Pariante et al., 1997; Vedder et al., 1999) or on other molecular systems (Chen & Rasenick, 1995; Szabo et al., 1999; Varga et al., 1996; Xia et al., 1999). In one series of experiments we tested clomipramine (100 μM and 1 mM) and found evidence of toxicity (reduced cells proliferation and survival); therefore, we did not use concentrations higher than 10 μM.
Assessment of GR-mediated gene transcription
Cell culture conditions and drug treatments
The stably transfected CAT reporter cell line LMCAT [derived from L929 (Sanchez et al., 1994)] was generously provided by E.R. Sanchez (Department of Pharmacology, Medical College of Ohio, Toledo, OH, U.S.A.). Cells were maintained in 175-cm2 flasks at 37°C with a 5% CO2 and 95% air atmosphere. The culture medium was DMEM with 10% (v v−1) charcoal-stripped, delipidated, heat-inactivated (56°, 30 min) bovine calf serum (from SIGMA) and 0.2 mg ml−1 G418 (Geneticin) antibiotic. For the CAT-assay, LMCAT cells were subcultured in 6-well plates and grown for 48 – 72 h (final confluency 95%) prior to drug treatment. Treatment consisted of incubation with fresh medium containing vehicle (ETOH, final concentration <0.04%, or water), or final concentrations of drugs as described in the Results section. For verapamil treatments, cells were preincubated with verapamil (100 μM) for 3 h, followed by 24 h coincubation of verapamil (100 μM) and glucocorticoids or antidepressants.
CAT reporter cell line and CAT assay
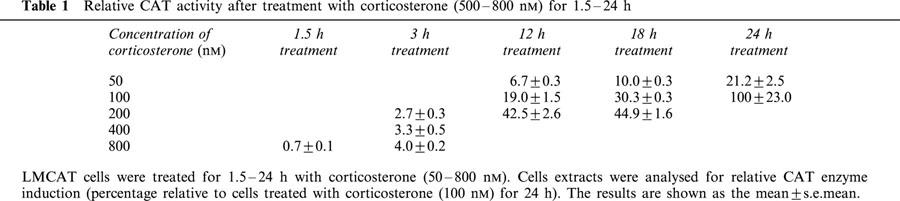
The LMCAT cell line is stably transfected with the mouse mammary tumour virus-chloramphenicol acetyltransferase (MMTV-CAT) reporter gene. Expression of CAT by these cells is under hormonal control by virtue of several GREs residing within the MMTV promoter, which lies upstream of the CAT reporter gene (Sanchez et al., 1994)._ The expression of CAT is dependent on the type of glucocorticoid hormone as well as the concentration and the duration of the incubation. As shown in Figure 1A,B, Dex, cortisol and corticosterone showed different potency, probably reflecting the different affinity for the GR (Dex has a much higher affinity than cortisol and corticosterone (de kloet et al., 1998; Rupprecht et al., 1993)) as well as the different ability to be expelled by the LMCAT membrane steroid transporter (see below in the Results section). We have previously described that 24 h treatment with Dex (both at 10 nM and 10 μM) gives approximately a 10 fold induction compared with 1.5 h treatment with the same concentration of Dex (Pariante et al., 1997). Corticosterone-induced CAT activity was also dependent on the duration of the incubation (see Table 1).
Figure 1.
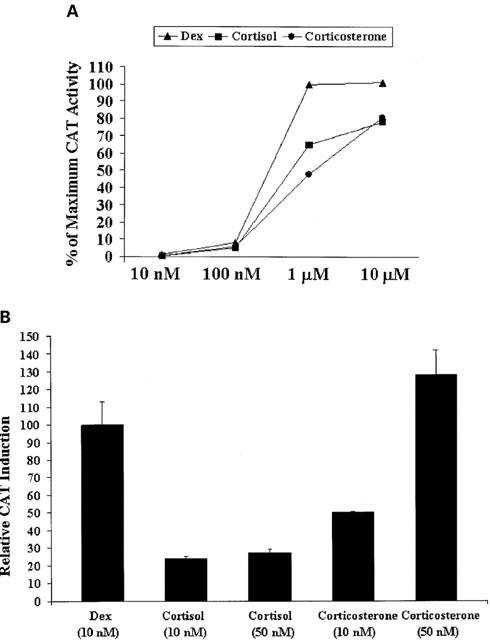
CAT induction after Dex, cortisol and corticosterone for 24 h. (A) LMCAT cells were treated for 24 h with Dex, cortisol or corticosterone (10 nM – 10 μM). Cells extracts were analysed for relative CAT enzyme induction (fold induction relative to maximal CAT activity). The results are shown as the mean±s.e.mean. (B). LMCAT cells were treated for 24 h with Dex (10 and 50 nM), or corticosterone (10 and 50 nM). Cells extracts were analysed for relative CAT enzyme induction (fold induction relative to Dex-treated cells). The results are shown as the mean±s.e.mean.
Table 1.
Relative CAT activity after treatment with corticosterone (500 – 800 nM) for 1.5 – 24 h
Measurement of CAT was performed using two different methods: a previously described liquid scintillation counting procedure (Promega, Madison, WI, U.S.A.) (Pariante et al., 1997; 1999; 2001), and a recently developed colorimetric enzyme immunoassay (Roche Diagnostic, U.K.), both according to the manufacturer's instructions. Most of the experiments were conducted using the colorimetric enzyme immunoassay or replicated with both techniques: no discrepancies in results between the two assays were found. For the liquid scintillation counting detection system, cell extracts were obtained by incubation/scraping cells in the manufacturer's lysis buffer, followed by 60°C heating for 10 min to inactivate endogenous deacetylase activity. After centrifugation (20,000×g for 5 min) supernatants were transferred to fresh tubes and processed for CAT enzyme activity. Each reaction was initiated by adding the cofactor n-butyryil Coenzyme A to tubes containing cell extracts and radiolabelled 3H-chloramphenicol. The CAT reaction was stopped and the butyrylated forms of 3H-chloramphenicol were separated by two consecutive extractions with mixed xylene. The extracts were transferred to vials for liquid scintillation counting. The c.p.m. measured in each sample represents the butyrylated fraction of the 3H-chloramphenicol and is directly proportional to CAT gene expression (as determined by a standard curve). The colorimetric enzyme immunoassay is based on the sandwich ELISA principle, and antibodies to CAT are prebound to the surface of the microtiter plate modules (MTP). Cell extracts were obtained by incubation in the manufacturer's lysis buffer. After centrifugation (20,000×g for 10 min) supernatants were transferred to the wells of the MTP (1 h, 37°C). Next, a digoxigenin-labelled antibody to CAT was added (1 h, 37°C), followed by an antibody to digoxigenin conjugated to peroxidase (1 h, 37°C). In the final step, the peroxidase substrate ABTS (with or without the substrate enhancer) was added, yielding a coloured reaction product. The absorbance of the sample was determined using the MRXII microplate reader (Dynex Technologies, Chantilly, VA, U.S.A.). The absorbance is directly correlated to the level of CAT present in the cell extract (as determined by a standard curve). Results were normalized with respect to cell number by measurement of metabolic activity by cleavage of the tetrazolium salt WST-1 (Roche Diagnostic, U.K.).
Statistical analysis
Data are presented as mean±standard error of the mean (s.e.m.) of three or more independent experiments, and were analysed using a one-way ANOVA. When the ANOVA revealed a significant main effect, the Student-Newman-Keuls post-hoc test was used for between-group comparisons. In the figures, * indicates P<0.05 using ANOVA (for two-groups comparison) or ANOVA followed by Student-Newman-Keuls post-hoc test.
Results
Effects of the antidepressants on GR-mediated gene transcription in the presence of Dex
To confirm and extend the previously described desipramine-induced enhancement of GR-mediated gene transcription in the presence of Dex (Pariante et al., 1997), we coincubated cells simultaneously for 24 h with Dex (10 nM) and amitriptyline (10 μM), clomipramine (10 μM), paroxetine (10 μM), citalopram (10 μM), or fluoxetine (10 μM). The results are presented in Figure 2 and expressed as percentage of the effect with Dex (10 nM). For all antidepressants, except for fluoxetine, simultaneous co-treatment with Dex (10 nM) induced an increase in GR-mediated gene transcription compared to cells treated with Dex (10 nM) alone. This potentiation was particularly intense in cells treated with Dex plus clomipramine (more than 10 fold) while was approximately +70 to +140% for the other antidepressants. With desipramine (10 μM), we also found approximately +80% of potentiation ((Pariante et al., 1997) and independently replicated for this study). Fluoxetine gave approximately 25% inhibition. We also examined amitriptyline (1 μM), clomipramine (1 μM) or paroxetine (1 μM), together with Dex (10 nM) for 24 h, and – as for desipramine (Pariante et al., 1997) – we found a smaller potentiation of GR-mediated gene transcription (+21±10% for amitriptyline;+22±3% for clomipramine; and+20±1% for paroxetine).
Figure 2.
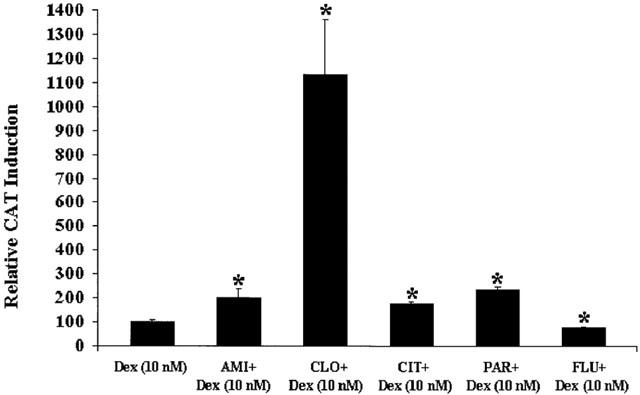
GR-mediated gene transcription after treatment with antidepressants in coincubation with Dex (10 nM). LMCAT cells were treated for 24 h with Dex (10 nM) alone, or in coincubation with the antidepressants, amitriptyline (10 μM) (AMI), clomipramine (10 μM) (CLO), citalopram (10 μM) (CIT), paroxetine (10 μM) (PAR), or fluoxetine (10 μM) (FLU). Cells extracts were analysed for relative CAT enzyme induction (fold induction relative to Dex-treated cells). The results are shown as the mean±s.e.mean. *Indicates significant (P<0.05) difference vs Dex (10 nM).
Finally, we tested clomipramine (10 μM) together with lower concentrations of Dex: (2.5 nM) and (0.625 nM) for 24 h. We found a clear clomipramine-induced potentiation of GR-mediated gene transcription with Dex 2.5 nM (+180±9%) but only a minimal effect with Dex 0.625 nM (+13±2%).
As we have previously reported for desipramine (Pariante et al., 1997), all antidepressants alone had no basal effect GR-mediated gene transcription (CAT levels equal to vehicle treatment).
Effects of verapamil and H-89 on GR-mediated gene transcription in the presence of Dex, cortisol or corticosterone
Although it is well known that the MDR p-glycoprotein and similar membrane steroid transporters in a variety of tissues expel Dex and cortisol, but not corticosterone, out of the cells (Krishna & Mayer, 2000; Ueda et al., 1992), we have limited data in L929 cells. Therefore, we wanted to test whether Dex and cortisol, but not corticosterone, are excreted by the L929 cells steroid transporter. We used two different approaches: blocking the function of the membrane steroid transporter with verapamil, and downregulating the expression of the membranes steroid transporter with H-89. Blocking or downregulating the membrane steroid transporter would lead to higher intracellular concentrations of transported glucocorticoids (in our hypothesis, Dex and cortisol, but not corticosterone) and therefore to higher levels of GR-mediated gene transcription. While we and others have previously demonstrated that verapamil blocks the L929 membrane steroid transporter and thereby increases Dex intracellular accumulation (Marsaud et al., 1998) as well as GR translocation and GR-mediated gene transcription in the presence of Dex (Medh et al., 1998; Pariante et al., 2001), H-89 effects have only been described for the MDR-p-glycoprotein (Wartenberg et al., 2000) and have not been examined in L929 cells.
Interestingly, corticosterone and cortisol have similar affinities for the GR (de kloet et al., 1998; Rupprecht et al., 1993), but cortisol gave smaller CAT induction than corticosterone (see Figure 1B). Based on the following results, we have interpreted this finding as being secondary to the fact that cortisol, but not corticosterone, is expelled by the membrane steroid transporter. In fact, 24 h coincubation of cells with verapamil (100 μM) and Dex (10 nM) or cortisol (50 nM) led to larger GR-mediated gene transcription compared to cells treated with these glucocorticoids alone: 100(±4) fold potentiation for Dex (10 nM) (thus reaching maximal CAT induction; see Figure 1A); and 19(±1) fold potentiation for cortisol (50 nM). However, verapamil (100 μM) did not enhance GR-mediated gene transcription in the presence of corticosterone (50 nM). In fact, coincubation of cells with verapamil (100 μM) and corticosterone (50 nM) led to 60(±3)% inhibition of GR-mediated gene transcription compared to cells treated with corticosterone (50 nM) alone. Similarly, 24 h coincubation of cells with H-89 (1 μM) and Dex (10 nM) or cortisol (50 nM) led to larger GR-mediated gene transcription compared to cells treated with these glucocorticoids alone: 45(±0.5) fold potentiation for Dex (10 nM) and 22(±0.9) fold potentiation for cortisol (50 nM). However, H-89 (1 μM) had no effect on GR-mediated gene transcription in the presence of corticosterone (50 nM) (+18±24%). Both verapamil and H-89 had no effect on GR-mediated gene transcription in the absence of steroids (CAT levels equal to vehicle treatment).
Effects of desipramine, clomipramine and paroxetine on GR-mediated gene transcription in the presence of cortisol or corticosterone
To assess whether the effects of antidepressants on GR-mediated gene transcription is mediated by the L929 membrane steroid transporter, we coincubated an antidepressant with steroid hormones that are differentially affected by the transporter. Since we found evidence that Dex and cortisol are expelled by the L929 cells steroid transporter, while corticosterone is not (see above), we decided to assess the effects of coincubation of an antidepressant with cortisol and corticosterone. If the antidepressants enhance GR-mediated gene transcription in the presence of Dex by inhibiting the membrane steroid transporter, and therefore increasing Dex intracellular concentrations, we would have seen these effects also in the presence of cortisol, but not in the presence of corticosterone. We focused these experiments on three antidepressants: desipramine (a tricyclic antidepressant and noradrenaline reuptake inhibitor), clomipramine (a tricyclic antidepressant and serotonin reuptake inhibitor) and paroxetine (a bicyclic antidepressant that inhibits both noradrenaline and serotonin reuptake (Owens et al., 2000)).
First, we coincubated cells simultaneously with desipramine (10 μM), clomipramine, (10 μM), or paroxetine (10 μM), plus cortisol (50 nM) for 24 h. The results are presented in Figure 3 and are expressed as percentage of the effect with cortisol (50 nM). We found that all antidepressants strongly potentiated GR-mediated gene transcription in the presence of cortisol (50 nM). Moreover, clomipramine, which gave the strongest potentiation of GR-mediated gene activity in the presence of Dex, also gave the strongest potentiation in the presence of cortisol (approximately 4 fold).
Figure 3.
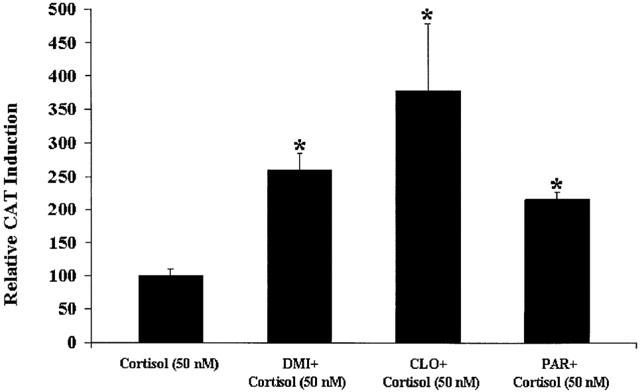
GR-mediated gene transcription after treatment with antidepressants in coincubation with cortisol (50 nM). LMCAT cells were treated for 24 h with cortisol (50 nM) alone, or in coincubation with the antidepressants, desipramine (10 μM) (DMI), clomipramine (10 μM) (CLO), or paroxetine (10 μM) (PAR). Cells extracts were analysed for relative CAT enzyme induction (fold induction relative to cortisol-treated cells). The results are shown as the mean±s.e.mean. *Indicates significant (P<0.05) difference vs cortisol (50 nM).
Secondly, we coincubated cells simultaneously with desipramine (10 μM), clomipramine, (10 μM), or paroxetine (10 μM), plus corticosterone (50 nM), for 24 h. The results are presented in Figure 4 and are expressed as percentage of the effect with corticosterone (50 nM). We found no evidence of antidepressant-induced potentiation of GR-mediated gene transcription in the presence of corticosterone (50 nM). In fact, we found that all antidepressants induced less GR-mediated gene transcription in the presence of corticosterone (50 nM) (with desipramine only reaching P=0.1 statistical significance), compared to cells treated with corticosterone (50 nM) alone. Interestingly, clomipramine, gave the strongest potentiation of GR-mediated gene activity in the presence of Dex and cortisol, and also gave the strongest inhibition in the presence of corticosterone (approximately 35% inhibition). To corroborate these findings, we conducted some more experiments in which clomipramine (10 μM) was coincubated with different concentrations of corticosterone. We never found evidence of clomipramine-induced potentiation of GR-mediated gene transcription in the presence of corticosterone [with corticosterone (10 nM) for 24 h: +2±4%; with corticosterone (1 μM) for 24 h: +4±12%; and with corticosterone (500 nM) for 3 h: −51+18%].
Figure 4.
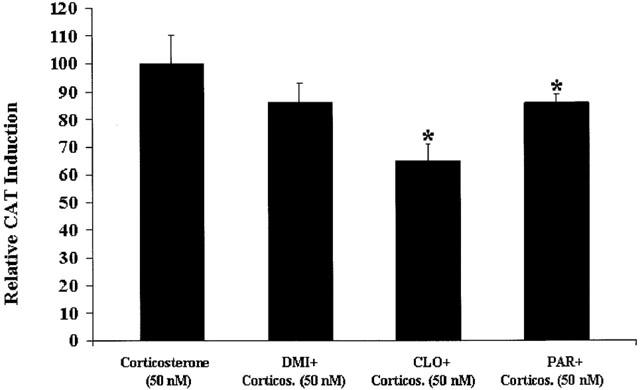
GR-mediated gene transcription after treatment with antidepressants in coincubation with corticosterone (50 nM). LMCAT cells were treated for 24 h with corticosterone (50 nM) alone, or in coincubation with the antidepressants, desipramine (10 μM) (DMI), clomipramine (10 μM) (CLO), or paroxetine (10 μM) (PAR). Cells extracts were analysed for relative CAT enzyme induction (fold induction relative to corticosterone-treated cells). The results are shown as the mean±s.e.mean. *Indicates significant (P<0.05) difference vs corticosterone (50 nM).
Effects of clomipramine on GR-mediated gene transcription in the presence of Dex and verapamil together
The previous experiments had supported our original hypothesis that the mechanisms by which desipramine and some other antidepressants increase GR-mediated gene transcription in L929 cells is by inhibiting the membrane steroid transporter and thus leading to increased intracellular concentrations of steroids – Dex or cortisol – in the cells. To corroborate these findings further, we examined the effects of clomipramine on GR-mediated gene transcription in the presence of Dex plus the steroid transporter inhibitor, verapamil. If inhibition of the steroid transporter is the mechanism by which antidepressants increase GR-mediated gene transcription in the presence of Dex, this effect should disappear in the presence of verapamil.
In a first series of experiments, we treated cells (for 24 h) with Dex (10 nM) plus verapamil (100 μM) or Dex (10 nM) plus verapamil (100 μM) plus clomipramine (10 μM), and found no potentiation of GR function by clomipramine (Figure 5, first three columns). However, because Dex (10 nM) plus verapamil induced maximum CAT induction (see above), this could have explained the lack of a further enhancing effect by clomipramine. Indeed, we found no potentiation of GR function by clomipramine in the presence of Dex (1 μM), which also induced maximum CAT induction (specifically, we had a small inhibition; see Figure 5, fourth and fifth columns. Therefore, we conducted a second series of experiment using Dex (2.5 nM), which alone gave a small but reliable CAT-induction (30% of the effect of Dex (10 nM)), showed potentiation by clomipramine (see above), and in the presence of verapamil (100 μM) induced levels of GR-mediated gene transcription similar to Dex (10 nM) alone (see Figure 6). We treated cells (for 24 h) with Dex (2.5 nM) plus verapamil (100 μM), or with Dex (2.5 nM) plus verapamil (100 μM) plus clomipramine (10 μM). As hypothesized, we found that clomipramine failed to potentiate GR-mediated gene transcription in the presence of Dex, after the membrane steroid transporter was blocked by verapamil. In fact, we found that clomipramine (10 μM) led to a approximately 40% decrease in GR-mediated gene transcription in the presence of Dex (2.5 nM) plus verapamil, compared to cells treated with Dex (2.5 nM) plus verapamil alone (Figure 6).
Figure 5.
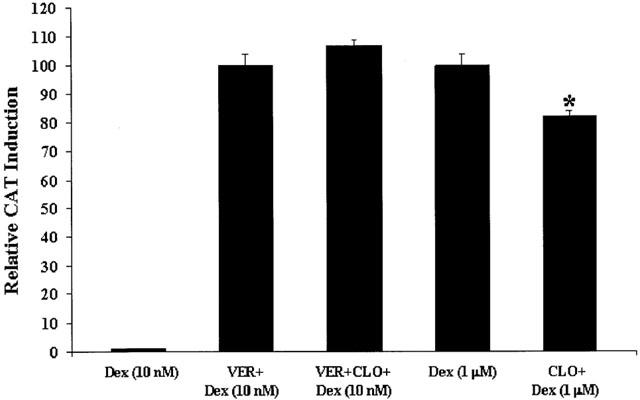
Effects of clomipramine on maximal CAT induction. LMCAT cells were treated for 24 h with: (1) Dex (10 nM) in coincubation with verapamil (VER) (100 μM); (2) Dex (10 nM) in coincubation with verapamil (100 μM) and clomipramine (CLO) (10 μM); (3) Dex (1 μM) alone; or (4) Dex (1 μM) in coincubation with clomipramine (10 μM). CAT induction after treatment with Dex (10 nM) is also presented. Cells extracts were analysed for relative CAT enzyme induction [fold induction relative to cells treated with Dex (1 μM)). The results are shown as the mean±s.e.mean. *Indicates significant (P<0.05) difference vs Dex (1 μM).
Figure 6.
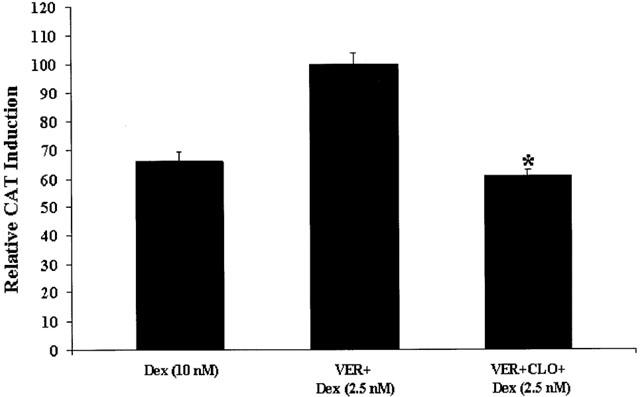
GR-mediated gene transcription after treatment with clomipramine in coincubation with verapamil and dex (2.5 nM). LMCAT cells were treated for 24 h with: (1) Dex (2.5 nM) in coincubation with verapamil (VER) (100 μM); or (2) Dex (2.5 nM) in coincubation with verapamil (VER) (100 μM) and clomipramine (CLO) (10 μM). CAT induction after treatment with Dex (10 nM) is also presented. Cells extracts were analysed for relative CAT enzyme induction (fold induction relative to cells treated with Dex (2.5 nM) plus verapamil). The results are shown as the mean±s.e.mean. *Indicates significant (P<0.05) difference vs Dex (2.5 nM) plus verapamil.
Discussion
Separate research groups in the recent years have reported that antidepressants increase GR function and GR expression in neuronal and non-neuronal cell cultures (Hery et al., 2000; Okugawa et al., 1999; Pariante et al., 1997; Pepin et al., 1992; Vedder et al., 1999). We have previously demonstrated that desipramine increases Dex-induced GR-mediated gene transcription in L929 mouse fibroblast cells (Pariante et al., 1997); and that these cells express a membrane steroid transporter that actively lowers intracellular concentration of Dex (Pariante et al., 2001). We now present novel evidence suggesting that the effects of desipramine on the GR are shared by some other chemically unrelated antidepressants, and that one molecular mechanism behind these effects is the inhibition of membrane steroid transporters, leading to increased intracellular concentrations of steroids. In summary: (1) clomipramine, amitriptyline, citalopram and paroxetine increased GR-mediated gene transcription in the presence of Dex; (2) desipramine, clomipramine and paroxetine increased GR-mediated gene transcription in the presence of cortisol (the endogenous glucocorticoid in humans) but not of corticosterone; (3) verapamil and H-89 (two drugs that inhibit the function of the membrane steroid transporters), also increased GR-mediated gene transcription in the presence of Dex or cortisol, but not of corticosterone; (4) clomipramine failed to have any effect in the presence of Dex, after blocking the membrane steroid transporter with verapamil; and (5) fluoxetine, an antidepressant that does not seem to interact with the MDR-p-glycoprotein (Uhr et al., 2000), did not enhance GR-mediated gene transcription in the presence of Dex.
Interestingly, the antidepressants shown in the present study to increase GR-mediated gene transcription have structural features associated with the ability to block the MDR p-glycoprotein (Ford, 1996): amitriptyline, clomipramine and desipramine have a tricyclic ring structure, with a tertiary amino group (amitriptyline, clomipramine) or a heterocyclic ring structure containing a nitrogen group (clomipramine, desipramine); citalopram and paroxetine have a bicyclic, heterocyclic, ring structure, with a tertiary amino group (citalopram) or containing a nitrogen group (paroxetine); clomipramine, which has the strongest effect on GR-mediated gene transcription, is the only compound that has both the heterocyclic ring structure containing a nitrogen group and the tertiary amino group; fluoxetine, which has no effect on GR-mediated gene transcription, has none of these features, as it is a monocyclic antidepressant with a secondary amino group.
Previous studies have suggested that some antidepressants interact with membrane steroid transporters like the MDR p-glycoprotein in vivo and in vitro. For example, clomipramine at 10 mg kg day−1 for 2 days completely overcomes resistance to anticancer drugs of subcutaneous tumours in mice (Merry et al., 1991). Of note is that the dose used in this study is within the range (10 – 20 mg kg day−1) used in most animal studies showing GR upregulation by tricyclic antidepressants (Pariante & Miller, 2001). In vitro, amitriptyline and desipramine (at concentrations similar to those used in the present study) block the MDR p-glycoprotein-mediated efflux of rhodamine 123 from human colon cancer cells (Varga et al., 1996), cells from a leukaemia cell line (Szabo et al., 1999) and peripheral blood mononuclear cells of treatment-resistant leukaemic patients (Szabo et al., 1999).
In this study we also confirmed previous findings showing that verapamil and H-89 inhibit the function of membrane steriod transporters like the MDR-p-glycoprotein (Ford, 1996; Krishna & Mayer, 2000; Wartenberg et al., 2000). Verapamil is also a well-known blocker of the L-type calcium channel, and L929 cells do show verapamil-sensitive calcium fluxes (Kong et al., 1997). However, verapamil seems to inhibit the MDR p-glycoprotein by competitive antagonism of the binding of substrates to the transporter, an effect that is independent from the block of calcium channels (Ford, 1996). This is confirmed by the fact that both the S- and the R-enantiomers of verapamil are inhibitors of the MDR p-glycoprotein, while only the S-enantiomer binds to calcium channels (Ford, 1996). Moreover, tests conducted on several channel blockers have found no correlation between the calcium channel blocking activity and the anti-MDR activity (Ford, 1996). On the contrary, H-89 seems to act on the MDR p-glycoprotein by reducing its expression, an effect mediated by its known ability to inhibit protein kinase A (PKA) (Wartenberg et al., 2000). In fact, while PKA stimulation induces an increase in the p-glycoprotein expression, PKA inhibition by H-89 induces down-regulation of p-glycoprotein expression (Wartenberg et al., 2000).
Interestingly, the relationship between the chemical structure of an antidepressant, the known pharmacological mechanism and the effects on the GR has yet to be clarified, although the inhibition of catecholamine uptake – the mechanism currently believed to be crucial in the antidepressant action of these drugs – seems not relevant (Pariante & Miller, 2001). For example, in animal studies, GR upregulation in the brain is induced by tricyclics (Eiring & Sulser, 1997; Reul et al., 1993; Rossby et al., 1995; Seckl & Fink, 1992) but not by SSRIs (Seckl & Fink, 1992). Moreover, the noradrenaline reuptake inhibitor, oxaprotiline, has shown no effects on GR expression (Eiring & Sulser, 1997), and desipramine has been shown to induce GR upregulation even following neurotoxic lesioning of noradrenergic neurons with DSP4 (Rossby et al., 1995). Within in vitro experimental systems, which are likely to be devoid of catecholamine synthesis or of catecholamine reuptake sites within synaptic connections, GR upregulation or increased GR function had been described with tricyclics, mianserin (another antidepressant with a heterocyclic ring structure containing a nitrogen group), paroxetine and citalopram, but not fluoxetine (Hery et al., 2000; Okugawa et al., 1999; Pariante et al., 1997; Pepin et al., 1992; Vedder et al., 1999; this study). Our hypothesis – that modulation of the membrane steroid transporters is important for the effects of antidepressants on the GR – is a potential explanation of how chemically and pharmacologically unrelated drugs may have similar effects on the GR. In fact, antidepressants, as other membrane steroid transporters inhibitors, seem to modulate MDR by interacting directly with the membrane phospholipids, an effect that is not receptor-mediated and is related to the drugs physiochemical properties, that is, lipophilicity and electric charge (Castaing et al., 2000) (although interaction with the adenosine triphosphatase activity (Watanabe et al., 1997) and direct binding to the transporters (Martin et al., 1999) by MDR modulators have also been described).
As discussed above, we have previously demonstrated that preincubation with desipramine leads to reduced GR-mediated gene transcription induced by a subsequent treatment with Dex (Pariante et al., 1997). This is not due to a direct effect of desipramine on the capability of the GR to bind Dex: we have examined cytosolic extracts of untreated L929 cells in an exchange assay with 3H-Dex in the presence or absence of a 1000 fold excess of desipramine, and have found no difference in GR binding in incubates containing desipramine compared with incubates without desipramine (Pariante et al., 1997). Therefore, we have hypothesized that the reduced GR-mediated gene transcription induced by preincubation is due to the fact that the antidepressant causes GR translocation to the nucleus (Pariante et al., 1997). Antidepressant-translocated GR does not show any functional activity, as shown by the lack of effects on GR-mediated gene transcription. Moreover, once in the nucleus the GR is incapable of rebinding ligand (Scherrer et al., 1990). Thus, if the antidepressant is given prior to steroid exposure, less receptor capable of binding ligand is available in the cytoplasm, and an inhibitory effect is revealed. The findings in this study confirm and extend this model. In fact, we can now hypothesise that treatment of cells with an antidepressant in the presence of Dex or cortisol leads to enhanced GR-mediated gene transcription by increasing the intracellular concentrations of these hormones. However, antidepressants cannot increase the intracellular concentrations of corticosterone – or of Dex in the presence of verapamil – and therefore in these conditions the inhibitory effect is revealed, possibly because the GR is progressively sequestrated in the nucleus. This model is consistent with the recent study by Budziszewska et al. (2000), who also showed that preincubation with several antidepressants leads to reduced GR-mediated gene transcription induced by a subsequent treatment with Dex or corticosterone. Moreover, these authors also showed that the tricyclic antidepressant, imipramine, added to hippocampal cytosol, decreases binding of the corticosterone-receptor complex to DNA. Because the antidepressants-translocated GR is unable to activate GR-mediated gene transcription, it is possible that antidepressants induce an incorrect conformational change of the GR, associated with a lower affinity to DNA, as it has been reported in the presence of other compounds that activate GR translocation but not GR function (Bourgeois et al., 1984; Pariante et al., 2001; Wagner et al., 1999; Yu et al., 1995).
Clearly, further studies are needed to elucidate the molecular interactions between antidepressants and the GR. However, it is important to remind that all in vivo evidence, in humans and animals, support the notion that antidepressant treatment increases, rather than decreases, GR function (Pariante & Miller, 2001). Specifically, antidepressant treatment in patients with major depression increases GR sensitivity to glucocorticoids in lymphocytes (Wodarz et al., 1992) and potentiates GR-mediated negative feedback on the HPA by glucocorticoids (Heuser et al., 1996). Moreover, animal studies have shown that antidepressants increase GR-mediated negative feedback on the HPA axis, even in the absence of GR upregulation (Montkowski et al., 1995; Reul et al., 1993). Therefore, it seems plausible that the effects of antidepressants in vivo are mainly related to the effects on the membrane steroid transporters, leading to increased GR function. To this regard, it is of note that pre-treatment of rats with nifedipine (a MDR p-glycoprotein inhibitor) prevents the hippocampal GR upregulation induced by chronic treatment with desipramine, amitriptyline or electroconvulsive shock (Przegalinski et al., 1993).
Interestingly, membrane steroid transporters like the MDR p-glycoprotein have been described in normal animal and human tissues, including epithelial cells in the blood-brain barrier where they limit the access of cortisol to the brain (de kloet et al., 1998; Karssen et al., 2001; Meijer et al., 1998). Therefore, it is possible to hypothesize that inhibition of the membrane steroid transporters (most notably, in the blood-brain barrier) represents the mechanism by which antidepressants increase activation of the GR in the brain, enhance GR-mediated negative feedback on the HPA axis, and normalize HPA hyperactivity in depressed patients.
In conclusion, we have demonstrated, in LMCAT cells, that GR function is increased in vitro by several antidepressants, in the presence of Dex or cortisol, but not of corticosterone. The fact that Dex and cortisol, but not corticosterone, are actively extruded from the LMCAT cells by a membrane steroid transporter, has led us to hypothesize that these effects are due to the antidepressants inhibiting the membrane steroid transporter, thus causing increased intracellular concentration of Dex or cortisol, but not corticosterone. This is further confirmed by the fact that clomipramine fails to enhance GR function in the presence of Dex when the membrane steroid transporter is blocked by verapamil. We propose that membrane steroid transporters, especially those regulating access of glucocorticoids to the brain in vivo like the MDR p-glycoprotein, could be a fundamental target for antidepressant action.
Acknowledgments
This research was funded by a U.K. Medical Research Council (MRC) Clinical Training Fellowship to C.M. Pariante. Citalopram was a kind gift from Lundbeck (Denmark) and paroxetine was a kind gift from SmithKline Beecham (U.K.).
Abbreviations
- Dex
dexamethasone
- GR
glucocorticoid receptor
- GREs
glucocorticoid response elements
- h
hour
- HPA axis
hypothalamic-pituitary-adrenal axis
- LMCAT cells
L929 cells stably transfected with the mouse mammary tumour virus-chloramphenicol acetyltransferase reporter gene
- MDR
multiple drug resistance
- MMTV-CAT
mouse mammary tumour virus-chloramphenicol acetyltransferase reporter gene
- MTP
microtiter plate
- SSRI
selective serotonin reuptake inhibitor
References
- BARDEN N. Regulation of corticosteroids receptor gene expression in depression and antidepressant action. J. Psychiatry Neurosci. 1999;24:25–39. [PMC free article] [PubMed] [Google Scholar]
- BOURGEOIS S., PFAHL M., BAULIEU E.-E. DNA binding properties of glucocorticosteroid receptors bound to the steroid antagonist RU-486. EMBO J. 1984;3:751–755. doi: 10.1002/j.1460-2075.1984.tb01879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUDZISZEWSKA B., JAWORSKA-FEIL L., KAJTA M., LASON W. Antidepressant drugs inhibit glucocorticoid receptor mediated gene transcription–a possible mechanism. Br. J. Pharmacol. 2000;130:1385–1393. doi: 10.1038/sj.bjp.0703445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BURKE M.J., PRESKORN S.H.Short-term treatment of mood disorders with standard antidepressants Psychopharmacology. The fourth generation of progress 1995New York: Raven Press; 1053–1065.ed. Bloom, F.E. & Kupfer, D.J. pp [Google Scholar]
- CASTAING M., BROUANT P., LOISEAU A., SANTELLI-ROUVIER C., SANTELLI M., ALIBERT-FRANCO S., MAHAMOUD A., BARBE J. Membrane permeation by multidrug-resistance-modulators and non-modulators: effects of hydrophobicity and electric charge. J. Pharm. Pharmacol. 2000;52:289–296. doi: 10.1211/0022357001773977. [DOI] [PubMed] [Google Scholar]
- CHEN J., RASENICK M.M. Chronic treatment of C6 glioma cells with antidepressant drugs increases functional coupling between a G protein (Gs) and adenylyl cyclase. J. Neurochem. 1995;64:724–732. doi: 10.1046/j.1471-4159.1995.64020724.x. [DOI] [PubMed] [Google Scholar]
- DE KLOET E.R., VREUGDENHIL E., OITZL M.S., JOELS M. Brain corticosteroid receptor balance in health and disease. Endocr. Rev. 1998;19:269–301. doi: 10.1210/edrv.19.3.0331. [DOI] [PubMed] [Google Scholar]
- EIRING A., SULSER F. Increased synaptic availability of norepinephrine following desipramine is not essential for increases in GR mRNA. J. Neural. Transm. 1997;104:1255–1258. doi: 10.1007/BF01294725. [DOI] [PubMed] [Google Scholar]
- FORD J.M. Experimental reversal of p-glycoprotein-mediated multidrug resistance by pharmacological chemosensitisers. Eur. J. Cancer. 1996;32A:991–1001. doi: 10.1016/0959-8049(96)00047-0. [DOI] [PubMed] [Google Scholar]
- GLOTZBACH R.K., PRESKORN S.H. Brain concentration of tricyclic antidepressants: single-dose kinetics and relationship to plasma concentrations in chronically dosed rats. Psychopharmacology. 1982;78:25–27. doi: 10.1007/BF00470582. [DOI] [PubMed] [Google Scholar]
- HERY M., SEMONT A., FACHE M.-P., FAUDON M., HERY F. The effects of serotonin on glucocorticoid receptor binding in rat raphe nuclei and hippocampal cells in culture. J. Neurochem. 2000;74:406–413. doi: 10.1046/j.1471-4159.2000.0740406.x. [DOI] [PubMed] [Google Scholar]
- HEUSER I.J.E., SCHWEIGER U., GOTTHARDT U., SCHMIDER J., LAMMERS C.H., DETTLING M., YASSOURIDIS A., HOLSBOER F. Pituitary-adrenal-system regulation and psychopathology during amitriptyline treatment in elderly depressed patients and normal comparison subjects. Am. J. Psychiatry. 1996;153:93–99. doi: 10.1176/ajp.153.1.93. [DOI] [PubMed] [Google Scholar]
- HOLSBOER F. The corticosteroids receptor hypothesis of depression. Neuropsychopharmacology. 2000;23:477–501. doi: 10.1016/S0893-133X(00)00159-7. [DOI] [PubMed] [Google Scholar]
- HRDINA P.D., DUBAS T.C. Brain distribution and kinetics of desipramine in the rat. Can. J. Physiol. Pharmacol. 1981;59:163–167. doi: 10.1139/y81-027. [DOI] [PubMed] [Google Scholar]
- KARSSEN A.M., MEIJER O.C., VAN DER SANDT I.C.J., LUCASSEN P.J., DE LANGE E.C.M., DE BOER A.G., DE KLOET E.R. Multidrug resistance p-glycoprotein hampers the access of cortisol but not corticosterone to mouse and human brain. Endocrinology. 2001;142:2686–1294. doi: 10.1210/endo.142.6.8213. [DOI] [PubMed] [Google Scholar]
- KONG S.K., FUNG K.P., CHOY Y.M., LEE C.Y. Slow increase in intranuclear and cytosolic free calcium concentrations in L929 cells is important in tumour necrosis factor-alpha-mediated cell death. Oncology. 1997;54:55–62. doi: 10.1159/000227662. [DOI] [PubMed] [Google Scholar]
- KRALLI A., YAMAMOTO K.R. An FK506-sensitive transporter selectively decreases intracellular levels and potency of steroid hormones. J. Biol. Chem. 1996;271:17152–17156. doi: 10.1074/jbc.271.29.17152. [DOI] [PubMed] [Google Scholar]
- KRISHNA R., MAYER L.D. Multidrug resistance (MDR) in cancer. Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur. J. Pharm. Sci. 2000;11:265–283. doi: 10.1016/s0928-0987(00)00114-7. [DOI] [PubMed] [Google Scholar]
- MARSAUD V., MERCIER-BODARD C., FORTIN D., LE BIHAN S., RENOIR J.-M. Dexamethasone and triamcinolone acetonide accumulation in mouse fibroblasts is differently modulated by the immunosuppressants cyclosporin A, FK506, rapamycin and their analogues, as well as by other p-glycoprotein ligands. J. Steroid. Biochem. Molec. Biol. 1998;66:11–25. doi: 10.1016/s0960-0760(98)00008-9. [DOI] [PubMed] [Google Scholar]
- MARTIN C., BERRIDGE G., MISTRY P., HIGGINS C., CHARLTON P., CALLAGHAN R. The molecular interaction of the high affinity reversal agent XR9576 with P-glycoprotein. Br. J. Pharmacol. 1999;128:403–411. doi: 10.1038/sj.bjp.0702807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCQUADE R., YOUNG A.H. Future therapeutic targets in mood disorders: the glucocorticoid receptor. Brit. J. Psychiatry. 2000;177:390–395. doi: 10.1192/bjp.177.5.390. [DOI] [PubMed] [Google Scholar]
- MEDH R.D., LAY R.H., SCHMIDT T.J. Agonist-specific modulation of glucocorticoid receptor-mediated transcription by immunosuppressants. Mol. Cell. Endocrinol. 1998;138:11–23. doi: 10.1016/s0303-7207(98)00055-0. [DOI] [PubMed] [Google Scholar]
- MEIJER O.C., DE LANGE E.C.M., BREIMER D.D., DE BOER A.G., WORKEL J.O., DE KLOET E.R. Penetration of dexamethasone into brain glucocorticoid targets is enhanced in mdr1A p-glycoprotein knockout mice. Endocrinology. 1998;139:1789–1793. doi: 10.1210/endo.139.4.5917. [DOI] [PubMed] [Google Scholar]
- MERRY S., HAMILTON T.G., FLANIGAN P., FRESHNEY R.I., KAYE S.B. Circumvention of pleiotropic drug resistance in subcutaneous tumours in vivo with verapamil and clomipramine. Eur. J. Cancer. 1991;27:31–34. doi: 10.1016/0277-5379(91)90054-h. [DOI] [PubMed] [Google Scholar]
- MONTKOWSKI A., BARDEN N., WOTJAK C., STEC I., GANSTER J., MEANEY M., ENGELMANN M., REUL J.M.H.M., LANDGRAF R., HOLSBOER F. Long-term antidepressant treatment reduces behavioural deficits in transgenic mice with impaired glucocorticoid receptor function. J. Neuroendocrinol. 1995;7:841–845. doi: 10.1111/j.1365-2826.1995.tb00724.x. [DOI] [PubMed] [Google Scholar]
- NEMEROFF C.B. The corticotropin-releasing factor (CRF) hypothesis of depression: new findings and new directions. Mol. Psychiat. 1996;1:336–342. [PubMed] [Google Scholar]
- OKUGAWA G., OMORI K., SUZUKAWA J., FUJISEKI Y., KINOSHITA T., INAGAKI C. Long-term treatment with antidepressants increases glucocorticoid receptor binding and gene expression in cultured rat hippocampal neurones. J. Neuroendocrinol. 1999;11:887–895. doi: 10.1046/j.1365-2826.1999.00405.x. [DOI] [PubMed] [Google Scholar]
- OWENS M.J., KNIGHT D.L., NEMEROFF C.B. Paroxetine binding to the rat norepinephrine transporter in vivo. Biol. Psychiatry. 2000;47:842–845. doi: 10.1016/s0006-3223(99)00314-5. [DOI] [PubMed] [Google Scholar]
- PARIANTE C.M., MILLER A.H. Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol. Psychiatry. 2001;49:391–404. doi: 10.1016/s0006-3223(00)01088-x. [DOI] [PubMed] [Google Scholar]
- PARIANTE C.M., PEARCE B.D., PISELL T.L., OWENS M.J., MILLER A.H. Steroid-independent translocation of the glucocorticoid receptor by the antidepressant desipramine. Mol. Pharmacol. 1997;52:571–581. doi: 10.1124/mol.52.4.571. [DOI] [PubMed] [Google Scholar]
- PARIANTE C.M., PEARCE B.D., PISELL T.L., SANCHEZ C.I., PO C., SU C., MILLER A.H. The proinflammatory cytokine, interleukin-1 alpha, reduces glucocorticoid receptor translocation and function. Endocrinology. 1999;140:4359–4366. doi: 10.1210/endo.140.9.6986. [DOI] [PubMed] [Google Scholar]
- PARIANTE C.M., PEARCE B.D., PISELL T.L., SU C., MILLER A.H. The steroid receptor antagonists, RU486 and RU40555, activate glucocorticoid receptor translocation and are not excreted by the steroid hormone transporter in L929 cells. J. Endocrinology. 2001;169:309–320. doi: 10.1677/joe.0.1690309. [DOI] [PubMed] [Google Scholar]
- PEPIN M.-C., GOVINDAN M.V., BARDEN N. Increased glucocorticoid receptor gene promoter activity after antidepressant treatment. Mol. Pharmacol. 1992;41:1016–1022. [PubMed] [Google Scholar]
- PRZEGALINSKI E., BUDZISZEWSKA B., SIWANOWICZ J., JAWORSKA L. The effect of repeated combined treatment with nifedipine and antidepressant drugs or electroconvulsive shock on the hippocampal corticosteroid receptors in rats. Neuropharmacology. 1993;32:1397–1400. doi: 10.1016/0028-3908(93)90036-3. [DOI] [PubMed] [Google Scholar]
- REUL J.M.H.M., STEC I., SODER M., HOLSBOER F. Chronic treatment of rats with the antidepressant amitriptyline attenuates the activity of the hypothalamic-pituitary-adrenocortical system. Endocrinology. 1993;133:312–320. doi: 10.1210/endo.133.1.8391426. [DOI] [PubMed] [Google Scholar]
- ROSSBY S.P., NALEPA I., HUANG M., PERRIN C., BURT A.M., SCHMIDT D.E., GILLESPIE D.D., SULSER F. Norepinephrine-independent regulation of GRII mRNA in vivo by a tricyclic antidepressant. Brain. Res. 1995;687:79–82. doi: 10.1016/0006-8993(95)00459-4. [DOI] [PubMed] [Google Scholar]
- RUPPRECHT R., REUL J.M.H.M., VAN STEENSEL B., SPENGLER D., SODER M., BERNING B., HOLSBOER F., DAMM K. Pharmacological and functional characterization of human mineralocorticoid and glucocorticoid receptor ligands. Eur. J. Pharmacol. 1993;247:145–154. doi: 10.1016/0922-4106(93)90072-h. [DOI] [PubMed] [Google Scholar]
- SANCHEZ E.R., HU J.-L., ZHONG S., SHEN P., GREENE M.J., HOUSLEY P.R. Potentiation of glucocorticoid receptor-mediated gene expression by heat and chemical shock. Mol. Endocrinol. 1994;8:408–421. doi: 10.1210/mend.8.4.8052262. [DOI] [PubMed] [Google Scholar]
- SCHERRER L.C., DALMAN F.C., MASSA E., MESHINCHI S., PRATT W.B. Structural and functional reconstitution of the glucocorticoid receptor-hsp90 complex. J. Biol. Chem. 1990;265:21397–21400. [PubMed] [Google Scholar]
- SECKL J.R., FINK G. Antidepressants increase glucocorticoid and mineralocorticoid receptor mRNA expression in rat hippocampus in vivo. Neuroendocrinology. 1992;55:621–626. doi: 10.1159/000126180. [DOI] [PubMed] [Google Scholar]
- SZABO D., SZABO G., JR, OCSOVSZKI I., ASZALOS A., MOLNAR J. Anti-psychotic drugs reverse multidrug resistance of tumor cell lines and human AML cell ex-vivo. Cancer Lett. 1999;139:115–119. doi: 10.1016/s0304-3835(99)00020-8. [DOI] [PubMed] [Google Scholar]
- UEDA K., OKAMURA N., HIRAI M., TANIGAWARA Y., SAEKI T., KIOKA N., KOMANO T., HORI R. Human P-glycoprotein transports cortisol, aldosterone, and dexamethasone, but not progesterone. J. Biol. Chem. 1992;267:24248–24252. [PubMed] [Google Scholar]
- UHR M., STECKLER T., YASSOURIDIS A., HOLSBOER F. Penetration of amitriptyline, but not fluoxetine, into brain is enhanced in mice with blood-brain barrier deficiency due to Mdr1a p-glycoprotein gene disruption. Neuropsychopharmacology. 2000;22:380–387. doi: 10.1016/S0893-133X(99)00095-0. [DOI] [PubMed] [Google Scholar]
- VARGA A., NUGEL H., BAEHR R., MARX U., HEVER A., NACSA J., OCSOVSZKY I., MOLNAR J. Reversal of multidrug resistance by amitriptyline in vitro. Anticancer Res. 1996;16:209–212. [PubMed] [Google Scholar]
- VEDDER H., BENING-ABU-SHACH U., LANQUILLON S., KRIEG J.-K. Regulation of glucocorticoid receptor-mRNA in human blood cells by amitriptyline and dexamethasone. J. Psychiat. Res. 1999;33:303–308. doi: 10.1016/s0022-3956(99)00006-0. [DOI] [PubMed] [Google Scholar]
- WAGNER B.L., POLLIO G., GIANGRANDE P., WEBSTER J.C., BRESLIN M., MAIS D.E., COOK C.E., VEDECKIS W.V., CIDLOWSKY J.A., MCDONNEL D.P. The novel progesterone receptor antagonists RTI 3021-012 and RTI 3021-022 exhibit complex glucocorticoid receptor antagonist activities: implications for the development of dissociated antiprogestins. Endocrinology. 1999;140:1449–1458. doi: 10.1210/endo.140.3.6581. [DOI] [PubMed] [Google Scholar]
- WARTENBERG M., FISCHER K., HESCHELER J., SAUER H. Redox regulation of p-glycoprotein-mediated multidrug resistance in multicellular prostate tumor spheroids. Int. J. Cancer. 2000;85:267–274. [PubMed] [Google Scholar]
- WATANABE T., KOKUBU N., CHARNICK S.B., NAITO M., TSURUO T., COHEN D. Interaction of cyclosporin derivatives with the ATPase activity of human P-glycoprotein. Br. J. Pharmacol. 1997;122:241–248. doi: 10.1038/sj.bjp.0701377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WODARZ N., RUPPRECHT R., KORNHUBER J., SCHMITZ B., WILD K., RIEDERER P. Cell-mediated immunity and its glucocorticoid-sensitivity after clinical recovery from severe major depressive disorder. J. Affect. Disord. 1992;25:31–38. doi: 10.1016/0165-0327(92)90090-s. [DOI] [PubMed] [Google Scholar]
- XIA Z., BERGSTRAND A., DEPIERRE J.W., NASSBERGER L. The antidepressants imipramine, clomipramine, and citalopram induce apoptosis in human acute myeloid leukemia HL-60 cells via caspase-3 activation. J. Biochem. Mol. Toxicol. 1999;13:338–347. doi: 10.1002/(sici)1099-0461(1999)13:6<338::aid-jbt8>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- YU C., WARRIAR N., GOVINDAN M.V. Cysteines 638 and 665 in the hormone binding domain of human glucocorticoid receptor define the specificity to glucocorticoids. Biochemistry. 1995;34:14163–14173. doi: 10.1021/bi00043a022. [DOI] [PubMed] [Google Scholar]