

Abstract
Searching for the structural requirements improving the potency and the stereoselectivity of Na+ channel blockers as antimyotonic agents, new derivatives of tocainide, in which the chiral carbon atom is constrained in a rigid α-proline or pyrrolo-imidazolic cycle, were synthesized as pure enantiomers.
Their ability to block Na+ currents, elicited from −100 to −20 mV at 0.3 Hz (tonic block) and 2 – 10 Hz (use-dependent block) frequencies, was investigated in vitro on single fibres of frog semitendinosus muscle using the vaseline-gap voltage-clamp method.
The α-proline derivative, To5, was 5 and 21 fold more potent than tocainide in producing tonic and 10 Hz-use-dependent block, respectively. Compared to To5, the presence of one methyl group on the aminic (To6) or amidic (To7) nitrogen atom decreased use-dependence by 2- and 6-times, respectively. When methylene moieties were present on both nitrogen atoms (To8), both tonic and use-dependent block were reduced.
Contrarily to tocainide, all proline derivatives were stereoselective in relation to an increased rigidity. A further increase in the molecular rigidity as in pyrrolo-imidazolic derivatives markedly decreased the drug potency with respect to tocainide.
Antimyotonic activity, evaluated as the shortening of the time of righting reflexes of myotonic adr/adr mice upon acute drug in vivo administration was 3 fold more effective for R-To5 than for R-Tocainide.
Thus, constraining the chiral centre of tocainide in α-proline cycle leads to more potent and stereoselective use-dependent Na+ channel blockers with improved therapeutic potential.
Keywords: Na+ channel, native frog skeletal muscle fibres, vaseline-gap voltage-clamp, tocainide derivatives, use-dependent block, stereoselectivity, antimyotonic activity, adr/adr mice
Introduction
Tocainide is an orally effective lidocaine derivative, able to block the voltage-gated Na+ channels in a use-dependent manner, a feature that enhances its potency in situations of prolonged depolarization and/or excessive firing of action potentials with respect to a physiological excitability (Catterall, 1987; de luca et al., 1997b; Conte Camerino et al., 2000). Thus, tocainide is actually among the few drugs clinically used for the symptomatic treatment of hyperexcitability of myotonic syndromes, hereditary disorders of skeletal muscle due to genetic mutations on either sodium or chloride channel genes (Streib, 1986; Kwiecinski et al., 1992, Rüdel et al., 1994; Lehmann-Horn & Rüdel, 1996; Cannon, 1997; Ptácek, 1998). However, its clinical use presents some disadvantages, due to the high doses required for its antimyotonic effectiveness with the risk of a greater incidence of serious side effects, especially at the cardiac, haematopoietic and central nervous system levels (Roden & Woosley, 1986; Puniani & Bertorini, 1991; Rüdel et al., 1994).
The use-dependent inhibition of Na+ channels by tocainide and other local anaesthetic (LA) drugs relies on the higher affinity of these drugs for their receptor site on the α-subunit of the channel when this latter is open and/or inactivated, and to a slow recovery from inactivation of the drug-bound channels during membrane repolarization (Catterall, 1987; de luca et al., 1997a). At the resting state, the LA interacts with its presumed receptor through hydrophobic interactions with two aromatic amino acid residues at the D4 – S6 segment (Ragsdale et al., 1994; Wright et al., 1998). Gating movements due to the channel opening may then allow further drug interactions with other residues in the pore that could stabilize the drug-bound channel in an inactivated state (Wang et al., 2000; Yarov-Yarovoy et al., 2001). Furthermore, a site in D1 – S6 segment would also be involved in determining the drug stereoselectivity (Nau et al., 1999). This feature is important because tocainide and mexiletine, another LA-like compound, have a chiral carbon atom that contributes to the potency and the stereoselectivity of the drug (Tricarico et al., 1991; de luca et al., 1997a; 2000; Desaphy et al., 1999). In particular, we found that the introduction of an aromatic lipophilic group on the chiral centre of mexiletine highly increases the drug potency suggesting the establishment of a strong two-points interaction with the two hydrophobic pockets of the receptor in D4 – S6 segment (Wright et al., 1998; de luca et al., 2000). Furthermore, the nature of the group on the stereogenic centre is important for the drug stereoselectivity because the introduction of an unbending lipophilic group such as the isopropyl one enhanced the stereoselectivity, likely due to the occurrence of a three-points interaction of this more rigid structure with the receptor (de luca et al., 1997a; 2000). In agreement with previous data, we confirmed that a higher basicity in the molecule determines a greater use-dependent behaviour (de luca et al., 1997a).
To go further in the study of the structure-activity relationship and to better evaluate the role of the chiral centre in the potency and/or stereoselectivity of drug for blocking Na+ channels, the effects of the pure enantiomers of a new derivative of tocainide, in which the asymmetric carbon atom is constrained in a rigid α-proline cycle (To5, Figure 1) were evaluated on Na+ currents of native frog skeletal muscle fibres. In addition, by introducing methyl groups on one or both of the nitrogen atoms of the proline derivative (as in To6, To7 and To8, Figure 1) or by constraining the chiral centre in a pyrrolo-imidazolic cycle (To3 and To4, Figure 1), we have investigated whether a further decrease in the ability of the molecule to transit through various conformations could modify the drug potency and/or stereoselectivity, and consequently the interaction with the receptor. The results indicate that constraining the stereogenic centre in a α-proline cycle, as in To5, highly improves potency, stereoselectivity and use-dependent behaviour of Na+ channel blockers. Accordingly, an acute in vivo administration of tocainide and To5 in myotonic adr/adr mice showed that To5 was 3 fold more effective than its parent compound in decreasing the time of righting reflex of myotonic mice. On the basis of these data, we can conclude that the newly synthesized proline derivative of tocainide, To5, may be considered as a potential therapeutic agent in the treatment of myotonic syndromes.
Figure 1.
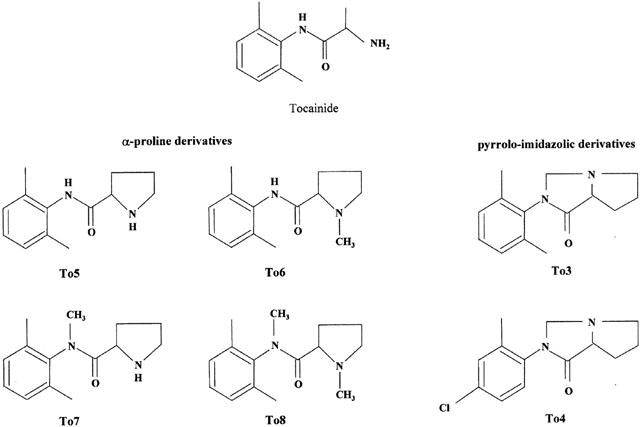
Chemical structure of tocainide and its newly synthesized α-proline and pyrrolo-imidazolic analogues.
Methods
Fibre preparation and voltage clamp apparatus
Segments of undamaged single muscle fibres (about 1 cm in length) were obtained by microsurgery (plucking procedure) from the ventral branch of the semitendinosus muscle of Rana Esculenta bathed in normal physiological solution at room temperature. The cut-end fibre was then superfused with an internal solution and mounted across three chamber partitions, which delineated the four pools. Three strips of vaseline were applied over the fibre and carefully sealed to the fibre to reduce leakage. The width of the gaps of the central pools (A and B) had been previously set to 70 – 100 μM and 200 μM, respectively. Four KCl/agar bridges electrodes connected the recording chamber to the voltage clamp amplifier based on methods described by Hille & Campbell (1976) and detailed elsewhere (de luca et al., 1995; 1997a). When the solution level was lowered below the vaseline strips, the four pools were physically and electrically independent from each other: this was confirmed by verifying that no leak current was flowing upon increasing the amplifier gain. Then the solution in the pool A was replaced with the external solution and after about 10 min of equilibration the recordings were performed at 10°C. The usual holding potential (h.p.) was −100 mV.
Sodium currents were recorded using an amplifier connected via a A/D and D/A Digidata 1200 Interface (Axon Instruments, Foster City, CA., U.S.A.) to a 486 DX2/66 personal computer and stored on the hard disk. The stimulation protocols and data acquisition were driven by the Clampex program (pClamp6 software package; Axon Instruments). The currents flowing in response to depolarizing command voltages were low pass filtered at 10 kHz (Frequency Devices, U.S.A.), visualized on an oscilloscope and sampled at 20 kHz. When necessary, leak and capacities currents were subtracted by P/4 method. The acquired traces were analysed later using Clampfit program (pClamp6 software package; Axon Instruments).
Drugs and solutions
The following solutions (mM) were used: Normal physiological solution: NaCl 115, KCl 2.5, CaCl2 1.8, Na2HPO4 2.15, NaH2PO4 0.85; External solution: NaCl 77, Choline-Cl 38, KCl 2.5, CaCl2 1.8, Na2HPO4 2.15, NaH2PO4 0.85; Internal solution: CsF 105, MOPS 5, MgSO4 2, EGTA 5, Na2ATP 0.55. The pH was adjusted at 7.2 with a standard NaOH concentrated (5N) solution.
The compounds tested and shown in Figure 1 were Tocainide; Proline derivatives: N-[(2,6-dimethylphenyl)-2-pyrrolidinecarboxamide, To5; N-[(2,6-dimethylphenyl)-1-methyl-2-pyrrolidinecarboxamide, To6; N-[(2,6-dimethylphenyl)-N-methyl-2-pyrrolidinecarboxamide, To7; N-[(2,6-dimethylphenyl)-N,1-dimethyl-2-pyrrolidinecarboxamide, To8; Pyrrolo-imidazolic derivatives: 2-(2,6-dimethylphenyl)-hexahydro-1H-pyrrolo[1.2-c]imidazol-1-one, To3; 2-(2-methyl-4-chorophenyl)-hexahydro-1H-pyrrolo[1.2-c]imidazol-1-one, To4. All the compounds were prepared in our laboratories, as pure or highly enriched enantiomeric forms (enantiomeric purity ∼93%), as hydrochloride or hydroiodide salts according to procedures described in details elsewhere (Loughhead et al., 1999; Franchini et al., 2000). Stock solutions were prepared by dissolving the compounds in external solution, that for To4 contained DMSO (<1%). DMSO, at the highest final concentration used (0.2%), was without effect on the parameters recorded. All other chemicals used were of analytical grade and obtained from Sigma Chemical Company (St. Louis, MO, U.S.A.).
Pulse protocols
The curves describing the voltage dependence of sodium current (I – V curve; not shown) were constructed with a cycle of 10 ms test pulses from the h.p. of −100 mV to increasing potentials (from −70 to +75 mV). The intervals between each test pulse were long enough (∼3 s) to allow complete recovery of sodium channel from inactivation. The exact value of membrane potential at which INa peaked its maximum (INa,max), calculated from the I – V curves, was −37.6±0.6 mV (n=62). Thus, the test pulse used for evaluating tonic block exerted by each test compound (see below) was −20 mV since this membrane potential value is in the linear part of the I – V curve, when the dynamic process of activation is complete. At −140 mV all the channels are virtually in the resting ‘activatable' state, and the INa reaches its maximal value. No difference in peak INa was observed when the h.p. was −100 mV, suggesting that at this potential the channels are mostly in the resting state. Thus the drug-induced reduction of peak INa at −100 mV was considered mostly as tonic block (block of the channel in the resting state). The use-dependent behaviour of each compound was evaluated with 30 s trains of test pulses from the h.p. of −100 to −20 mV at the frequency of 2 and 10 Hz. With this protocol in the presence, but not in the absence, of use-dependent compounds a further reduction in peak INa over the tonic block was observed, that progressively cumulated until a new equilibrium was reached. The value of the current at the equilibrium normalized with respect to the current in the absence of drug was used to calculate the potency of the drug for blocking the channels under conditions of excessive stimulation (e.g. high-frequency firing). Steady-state inactivation (h∞) curves were determined by cyclic protocol of pulse sequences. Each sequence consisted of a conditioning pulse to −140 mV for 500 ms (to have most of the sodium channels in the ‘activatable' state), a prepulse of variable potential of either 50 or 1000 ms duration and the 10 ms test pulse to −20 mV; after a pause of 1 s the sequence was cyclically repeated 18 – 20 times with the prepulse potential value increased each time by 5 mV steps (de luca et al., 1995; 1997a).
The data were expressed as mean±standard error of the mean (s.e.mean) and statistical significance of differences between mean values has been estimated by unpaired Student's t-test. The estimates of s.e.mean of normalized INa values have been obtained as described previously (de luca et al., 1995). Molar concentrations of the drugs tested producing a 50% block of INa (IC50) were determined by using a non-linear least squares fit of the concentration-response curves to the following logistic equation: Effect=−100/{1+(K/[drug])n}, where Effect is the per cent change of INa, −100 is the maximal per cent block of INa, K is the IC50, n is the logistic slope factor, and [drug] is the molar concentration of the compound. The hh∞ curves have been fitted with a single Boltzmann distribution and the potential at which 50% of the sodium channels were inactivated (Vh1/2) was calculated at the inflection point of the curves (de luca et al., 1991).
Conformational study
A systematic conformational search on tocainide and its proline derivatives (To5, To6, To7, To8) was performed by using the semi-empirical AM1 method of calculation implemented in PC Spartan Pro (Wavefunction Inc.) (Dewar et al., 1985). For each compound, clusters of conformations were located in the range of 3 Kcal/mol from global minimum, on the basis of the values presented by the dihedral related to the bond connecting the pyrrolidine ring to the xylylamino carbonyl moiety (N1-C2-CO-O). Two conformers were considered as belonging to the same cluster when they shared the same amide configuration (E/Z) and the respective dihedrals differed by no more than 5°. Rotatable bonds were varied in increments of 60°. The amide bond was assumed to be planar or near planar and it was allowed to vary in increments of 180°. For each compound, the number of conformer clusters was plotted against the eudismic ratio, evaluated as [IC50 distomer/IC50 eutomer], for the tonic block and the points were fitted by using a linear regression correlation.
In vivo studies
Two-month-old myotonic adr/adr mice were used in this study. The tested drugs (tocainide and To5) were injected subcutaneously and their effects were evaluated on the righting reflex time of the mice (TRR), the time the mouse needed to right itself on the four legs after it was turned on its back, monitored at various intervals after the drug administration (30 s, 1 h, 2 h, 3 h and 4 h). At each time, five measurements were taken from each mouse and the maximum value (t) recorded was used for statistical analysis. The drug effect was estimated as the ability of the tested compound to shorten the TRR with respect to the TRR before the administration.
Results
Tonic and use-dependent block of Na+ channels by tocainide derivatives
Constraining the stereogenic centre in a rigid α-proline cycle, as in To5 derivative, caused a marked increase in the potency for producing both tonic and use-dependent block of INa (elicited with test pulses from −100 to −20 mV at 0.3 Hz and 2 – 10 Hz frequencies, respectively) with respect to tocainide. Indeed, as illustrated in Figure 2, 100 μM of R-To5 produced a tonic block (47.9±8.7%, n=6) comparable to that observed with 500 μM of R-tocainide (42.6±4.2%, n=8). The higher potency of R-To5 versus R-tocainide was more evident during the stimulation at 10 Hz (Figure 2). Consequently, the concentration-response curves of R-To5 for both tonic and use-dependent block were shifted to the left with respect to those of R-tocainide (Figure 3A,B). As shown in Table 1, the concentrations of test compounds for the half-maximal block of INa (IC50 values) decreased upon increase of stimulation frequency. The IC50 values of To5 for tonic and 10 Hz-use-dependent blocks were 5 and 21 fold lower than those of R-tocainide, respectively.
Figure 2.
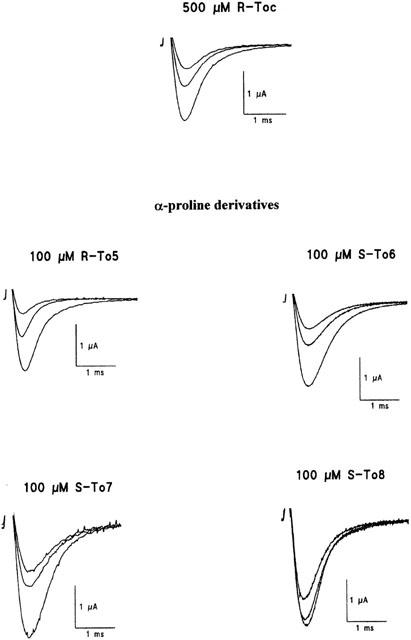
Na+ current transients recorded by the three vaseline gap voltage-clamp method from single fibres of frog semitendinosus muscle in the absence and presence of eutomers of tocainide (Toc) and of its proline-like derivatives without methyl groups (To5), with a methyl on the aminic (To6) or amidic (To7) nitrogen atom and with methylene moieties on both nitrogen atoms (To8). In particular, the effects produced by 500 μM of R-Toc were compared to those obtained in the presence of 100 μM of R-To5, S-To6, S-To7, and S-To8. In each group of traces, the control current (in the absence of drug) is the greatest one obtained with single depolarizing pulses from −100 to −20 mV for 10 ms. A similar depolarizing stimulus elicited around 10 min after the application of each compound allowed to estimate the tonic block exerted by the drug. Afterwards, the depolarizing stimulus was repetitively applied at a 10-Hz frequency for 20 – 30 s, producing a cumulative block over the tonic one due to a use-dependent behaviour, until a new equilibrium was reached. The smallest traces correspond to the residual current at the end of the 10 Hz stimulation protocol.
Figure 3.
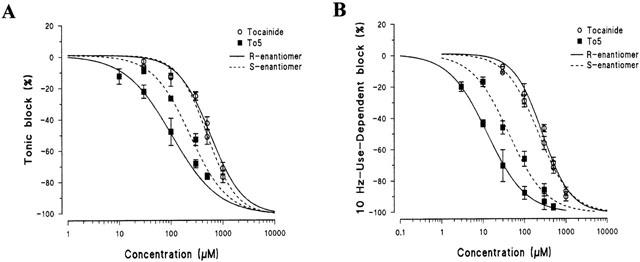
Concentration-response curves for tonic (A) and 10 Hz-use-dependent block (B) of Na+ currents obtained with R- and S- enantiomers of tocainide and its α-proline derivative, To5. The amount of block was calculated in conditions of single depolarizing 10-ms pulses from −100 to −20 mV (tonic block, A) or after 20 – 30 s of a 10-Hz train of similar pulses (use-dependent block, B). Each point shows the per cent block of INa,max observed in the presence of each concentration of drugs versus INa,max in the absence of drug in the same fibre and is the mean±s.e.mean from 4 – 6 fibres. The curves fitting the experimental points were obtained using the logistic function described in Methods.
Table 1.
Concentrations for half-maximal tonic and use-dependent block of sodium currents by tocainide and its analogues

It is noteworthy that under the present experimental conditions, tocainide showed almost no stereoselectivity for both tonic and use-dependent block, as can be seen in Figure 3, in which the dose-response curves of R- and S-enantiomers of tocainide are almost overlapping. By contrast, the R-enantiomer of To5 was more potent than the S-one and the eudismic ratio [IC50 distomer/IC50 eutomer] increased with the stimulation frequency, being 2, 2.8 and 3.4 for the tonic, 2 Hz- and 10 Hz-use-dependent blocks, respectively (Table 1). The stereoselectivity was maintained or even enhanced when the structure was made more rigid with the introduction of methylene moieties on one or both nitrogen atoms of the proline derivatives, as in To6, To7 and To8 (Figure 1 and Table 1). For these compounds, the S-enantiomers were more potent than the R-ones (Table 1). To verify if the increase in stereoselectivity was correlated with an increase in molecular rigidity, for each test compound, we plotted the eudismic ratio for the tonic block versus the number of conformer clusters evaluated by the semi-empirical AM1 method. As shown in Figure 4, the points were highly correlated with a correlation coefficient r2=0.860 (P<0.05).
Figure 4.
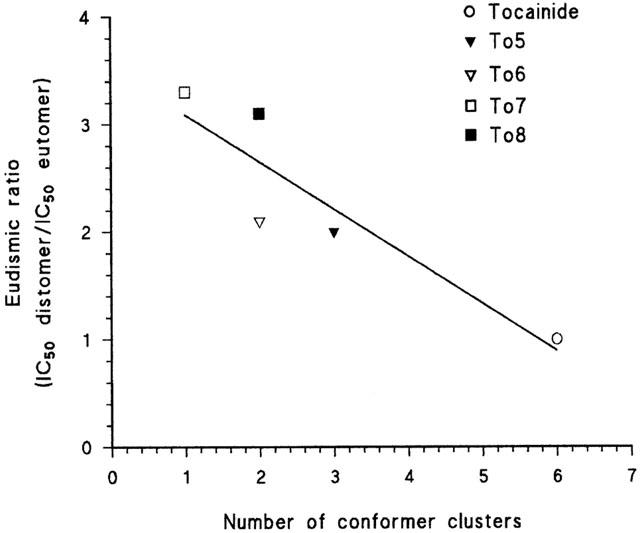
Relationship between the eudismic ratio (IC50 distomer/IC50 eutomer) obtained for the tonic block and the number of conformer clusters of tocainide and its proline derivatives. For each test compound, the number of conformer clusters was evaluated by using the semi-empirical AM1 method as described in Methods. For each drug, clusters of conformations were located in the range of 3 Kcal/mol from global minimum, on the basis of the values presented by the dihedral related to the bond connecting the pyrrolidine ring to the xylylamino carbonyl moiety (N1-C2-CO-O). The points were fitted by a linear regression equation, with r2=0.860 and P<0.05.
The S-To6 and S-To7 derivatives, in which one methyl group is introduced, produced a tonic block similar to that induced by R-To5; however, they were less effective in producing the use-dependent block (Figure 2 and Table 1). In fact, the dose-response curves of the methylated derivatives, S-To6 and S-To7, for the 10 Hz-use-dependent block were shifted to the right with respect to that of R-To5 (Figure 5). S-To7, having the methyl group on the amidic nitrogen, was even less use-dependent than S-To6, in which the methylene moiety is on the aminic nitrogen. The drug use-dependent behaviour could be appreciated by calculating the ratio [IC50 tonic block/IC50 10 Hz-use-dependent block]. This ratio was 4 and 1.5 for S-To6 and S-To7, respectively, these values being strongly reduced with respect to that of R-To5 (around 9) (Table 1). When two methyl groups were introduced in the proline derivative as in S-To8, both tonic and use-dependent block were decreased with respect to those of R-To5 (Figure 2). Among the methylated proline derivatives, S-To8 is the less potent for blocking sodium channels, particularly at high frequency of stimulation (Figure 5 and Table 1). Indeed, compared with R-To5, the tonic and the 10 Hz-use-dependent block of S-To8 are reduced by a factor of 2.5 and 15 respectively (Table 1). The use-dependent behaviour of S-To8, estimated by the ratio [IC50 tonic block/IC50 10 Hz-use-dependent block] (around 1.5) is similar to that of S-To7 (Table 1).
Figure 5.
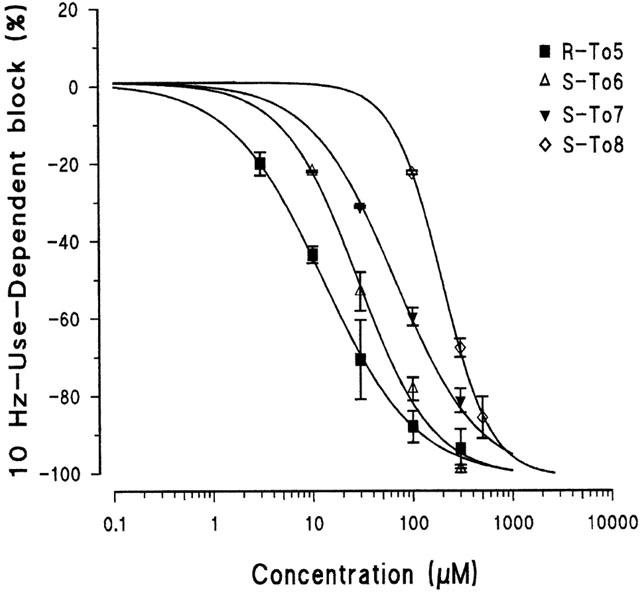
Concentration-response curves for the 10 Hz-use-dependent block of INa obtained with the eutomers of the proline derivatives of tocainide, R-To5, S-To6, S-To7 and S-To8. The amount of block was calculated after 20 – 30 s of a 10-Hz train of single depolarizing 10-ms pulses from −100 to −20 mV. Each value is the per cent block of INa,max observed in the presence of each concentration of drugs versus INa,max in the absence of the drug in the same fibre and is the mean±s.e.mean from 4 – 6 fibres. The curves fitting the experimental points were obtained using the logistic function described in Methods.
The To3 and To4 derivatives in which the chiral centre of tocainide is introduced in a pyrrolo-imidazolic cycle, more rigid than the proline one, were only available as R-enantiomers. Both pyrrolo-imidazolic derivatives showed a large reduction of the potency in producing both tonic and use-dependent block with respect to tocainide. Indeed, the IC50 values of R-To3 for both tonic block and 10 Hz-use-dependent block were around twice higher than those of R-tocainide and 10 to 50 fold higher than those of the proline derivative (Table 1). The R-To4 analogue, containing a chloride atom but with only a methyl moiety in the aromatic ring in ‘ortho' position, is even less potent in producing both tonic and use-dependent block (Table 1).
Effect of tocainide derivatives on steady-state inactivation
Tocainide has been previously shown to shift the steady-state inactivation curves (h∞ curves) toward more negative potentials in a manner dependent on the duration of the depolarizing prepulse, implying a voltage- and time-dependent reduction of the number of channels available for opening, principally due to a preferential binding of this drug to sodium channels in an inactivated state that, in turn, explains the use-dependence of tocainide (Fakler et al., 1990; Tricarico et al., 1991; de luca et al., 1991, 1997a).
All proline and pyrrolo-imidazolic derivatives of tocainide acted as inactivated Na+ channel blockers because they shifted the h∞ curves at 50 and 1000 ms prepulse duration towards more negative potentials. As can be seen in Figure 6, in which the effects of 100 and 300 μM of R-To5 and of S-To8 on h∞ curves at 1000 ms prepulse are compared, the shift was concentration-dependent. Furthermore, the left-shift in the h∞ curves produced by R-To5 was more pronounced than that produced by S-To8 (Figure 6, Table 2), as expected by taking into account the higher potency of R-To5.
Figure 6.
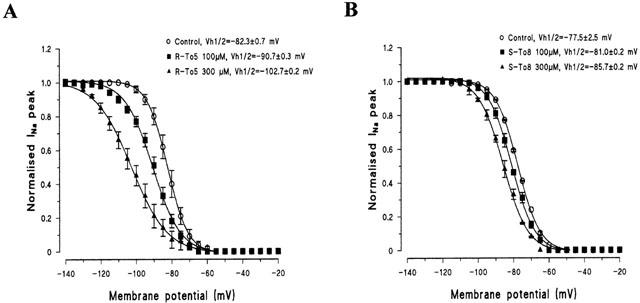
Steady-state inactivation curves (h∞ curves) constructed with a prepulse duration of 1000 ms and the effect of increasing concentration of R-To5 (A) and S-To8 (B). At each membrane potential is shown the current amplitude normalized to the INa,max value obtained at −140 mV. Because certain variability exists between controls, the effect of each drug has been compared to the control h∞ curves recorded in the same experiments. Each point is the mean±s.e.mean of 3 – 6 experiments. Curves were fitted by a single Boltzmann distribution as described in Methods that allowed to calculate the Vh1/2, value of membrane potential at which 50% of channels are inactivated.
Table 2.
Effects of new proline and pyrroloimidazolic derivatives of tocainide on the steady-state inactivation curves (h∞) of sodium channels

Contrarily to tocainide, the proline derivatives showed a certain stereoselective behaviour in shifting the h∞ curves, this feature being particularly evident at the 1000 ms prepulse duration (Table 2). Furthermore, for the eutomers of proline derivatives, the shift observed at 1000 ms was generally greater than that at 50 ms (Table 2). For R-To5 and S-To6 derivatives, this difference was significant at a concentration of 100 μM whereas the statistical significance was only reached at 300 μM for S-To7 and was never observed with S-To8. This could be due to the higher use-dependent behaviour of R-To5 and S-To6 for blocking Na+ currents (evaluated by the ratio (IC50 tonic block/IC50 10 Hz-use-dependent block)) with respect to that of S-To7 and S-To8 (Table 1).
Compared to tocainide, both pyrrolo-imidazolic derivatives, R-To3 and R-To4 which are less potent for blocking sodium channels are also less effective in shifting the steady-state inactivation curves towards more negative potentials. For example, at 1000 ms, 500 μM of these derivatives produced a shift in h∞ curves similar to 300 μM of tocainide (Table 2).
Effects of in vivo administration of tocainide and its proline derivative, To5, on the time of righting reflex (TRR) of myotonic adr/adr mice
The potency and the use-dependent behaviour of the Na+ channel blockers are two critical characteristics that are taken into account in the search of new antimyotonic agents. Then, regarding the results presented above, To5 may have a greater interesting therapeutic potential for the treatment of myotonia with respect to tocainide. Consequently, we have investigated the effects of To5 as a potential antimyotonic agent by evaluating in vivo its ability to shorten the time of righting reflex of myotonic adr/adr mouse, a phenotype with a severe recessive form of chloride channel-related myotonia (Gronemeier et al., 1994). The effect of this compound was compared with that of tocainide.
When healthy mice were turned on their back, the time needed to right themselves on the four legs (TRR) was around 0.5±0.1 s (n=12) while the TRR of myotonic adr/adr mice was 7.9±0.6 s (n=12). Acute subcutaneous administration to adr/adr mice of close-to-therapeutic doses of tocainide (20 μg/g) significantly decreased the TRR, the maximal reduction (57.6±6.4%, n=4), being reached at around 30 min-1 h after the drug administration. Comparable effects on the TRR of adr/adr mice were observed with 7 μg/g of R-To5 (55.9±4.1%, n=5) without manifest side effects. By contrast, the administration of a saline solution did not significantly modify the TRR of myotonic mice (TRR=7.5±0.8 s, n=3). Although a correlation between the in vivo studies and in vitro experiments is difficult due to the specific pharmaco-kinetic profile of each drug, the greater effectiveness in vivo of To5 with respect to tocainide strongly relies on the higher potency and use-dependent behaviour of the drug observed in the in vitro experiments.
Discussion
In the present work, we show that constraining the stereogenic centre of tocainide in a rigid proline-like cycle represents an important structural requirement for improving the potency and the stereoselectivity of the drug for blocking skeletal muscle Na+ channels. Compared to tocainide, the α-proline-like derivative, To5, always behaves as an inactivated Na+ channel blocker but it is 5 and 21 fold more potent in producing the tonic and the use-dependent block of Na+ currents, respectively. With respect to its parent compound, the proline derivative shows a greater lipophilicity due to the proline cycle and a greater basicity (a higher pKa value) due to the presence of a secondary amine group. Since drugs are applied outside the cell, a higher hydrophobicity favours the drug access to the receptor site by diffusion through membrane while a greater charged fraction of the drug in the cytoplasm, due to a higher pKa value, has access to the receptor through the hydrophilic pathway of the open channel. These results are supported by our previous structure-activity studies with mexiletine analogues showing that the drug potency is strongly correlated with a high lipophilicity of the group on the chiral carbon atom while a greater basicity in the drug is pivotal for a higher use-dependent block (de luca et al., 1997a; 2000; Desaphy et al., 1999). A higher drug lipophilicity and basicity are also critical features for a stronger drug interaction to its presumed receptor site in the D4-S6 segment of α-subunit. This latter has hydrophobic pockets formed by the presence of two aromatic amino acids lining the pore, Tyr and Phe, that could interact with the aromatic ring and the ionisable amino group of LA drugs, respectively (Ragsdale et al., 1994; 1996; Wright et al., 1998; Li et al., 1999). Due to the close position of the amino group to the asymmetric carbon atom, it is likely that constraining both of them in a lipophilic moiety, such as a proline-like cycle, could allow the establishment of a strong π-π or cation-π interaction of this part of the drug with the aromatic residue Phe, similarly to what previously observed with lipophilic mexiletine derivatives (Desaphy et al., 1999; de luca et al., 2000). This, along with the higher basicity, can explain the higher potency of To5, particularly evident during the use-dependent block of sodium currents.
Compared to To5, the introduction of a methylene group on either the aminic or amidic nitrogen atom, as in To6 and To7, respectively, induced a decrease in the use-dependent behaviour of drug without significantly affecting its ability to block the resting channels. By contrast, the introduction of a methyl group on both aminic and amidic nitrogen atoms, as in S-To8, strongly reduced the drug potency for both tonic and use-dependent block of sodium channel. Concerning the To6 derivative, the presence of a methyl group on the aminic nitrogen atom could induce little changes in the planarity and/or the rigidity of the molecule that could lead to little or no modification in the interactions between the proline cycle and the receptor sites. With regard to the derivatives S-To7 and S-To8, the presence of methylene moieties on the amidic nitrogen atom could distort the planar amidic link geometry, enhance the rigidity of the molecule and then destabilize the drug binding to the receptor, especially during the use-dependent block. This presumed relationship between the rigidity and the potency of the drug is further supported by the results obtained with the pyrrolo-imidazolic derivatives. These latter, more rigid than the proline derivatives, were even less potent than tocainide for both tonic and use-dependent block of Na+ channels.
According to this view, the proline-like derivatives are more stereoselective than tocainide. The structural requirements for the drug stereoselectively blocking Na+ channels are not yet well-known. However, Nau et al. (1999) have proposed that a third site of LA interaction in D1-S6 segment of α- subunit of skeletal Na+ channel could be involved in the drug stereoselectivity. It is possible to hypothesise that the proline derivatives, owing to their conformational constraint, would be able to establish such a third-point interaction, rising then a stereoselective behaviour. In fact, a conformational search by semi-empirical methods on tocainide and its proline derivatives gave a number of conformers, in the range of 3 kcal/mol from the lower energy conformation, that strongly correlated with the drug stereoselectivity (Figure 4).
A useful animal model for studying the potential antimyotonic effects of LA-like drugs is the adr/adr mouse that is characterized by a severe myotonic state due to a missense mutation on the skeletal muscle chloride channel gene. As a result, the macroscopic chloride conductance (gCl), the parameter that ensures the electrical stability of sarcolemma under resting conditions (Adrian & Bryant, 1974) is dramatically lowered, leading to a pathological hyperexcitability. Then, in myotonic adr/adr mice, the muscles spontaneously generate trains of action potentials triggering involuntary spasms, a clinical phenotype very similar to that of Na+ channel-related myotonic syndromes (Cannon, 1996; Cooper & Jan, 1999). Consequently, the adr/adr mouse model is useful to test in vivo and/or in vitro drugs potentially able to reduce the clinical symptoms of myotonic syndromes. Accordingly, in a previous work, we have demonstrated a correlation between the potency of mexiletine and some related compounds for blocking voltage-gated Na+ channels and their ability in vitro to solve the abnormal hyperexcitability of intercostal muscle fibers of myotonic adr/adr mice (de luca et al., 1997b). In the present study, we have taken a particular interest in the evaluation of the in vivo effects of tocainide and its more potent proline derivative, To5, on a peculiar myotonic sign of adr/adr phenotype, the abnormally increased time of righting reflex. Indeed, compared to healthy mice, the time of righting reflex of myotonic adr/adr mice was around 15 fold longer, probably due to the delayed relaxation caused by the low gCl-induced potassium accumulation in the T-tubules leading to the fibre depolarization (Cannon, 1996). Both compounds reduced the righting reflex time of myotonic adr/adr mice but To5, in line with its higher Na+ channel blocking activity, was around 3 fold more effective than tocainide. The stronger effectiveness of To5 in vitro versus in vivo may be related to specific pharmaco-kinetic aspects of each tested compound.
Taken together, the present results indicate that the presence of a proline-like cycle at the asymmetric centre level of Na+ channel blockers could represent an important structural requirement leading to more potent and stereoselective use-dependent drugs with an improved therapeutic potential of a great interest for the symptomatic treatment of myotonic syndromes.
Acknowledgments
This work was support by grants from Telethon-Italy to D. Conte Camerino (#1208).
Abbreviations
- adr
arrested development of righting response
- INa
sodium current
- LA
local anaesthetic
- h.p.
holding potential
- TRR
righting reflex time
References
- ADRIAN R.H., BRYANT S.H. On the repetitive discharge in myotonic muscle fibers. J. Physiol. (Lond.) 1974;258:125–143. doi: 10.1113/jphysiol.1974.sp010620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CANNON S.C. Ion-channel defects and aberrant excitability in myotonia and periodic paralysis. Trends Neurosci. 1996;19:3–10. doi: 10.1016/0166-2236(96)81859-5. [DOI] [PubMed] [Google Scholar]
- CANNON S.C. From mutation to myotonia in sodium channel disorders. Neuromuscul. Disord. 1997;7:241–249. doi: 10.1016/s0960-8966(97)00430-6. [DOI] [PubMed] [Google Scholar]
- CATTERALL W.A. Common modes of drug action on Na+ channels: local anesthesics, antiarrhythmics and anticonvulsants. Trends Pharmacol. Sci. 1987;8:57–65. [Google Scholar]
- CONTE CAMERINO D., PIERNO S., DE LUCA A., BRYANT S.H. Antimyotonic effects of tocainide enantiomers on skeletal muscle fibers of congenitally myotonic goats. Neuromuscul. Disord. 2000;10:160–164. doi: 10.1016/s0960-8966(99)00101-7. [DOI] [PubMed] [Google Scholar]
- COOPER E.C., JAN L.Y. Ion channel genes and human neurological disease: recent progress, prospects, and challenges. Proc. Natl. Acad. Sci. U.S.A. 1999;96:4759–4766. doi: 10.1073/pnas.96.9.4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE LUCA A., NATUZZI F., DESAPHY J.-F., LONI G., LENTINI G., FRANCHINI C., TORTORELLA V., CONTE CAMERINO D. Molecular determinants of mexiletine structure for potent and use-dependent block of skeletal muscle sodium channels. Mol. Pharmacol. 2000;57:268–277. [PubMed] [Google Scholar]
- DE LUCA A., NATUZZI F., FALCONE G., DURANTI A., LENTINI G., FRANCHINI C., TORTORELLA V., CONTE CAMERINO D. Inhibition of frog skeletal muscle sodium channels by newly synthesized chiral derivatives of mexiletine and tocainide. Naunyn-Schmiedeberg's Arch. Pharmacol. 1997a;356:777–787. doi: 10.1007/pl00005118. [DOI] [PubMed] [Google Scholar]
- DE LUCA A., NATUZZI F., LENTINI G., FRANCHINI C., TORTORELLA V., CONTE CAMERINO D. Stereoselective effects of mexiletine enantiomers on sodium currents and excitability characteristics of adult skeletal muscle fibers. Naunyn-Schmiedeberg's Arch. Pharmacol. 1995;352:653–661. doi: 10.1007/BF00171325. [DOI] [PubMed] [Google Scholar]
- DE LUCA A., PIERNO S., NATUZZI F., FRANCHINI C., DURANTI A., LENTINI G., TORTORELLA V., JOCKUSCH H., CONTE CAMERINO D. Evaluation of the antimyotonic activity of mexiletine and some new analogs on sodium currents of single muscle fibers and on the abnormal excitability of the myotonic ADR mouse. J. Pharmacol. Exp. Ther. 1997b;282:93–100. [PubMed] [Google Scholar]
- DE LUCA A., PRÖBSTLE T., BRINKMEIER H., RÜDEL R. The different use dependences of tocainide and benzocaine are correlated with different effects on sodium channel inactivation. Naunyn-Schmiedeberg's Arch. Pharmacol. 1991;344:596–601. doi: 10.1007/BF00170658. [DOI] [PubMed] [Google Scholar]
- DESAPHY J.-F., CONTE CAMERINO D., FRANCHINI C., LENTINI G., TORTORELLA V., DE LUCA A. Increased hindrance on the chiral carbon atom of mexiletine enhances the block of rat skeletal muscle Na+ channels in a model of myotonia induced by ATX. Br. J. Pharmacol. 1999;128:1165–1174. doi: 10.1038/sj.bjp.0702901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEWAR M.J.S., ZOEBISCH E.G., HEALY E.F., STEWART J.J.P. AM1: a new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 1985;107:3902–3909. [Google Scholar]
- FAKLER B., RUPPERSBERG J.P., SPITTELMEISTER W., RÜDEL R. Inactivation of human sodium channels and the effect of tocainide. Pflügers Arch. 1990;415:693–700. doi: 10.1007/BF02584007. [DOI] [PubMed] [Google Scholar]
- FRANCHINI C., CORBO F., LENTINI G., BRUNO G., SCILIMATI A., TORTORELLA V., CONTE CAMERINO D., DE LUCA A. Synthesis of new 2,6-Prolylxylide analogues of tocainide as stereoselective blockers of voltage-gated Na+ channel with increased potency and improved use-dependent activity. J. Med. Chem. 2000;43:3792–3798. doi: 10.1021/jm000931l. [DOI] [PubMed] [Google Scholar]
- GRONEMEIER M., CONDIE A., PROSSER J., STEINMEYER K., JENTSCH T.J., JOCKUSCH H. Nonsense and missense mutations in the muscular chloride channel gene Clc-1 of myotonic mice. J. Biol. Chem. 1994;259:5963–5967. [PubMed] [Google Scholar]
- HILLE B., CAMPBELL D.T. An improved vaseline-gap voltage-clamp for skeletal muscle fibers. J. Gen. Physiol. 1976;67:265–293. doi: 10.1085/jgp.67.3.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KWIECINSKI H., RYNIEWICZ B., OSTRZYCKI A. Treatment of myotonia with antiarrhythmic drugs. Acta. Neurol. Scand. 1992;86:371–375. doi: 10.1111/j.1600-0404.1992.tb05103.x. [DOI] [PubMed] [Google Scholar]
- LEHMANN-HORN F., RÜDEL R. Molecular pathophysiology of voltage-gated ion channels. Rev. Physiol. Biochem. Pharmacol. 1996;128:195–268. doi: 10.1007/3-540-61343-9_9. [DOI] [PubMed] [Google Scholar]
- LI H.L., GAULE A., MEADOWS A., RAGSDALE D.S. A molecular basis for different local anesthesic affinities of resting versus open and inactivated states of the sodium channels. Mol. Pharmacol. 1999;55:134–141. doi: 10.1124/mol.55.1.134. [DOI] [PubMed] [Google Scholar]
- LOUGHHEAD D.G., FLIPPIN L.A., WEIRKERT R.J. Synthesis of mexiletine stereoisomers and related compounds via SNAr nucleophilic substitution of a Cr(CO)3− complexed aromatic fluoride. J. Org. Chem. 1999;64:3373–3375. doi: 10.1021/jo982287c. [DOI] [PubMed] [Google Scholar]
- NAU C., WANG S.-Y., STRICHARTZ G.R., WANG G.K. Point-mutations at N434 in D1-S6 of μ 1 Na+ channels modulate binding affinity and stereoselectivity of local anesthesic enantiomers. Mol. Pharmacol. 1999;56:404–413. doi: 10.1124/mol.56.2.404. [DOI] [PubMed] [Google Scholar]
- PTÁCEK L.J. The familial periodic paralyses and nondystrophic myotonias. Am. J. Med. 1998;104:58–70. doi: 10.1016/s0002-9343(98)00123-5. [DOI] [PubMed] [Google Scholar]
- PUNIANI T.S., BERTORINI T.E. Tocainide therapy in muscle cramps and spasms due to neuromuscular disease. Muscle Nerve. 1991;14:280–285. doi: 10.1002/mus.880140312. [DOI] [PubMed] [Google Scholar]
- RAGSDALE D.S., MCPHEE J.C., SCHEUER T., CATTERALL W.A. Molecular determinants of state-dependent block of Na+ channels by local anesthesics. Science. 1994;265:1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- RAGSDALE D.S., MCPHEE J.C., SCHEUER T., CATTERALL W.A. Common molecular determinants of local anesthesic, antiarrhythmetic, and anticonvulsant block of voltage-gated Na+ channels. Proc. Natl. Acad. Sci. U.S.A. 1996;93:9270–9275. doi: 10.1073/pnas.93.17.9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RODEN D.M., WOOSLEY R.L. Drug therapy: tocainide. N. Engl. J. Med. 1986;315:41–45. doi: 10.1056/NEJM198607033150107. [DOI] [PubMed] [Google Scholar]
- RÜDEL R., LEHMANN-HORN F., RICKER K.The non-dystrophic myotonias Myology 1994New York: McGraw-Hill; 1291–1302.eds. Engel, A.G. & Franzini-Armstrong, C. pp [Google Scholar]
- STREIB E.W. Successful treatment with tocainide of recessive generalized congenital myotonia. Ann. Neurol. 1986;19:501–504. doi: 10.1002/ana.410190515. [DOI] [PubMed] [Google Scholar]
- TRICARICO D., FAKLER B., SPITTELMEISTER W., RUPPERSBERG J.-P., STUTZEL R., FRANCHINI C., TORTORELLA V., CONTE CAMERINO D., RÜDEL R. Stereoselective interaction of tocainide and its chiral analogs with the sodium channels in human myoballs. Pflügers Arch. 1991;415:234–237. doi: 10.1007/BF00370521. [DOI] [PubMed] [Google Scholar]
- WANG S.-Y., NAU C., WANG G.K. Residues in Na+ channel D3-S6 segment modulates both batrachotoxin and local anesthetic affinities. Biophys. J. 2000;79:1379–1387. doi: 10.1016/S0006-3495(00)76390-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WRIGHT S.N., WANG S.-Y., WANG G.K. Lysine point mutations in Na+ channel D4-S6 reduce inactivated channel block by local anesthesics. Mol. Pharmacol. 1998;54:733–739. [PubMed] [Google Scholar]
- YAROV-YAROVOY V., BROWN J., SHARP E.M., CLARE J.J., SCHEUER T., CATTERALL W.A. Molecular determinants of voltage-dependent gating and binding of pore-blocking drugs in transmembrane segment IIIS6 of the Na+ channel α subunit. J. Biol. Chem. 2001;276:20–27. doi: 10.1074/jbc.M006992200. [DOI] [PubMed] [Google Scholar]