

Abstract
The effects of ZD6169, a novel ATP-sensitive K+ channel (KATP channel) opener, were investigated on membrane currents in isolated myocytes using patch-clamp techniques. Tension measurement was also performed to study the effects of ZD6169 on the resting tone of pig urethral smooth muscle.
Levcromakalim was more potent than ZD6169 in lowering the resting urethral tone. Relaxation induced by low concentrations of ZD6169 (⩽3 μM) was completely suppressed by additional application of glibenclamide (1 μM). In contrast, glibenclamide (1 – 10 μM) only partially inhibited the relaxation induced by higher concentrations of ZD6169 (⩾10 μM).
Bay K8644 (1 μM) reduced the maximum relaxation produced by ZD6169 (⩾10 μM).
In whole-cell configuration, ZD6169 suppressed the peak amplitude of voltage-dependent Ba2+ currents in a concentration- and voltage-dependent manner, and at 100 μM, shifted the steady-state inactivation curve of the voltage-dependent Ba2+ currents to the left at a holding potential of −90 mV.
In cell-attached configuration, open probability of unitary voltage-dependent Ba2+ channels (27 pS, 90 mM Ba2+) was inhibited by 100 μM ZD6169 and by 10 μM nifedipine.
Reverse transcriptase-polymerase chain reaction (RT – PCR) analysis revealed the presence of the transcript of the α1C subunit of L-type Ca2+ channels in pig urethra.
These results demonstrate that ZD6169 causes urethral relaxation through two distinct mechanisms, activation of KATP channels at lower concentrations and inhibition of voltage-dependent Ca2+ channels at higher concentrations (about 10 μM).
Keywords: α1C subunit, Ca2+ channel blocker, K+ channel opener, voltage-dependent Ca2+ channels, ZD6169
Introduction
It has been reported that ZD6169, the S-enantiomer of the racemic compound N-(4-benzoylphenyl)-3,3,3-trifluoro-2-hydroxy-2-methylpropionamide, increases the capacity of the urinary bladder following oral administration in a dose which produces minimal cardiovascular effects in normal conscious rats and dogs (Howe et al., 1995). Several results from electrophysiological and functional studies suggest that ZD6169 may activate ATP-sensitive K+ channels (i.e. KATP channels) in detrusor smooth muscle, hyperpolarize the membrane and inhibit the contractile force (Li et al., 1995; Trivedi et al., 1995). Thus, a general agreement seems to have been reached that ZD6169 is a detrusor-selective KATP channel opener.
Recently, by use of single-channel recordings, we have been able to confirm that a glibenclamide-sensitive KATP channel is indeed the target K+ channel for ZD6169, and further demonstrate that it has a dual effect (activation and inhibition) on the activity of KATP channels in pig urethra (Teramoto et al., 2001a). We suggested that ZD6169 may be useful for the treatment of incontinence, not only abolishing unstable contractions of detrusor muscle, but also inhibiting the activation of KATP channels in the urethra at relatively high concentrations (⩾30 μM). However, the effects of ZD6169 on the resting urethral tone still remain to be elucidated. The purpose of the present study was to compare the relaxing potency of ZD6169 on the urethral tone with that of levcromakalim a known potent KATP channel opener, by use of tension recordings. Furthermore, in the present experiments, we found that glibenclamide partially inhibited the muscle relaxation induced by higher concentration of ZD6169 (⩾10 μM) and that Bay K8644 (1 μM) reduced the relaxing action of ZD6169. Therefore, we have investigated the effects of ZD6169 on voltage-dependent Ba2+ macroscopic and unitary currents which demonstrate electrophysiological and molecular properties of voltage-dependent Ca2+ channels in pig urethra. We found a glibenclamide-insensitive ZD6169-induced urethral relaxation which may be caused by inhibition of voltage-dependent Ca2+ channels in pig urethra. A preliminary account of the present results has been communicated to the 74th annual meeting of the Japanese Pharmacological Society (Teramoto et al., 2001b).v
Methods
Cell dispersion
Fresh urethras from female pigs were collected from a local abattoir. Pig urethral myocytes were freshly isolated by the gentle tapping method as described previously (Teramoto & Brading, 1998). Briefly, thin strips of smooth muscle (10 – 15 mm×2 – 4 mm) were dissected from the fresh proximal urethral wall and stored in nominally Ca2+-free solution (mM): Na+ 140, K+ 5, Mg2+ 0.5, Cl− 146, glucose 10, HEPES 10/Tris, titrated to pH 7.35 – 7.4, containing papain (17 u mg−1 protein, 0.3 – 0.4 mg ml−1) bubbled with O2 at 4 – 6°C for 20 min. The digested strips were washed in Ca2+-free solution containing 1 mg ml−1 bovine serum albumin (BSA), and preincubated in Ca2+-free solution at 35°C for 5 – 6 min. The strips were then incubated in Ca2+-free solution containing 0.3 – 0.4 mg ml−1 collagenase (Type I, Sigma Chemical K.K., Tokyo, Japan) at 35°C for 10 – 15 min. Relaxed spindle-shaped cells, with length varying between 400 and 500 μm, were isolated and stored at 4°C. The dispersed cells were used within 2 h for experiments.
Tension measurement and data analysis
For isometric tension recording, fine strips were prepared as described previously (Teramoto & Ito, 1999). An initial tension equivalent to 1 g weight was applied to each strip, which was then allowed to equilibrate for approximately 1 – 1.5 h until the basal urethral tone became stable (37°C). To prevent both noradrenaline outflow from sympathetic nerve terminals and ß-adrenoceptor stimulation, 3 μM guanethidine and 0.3 μM propranolol were present throughout the experiments. Data were recorded on a Macintosh computer, through ‘MacLab 3.5.6' (ADInstruments Pty Ltd., Castle Hill, Australia). The tension was expressed as g mg−1 of tissue.
Patch-clamp experiments recording procedure
Patch-clamp experiments were performed at room temperature (21 – 23°C) as described previously (Teramoto et al., 2001a). Junction potentials between bath and pipette solutions were measured with a 3 M KCl reference electrode and were <2 mV, so that correction for these potentials was not made. Capacitance noise was kept to a minimum by maintaining the test solution in the electrode as low as possible. At the beginning of each experiment, the series resistance was compensated.
Drugs and solutions
For tension measurement, modified Krebs solution was used (mM): 137 Na+, 5.9 K+, 1.2 Mg2+, 2.5 Ca2+, 133.7 Cl−, 15.4 HCO3−, 1.2 H2PO4− and 11.5 glucose which was bubbled with 97% O2 and 3% CO2. Occasionally, a proportion of external Ca2+ in the above modified Krebs solution was replaced with equimolar Na+, and 2 mM EGTA (pH 7.3) was added. For recording voltage-dependent Ba2+ currents in whole-cell configuration, high caesium pipette solution contained (mM): Cs+ 130, tetraethylammonium (TEA+) 10, Mg2+ 2, Cl− 144, glucose 5, EGTA 5, ATP 5, HEPES 10/Tris (pH 7.35 – 7.40). Ba2+ 10 mM bath solution contained (mM): Ba2+ 10, TEA+, 135, Cl− 155, glucose 10, HEPES 10/Tris (pH 7.35 – 7.40). For recording unitary Ba2+ current (cell-attached configuration), a pipette was filled with high Ba2+ solution (mM): Ba2+ 90, TEA+ 10, glucose 10, Cl− 190, HEPES 10, titrated to pH 7.35 – 7.40 with Tris base. In some experiments, 1 μM Bay K 8644 (methyl 1,4-dihydro-2,6-dimethyl -3 -nitro -4 -(2 -trifluoromethylphenyl)-pyridine-5-carboxylate) was included in the above pipette solution. High K+ bath solution contained (mM): K+ 142, glucose 10, Cl− 142, EGTA 5, HEPES 10, titrated to pH 7.35 – 7.40 with Tris base. The bath solution was superfused by gravity throughout the experiments at a rate of 2 ml min−1. The following chemicals were used: ATP, Bay K 8644, collagenase (type I), dimethylsulphoxide (DMSO), EGTA, glibenclamide, HEPES, papain, TEACl and Tris (Sigma Chemical K.K., Tokyo, Japan). Levcromakalim (SmithKline Beecham Pharmaceuticals, Harlow, U.K.), ZD6169 (AstraZeneca Pharmaceuticals, Cheshire, U.K.) and glibenclamide were prepared daily as 100 mM stock solutions in DMSO. Bay K 8644 was dissolved to 1 mM in DMSO. The final concentration of DMSO was less than 0.3%, and this concentration was shown not to affect urethral tone and membrane currents in pig urethra.
Data analysis and statistics
The whole-cell recording data were low-pass filtered at 500 Hz by an 8 pole Bessel filter (E-3201B, NF Electronic Instruments, Yokohama, Japan), sampled at either 1 ms (voltage-dependent Ba2+ currents) or 25 ms (glibenclamide-sensitive membrane currents) intervals and analysed on a computer (Macintosh PowerPC G3, Apple Computer Japan Limited, Tokyo, Japan) by the commercial software ‘MacLab 3.5.6' (ADInstruments Pty Ltd., Castle Hill, Australia). Statistical analyses were performed with Student's t-test for paired values. Changes were considered significant at P<0.01. Data are expressed as mean with the standard deviation (s.d.).
RNA preparation and reverse transcription-polymerase chain reaction analysis
For RNA isolation, total RNA from pig urethral smooth muscle was isolated using TRIzol reagent according to the manufacturer's instructions (RNeasy Mini Kit, QIAGEN K.K., Tokyo, Japan). First-strand synthesis of cDNA using random hexamers was prepared as follows: total RNA isolated from tissues was incubated with random hexamers at 70°C for 10 min and then with PCR buffer (20 mM Tris/HCl, pH 8.4, 50 mM KCl), 2.5 mM MgCl2, 0.5 mM deoxynucleoside-5′-triphosphate, and 10 mM dithiothreitol at 25°C for 5 min. RT – PCR was initiated by the addition of Superscript II RT (200 u) at 25°C for 10 min followed by incubation at 42°C for 50 min. The reaction was terminated by incubation at 70°C for 15 min, before chilling on ice. PCR was performed using 1 μl of cDNA in 20 μl reaction mixture containing 0.5 μM concentration of each primer, 200 μM concentration of each deoxynucleoside-5′-triphosphate, and 0.5 units of Taq polymerase (Takara Co. Ltd., Osaka, Japan). The cycling conditions were 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s for 40 cycles. An aliquot (10 μl) of the RT – PCR product was analysed on a 2% Tris-borate-EDTA polyacrylamide gel. Since no sequence information is available regarding the α1C subunit in pig, generic subunit-specific primers were designed based on information from rabbit sequences (GenBank DNA sequence X15539; Mikami et al., 1989). The sequences of the primers for amplification of the novel α1C subunit were as follows: 5′-GCTGTGGACCCCCTTCATCAAGTCC-3′ (forward) and 5′-ACCCTTGGCTTCAGGGTCATA-3′ (reverse). Control reactions were carried out where samples in the absence of reverse transcriptase were amplified to ensure that the detected product was not the result of possible DNA contamination and by use of corresponding templates as positive controls to ensure that the primers were annealing successfully. These primers gave products of the expected sizes that were confirmed by DNA sequence analysis.
Results
The effects of ZD6169 on the resting urethral tone
Figure 1a shows that cumulative application of ZD6169 produced a concentration-dependent relaxation of the resting urethral tone. After washing out the drug (⩽10 μM), the urethral tone recovered to the control level. Subsequently, 10 μM levcromakalim was applied in order to obtain the maximum relaxation in the urethral strips. Since the ZD6169 (⩾30 μM)-induced relaxation did not completely recover to the control level, on some occasions, 10 μM levcromakalim was applied before application of higher concentrations of ZD6169. Figure 1b shows the concentration-response curve of ZD6169, expressed relative to the maximum relaxation to 10 μM levcromakalim. The EC50 value for the ZD6169-induced relaxation was 1.6 μM.
Figure 1.
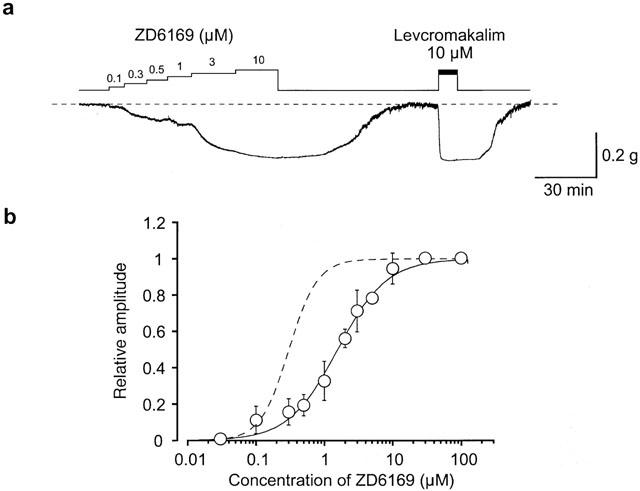
Identification of the glibenclamide-sensitive component of the ZD6169-induced relaxation
As shown in Figure 2a, ZD6169 (3 μM) produced a slow relaxation which reached a level similar to the maximal relaxation produced by 10 μM levcromakalim, and this relaxation was completely suppressed by application of 1 μM glibenclamide (n=10). However, the relaxation produced by higher concentrations of ZD6169 (⩾10 μM) were not completely suppressed by subsequent application of 1 μM glibenclamide. Figure 2b shows that glibenclamide caused a partial inhibition of the relaxation induced by 30 μM ZD6169 (0.41±0.12, n=6) and had no effect on the 100 μM ZD6169-induced relaxation (0.02±0.03, n=6, Figure 2c). Note that even application of higher concentration of glibenclamide (⩾10 μM) did not change the level of the 100 μM ZD6169-induced relaxation (data not shown). When tissues were pretreated with 1 μM glibenclamide for 11 min (Figure 2d), the time taken to reach peak relaxation in 100 μM ZD6169 was noticeably slower than that in the absence of 1 μM glibenclamide (in the absence of glibenclamide, 3 min 53 s±1 min 34 s, n=5, vs. in the presence of glibenclamide, 10 min 52 s±2 min 44 s, n=5). In contrast, 10 μM levcromakalim induced a much quicker relaxation which reached maximum in 2 min 15±59 s (n=10).
Figure 2.
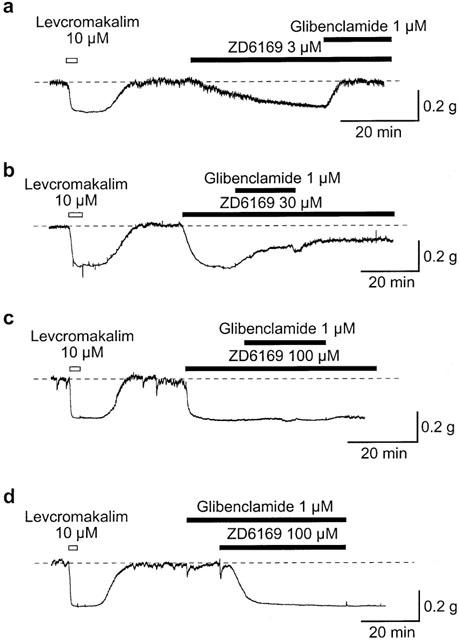
Glibenclamide-sensitive and -insensitive components of the ZD6169-induced urethral relaxation. The dashed line indicates the mean resting urethral tone. (a), (b), (c) The effects of 1 μM glibenclamide on the relaxation induced by 3 μM, (a); 30 μM, (b); 100 μM, (c); ZD6169. (d) ZD6169 (100 μM)-induced relaxation after 11 min pretreatments with 1 μM glibenclamide.
Bay K8644, a selective voltage-dependent L-type Ca2+ channel agonist, caused a dramatic increase in resting tone (Figure 3a), and the maximum level of relaxation achieved by 10 μM levcromakalim showed no significant difference in the presence and absence of 1 μM Bay K8644. In contrast, the relative ratio of the ZD6169-induced relaxation to the 10 μM levcromakalim-induced relaxation was significantly reduced in the presence of 1 μM Bay K8644 (0.78±0.08, n=4, 30 μM ZD6169; 0.71±0.02, n=4, 100 μM ZD6169) in comparison with that in the absence of Bay K8644 (0.95±0.09, n=6, 30 μM ZD6169; 1.02±0.02, n=6, 100 μM ZD6169) in Figure 3b. Figure 3b also shows the relationships between the relative ratio of the glibenclamide-sensitive ZD6169-induced relaxation and the concentration of ZD6169, resulting in a bell-shaped curve and that at higher concentrations relaxations to ZD6169 (⩾30 μM) is insensitive to 1 μM glibenclamide. When extracellular Ca2+ was removed and 2 mM EGTA was added in order to obtain zero Ca2+, the urethral tone was reversibly reduced (Figure 3c, n=16). Application of 1 μM nifedipine caused an irreversible urethral relaxation (Figure 3d, n=6). These results suggest that concentrations of ZD6169 (⩾30 μM) are likely to cause glibenclamide-insensitive urethral relaxation in pig urethra which may be related to voltage-dependent Ca2+ channels and that voltage-dependent Ca2+ channels may play an important role in regulating the resting tone of pig urethra.
Figure 3.
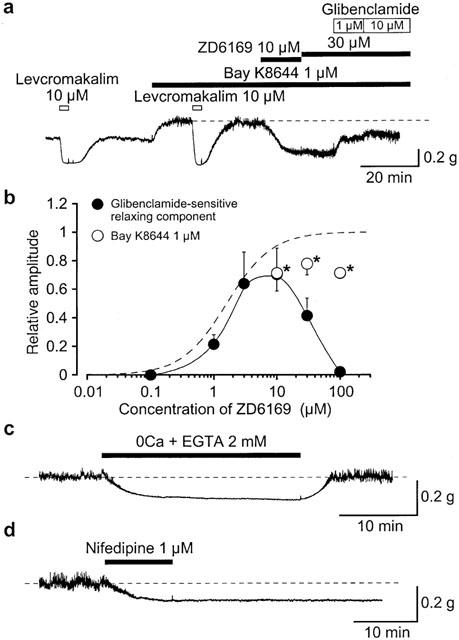
The relaxing effects of ZD6169, levcromakalim and nifedipine on the urethral tone. (a) The effects of ZD6169 and levcromakalim on the urethral tone in the absence and presence of 1 μM Bay K8644. The dashed line indicates the increased urethral tone in the presence of Bay K8644. (b) Relationships between the relative value of the glibenclamide-sensitive ZD6169-induced relaxation and the concentration of ZD6169. The peak amplitude of the 10 μM levcromakalim-induced relaxation was taken as one. Each symbol indicates mean with s.d. shown by vertical lines (n=4 – 7). The line was drawn by eye. The curve with the broken line (ZD6169) is obtained from Figure 1(b). *Significantly different from the relative value in the absence of Bay K8644 (t-test, P<0.01). (c) When extracellular Ca2+ was removed (and 2 mM EGTA added), the resting urethral tone was reversibly reduced. The dashed line indicates the mean resting urethral tone. (d) Applying 1 μM nifedipine caused an irreversible urethral relaxation. The dashed line indicates the mean resting urethral tone.
Actions of ZD6169 on voltage-dependent Ba2+ currents in pig urethra
In order to study further the mechanisms involved in the glibenclamide-insensitive relaxation, the effects of ZD6169 on voltage-dependent Ca2+ currents were investigated by use of conventional whole-cell configurations. When the bath solution was PSS, the peak amplitude of inward Ca2+ current was too small to analyse (less than 20 pA at 0 mV). In order to enhance the inward currents for reasonable analysis and to isolate voltage-dependent inward currents through Ca2+ channels by inhibiting other Ca2+-activated mechanisms (such as Ca2+-activated K+ currents and Ca2+-activated Cl− currents etc.), 10 mM Ba2+ bath solution containing 135 mM TEA+ was used and the pipette was filled with a Cs+ – TEA+ solution containing 5 mM EGTA. Figure 4a shows the time course of the effects of ZD6169 (100 μM) on the Ba2+ inward current evoked by a depolarizing pulse of +10 mV from a holding potential of −50 mV. The depolarizing pulses were applied every 15 s. When the peak amplitude of the Ba2+ inward current just before application of ZD6169 (control) was taken as one, ZD6169 reduced the peak amplitude of the Ba2+ inward current (0.2±0.1, n=8). On removal of ZD6169, the peak amplitude gradually recovered, but not to the control level. Subsequently, application of 100 μM calciseptine, a peptide which is a selective L-type Ca2+ channel blocker (Teramoto et al., 1996), greatly inhibited the Ba2+ inward currents. Similarly, 10 μM nifedipine completely suppressed the Ba2+ inward currents. The block produced by ZD6169 showed some use dependence. As shown in Figure 4a, when a depolarizing pulse was applied after an interval of 4 min in the presence of 100 μM ZD6169, the peak amplitude of the Ba2+ inward current was smaller than that observed before application of ZD6169 but consistently larger than that recorded at 4 min with repetitive application of the depolarizing pulses (single pulse, 0.79±0.09, n=6 vs repetitive pulse 0.32±0.1, n=4). Similar experiments were performed at three different holding potentials (−90 mV, 0.85±0.09, n=4; −100 mV, 0.87±0.05, n=4; −120 mV, 0.89±0.06, n=3). A depolarizing pulse to +10 mV produced the same amplitude of voltage-dependent Ba2+ currents in pig urethra at holding potentials more negative than −80 mV when the peak amplitude of voltage-dependent Ba2+ currents at −120 mV was taken as one (−90 mV, 0.98±0.03, n=4; −100 mV, 0.99±0.02, n=4). Figure 4b shows the relationships between the relative peak amplitude of Ba2+ inward currents and concentrations of ZD6169 at two different holding potentials (−50 mV and −90 mV). The Ba2+ inward currents were evoked by a depolarizing pulse to 10 mV applied every 15 s. ZD6169 inhibited the peak amplitude of the Ba2+ inward currents in a concentration-dependent manner (Ki; 55 μM, −50 mV; 122 μM, −90 mV).
Figure 4.
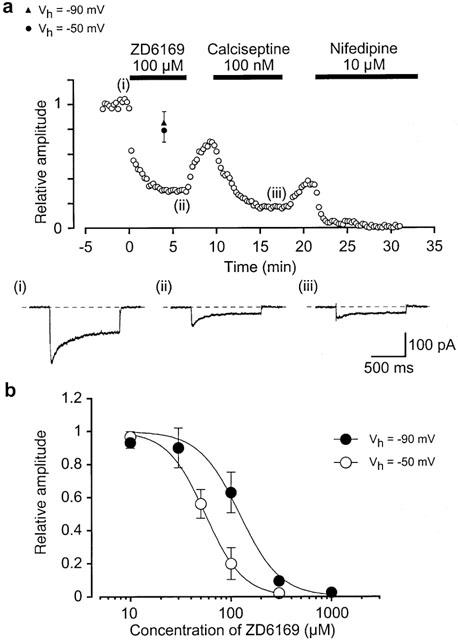
Electrophysiological properties of the voltage-dependent Ba2+ currents in pig urethra
In order to further characterize the electrophysiological properties of Ba2+ inward currents in pig urethra, depolarizing step pulses (10 mV increment from −40 to +40 mV for 1 s duration, every 15 s, whole-cell recording) were applied from a holding potential of −90 mV. At potentials more positive than −30 mV, an inward Ba2+ current was evoked (Figure 5a) which reached a peak and then gradually decayed. The maximum peak amplitude was obtained at approximately +10 mV and the amplitude was reduced at more positive potentials, demonstrating a voltage-dependency. At a holding potential of −40 mV, both the peak amplitude and the amplitude at the end of the command pulse were smaller, but the time course of the current decay was identical at both holding potentials (Figure 5a).
Figure 5.
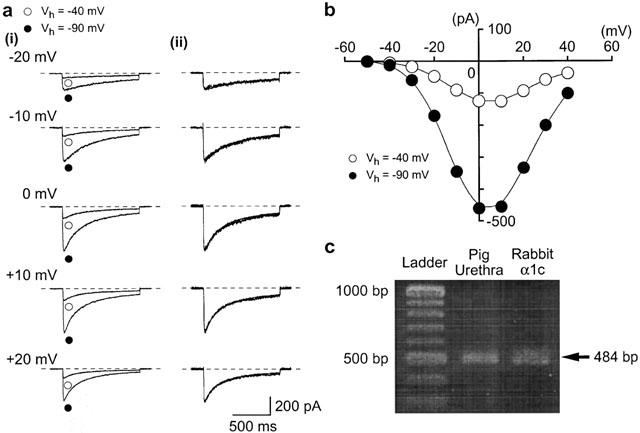
Inward Ba2+ currents recorded by application of depolarizing pulses at two different holding potentials (−90 and −40 mV). Whole-cell recording, pipette solution Cs+ – TEA+ solution containing 5 mM EGTA and the bath solution 10 mM Ba2+ containing 135 mM TEA+. (a)(i) Inward Ba2+ currents at each indicated depolarizing potential from both holding potentials superimposed. (ii) Inward Ba2+ current from (i) scaled to match their peak amplitudes and superimposed. (b) Current-voltage relationships of inward Ba2+ current obtained at −40 and −90 mV. The current amplitude was measured as the peak amplitude of inward Ba2+ current in each condition. The lines were drawn by eye. (c) RT – PCR detection of voltage-dependent Ca2+ channel α1C subunit mRNA. RT – PCR was performed as described in the Methods and size makers were used to indicate the size of the experimental fragments (lane 1). RT – PCR yielded visible amounts of α1C subunit (484 bp fragment) in mRNAs from pig urethra (lane 2) and rabbit cDNA of α1C subunit (lane 3).
Molecular expression of α1C subunit in pig urethra
The distribution of the α1C subunit in voltage-dependent Ca2+ channels in the tissue was examined by use of RT – PCR technique. Specific primers for the amplification of α1C subunit mRNA were designed to produce a cDNA fragment of 484 bp. As shown in Figure 5c, the α1C subunit mRNA was expressed in pig urethra and rabbit α1C cDNA with the expected fragment size (484 bp, see Methods). Similar observations were obtained in seven other urethral tissues. Expression of α1B or α1G subunit mRNAs were not detectable in pig urethra using set of specific primers (data not shown).
Voltage-dependent inhibitory effects of ZD6169 on voltage-dependent Ba2+ currents
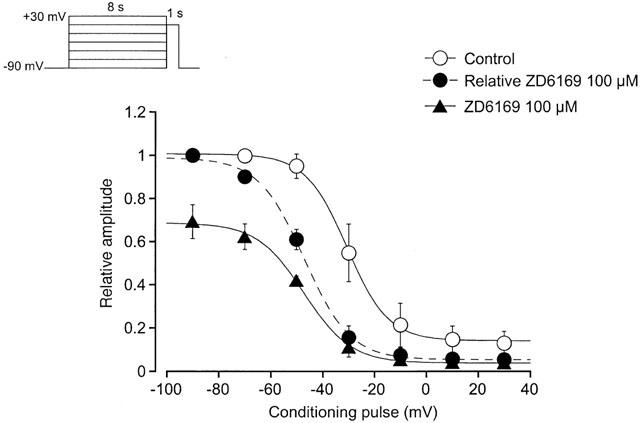
Figure 6b shows the current-voltage relationships in the absence and presence of 50 μM ZD6169. ZD6169 inhibited the peak amplitude of the Ba2+ currents evoked by depolarizing pulses (1 s duration) from a holding potential of −50 mV at levels more positive than −30 mV, and the inhibition showed some voltage-dependency (Figure 6c). This voltage-dependency was investigated before and after application of 100 μM ZD6169 using the experimental protocol shown in Figure 7 (conditioning pulse duration, 8 s; holding membrane potential, −90 mV). In the absence of ZD6169 (control), inactivation of the Ba2+ current occurred with depolarizing pulses positive to −50 mV. After application of 100 μM ZD6169 (approximately 5 min later), the voltage-dependent inactivation curve in the same cells was shifted to the left (Figure 7).
Figure 6.
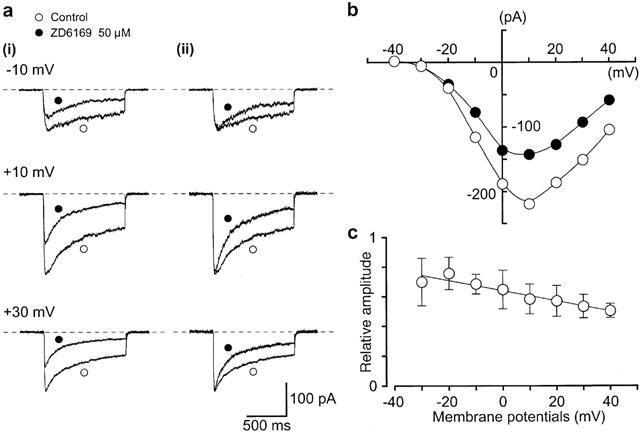
Effects of ZD6169 on voltage-dependent Ba2+ inward currents at a holding membrane potential of −50 mV in pig urethra. The pipette solution was Cs+ – TEA+ solution containing 5 mM EGTA and the bath solution was 10 mM Ba2+ containing 135 mM TEA+. (a) (i) Original current traces before (control) and after application of 50 μM ZD6169 at the indicated pulse potentials. (ii) Inward Ba2+ current from (i) scaled to match their peak amplitudes and superimposed. (b) Current-voltage relationships obtained in the absence (control) or presence of 50 μM ZD6169. The current amplitude was measured as the peak amplitude of the Ba2+ inward current in each condition. The lines were drawn by eye. (c) Relationship between the test potential and relative value of the Ba2+ inward currents inhibited by 50 μM ZD6169, expressed as a fraction of the peak amplitude of the Ba2+ inward current evoked by various amplitude of the depolarizing pulse in the absence of ZD6169. Each symbol indicates the mean of four observations with±s.d. shown by vertical lines. The line was drawn by eye.
Figure 7.
Effects of ZD6169 and nifedipine on unitary Ba2+ currents in pig urethra
Single-channel recordings in the cell-attached configuration (pipette solution, 90 mM Ba2+; bath solution, 142 mM K+) were used to investigate further the inhibitory effects of ZD6169 on voltage-dependent Ba2+ currents. At potentials more positive than −40 mV, depolarizing pulses (400 ms duration) from a holding potential of −90 mV elicited a unitary inward current. The current-voltage relationship for this current was linear as shown in Figure 8b. The conductance, obtained from the amplitude of the Ba2+ currents, was 27.3±1.9 pS (n=8) measured from the all-points amplitude histograms. Application of 10 μM nifedipine suppressed the unitary Ba2+ current activity at all of the depolarizing membrane potentials (n=5, data not shown). Bay K8644 (1 μM, included in the pipette solution) enhanced the channel activity. Under these experimental conditions, on some occasions (11 out of 31 patches), channel openings of a 1.9 pA unitary Ba2+ current were continuously observed at potentials more negative than −30 mV in cell-attached configuration (Figure 9a, NPo value, 0.055). Note that the channel activity could be observed for more than 20 min (n=4) and that the NPo value increased in a voltage-dependent manner (−40 mV, 0.02±0.01, n=7; −30 mV, 0.06±0.02, n=7). Application of 100 μM ZD6169 inhibited the channel activity to an NPo value of 0.005 without changing the amplitude of the unitary current (control, 1.9 pA; ZD6169 100 μM, 1.9 pA, n=5, in Figure 9b). When the NPo value just before application of ZD6169 (control) was taken as one, the relative NPo value was 0.11±0.05 (n=5) in the presence of 100 μM ZD6169, and 0.62±0.12, n=5 in the presence of 50 μM ZD6169 (Figure 9c). The channel activity did not recover to the control level on removal of ZD6169. Approximately 3 min later, bath application of nifedipine (10 μM) abolished the channel activity.
Figure 8.
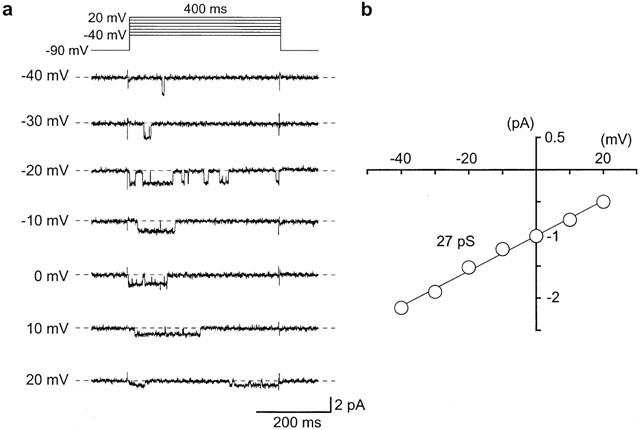
Unitary Ba2+ currents recorded in cell-attached patches of pig urethral myocytes. The pipette was filled with 90 mM Ba2+ solution and 142 mM K+ solution was superfused in the bath. (a) The unitary Ba2+ currents were obtained using a depolarizing pulse (400 ms duration, 10 s interval) from a holding potential of −90 mV to the indicated membrane potential (from −40 mV to +20 mV). The average currents from 10 null traces (the nearest five traces before and after the event in which the channel did not open) were subtracted. The dashed line indicates the current base line where the channel is not open. (b) Current-voltage relationships of the unitary Ba2+ current obtained from −40 mV to +20 mV. The amplitude of the Ba2+ channel currents was taken from the all-points amplitude histograms at each depolarizing potential. The line was fitted by the least-squares method. The channel conductance was 27 pS (27.3±1.9 pS, n=8).
Figure 9.
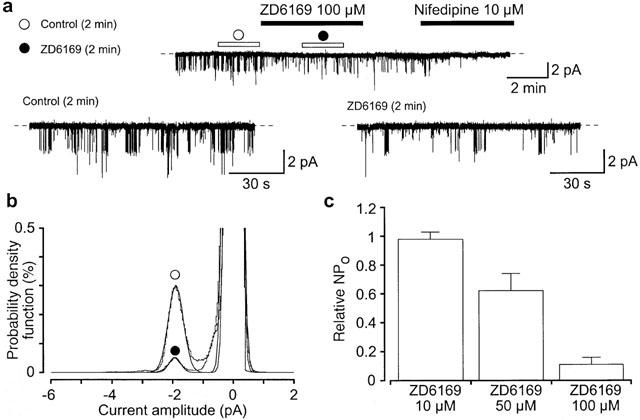
Effects of ZD6169 on the unitary Ba2+ current in pig urethra at a holding potential of −30 mV. High K+ (142 mM) solution was superfused in the bath and high Ba2+ (90 mM) solution in the pipette. Bay K8644 (1 μM) had been included in the pipette solution. (a) Application of 100 μM ZD6169 (5 min duration) reduced the activity of the 1.9 pA Ba2+ channel. After removal of ZD6169, 3 min later, application of nifedipine (10 μM) abolished channel activity. Lower traces show expansions of the upper trace (1 kHz filtration; 80 μs digital sampling interval). The open horizontal bars in the absence and presence of 100 μM ZD6169 indicate the duration (2 min) of the traces analysed in (b). The dashed line indicates the current when the channel is not open. (b) All-point amplitude histograms in the absence (obtained during the last 2 min just before application of ZD6169) and presence of 100 μM ZD6169 (obtained during 2 min of a 5 min application). Continuous lines in the histograms are theoretical curves fitted with the Gaussian distribution, by the least-squares method. The abscissa scales show the amplitude of the current (pA) and the ordinate scales show the percentage value of the probability density function (per cent) for the recording period (2 min). (c) Each column shows the relative level of the activity of the 1.9 pA Ba2+ channel (mean value with+s.d.) when the mean open probability of the channel activity was normalized as one just before application of each concentration of ZD6169 (n=5).
Discussion
Potency of ZD6169 in urethral smooth muscle
In the present experiments, we have been able to evaluate the relaxing efficacy of ZD6169 on spontaneous tone of urethra in the absence of either excess [K+]o (high KCl test protocol) or agonists (such as acetylcholine, serotonin etc.), comparing ZD6169 with levcromakalim (EC50 value; 1.6 μM, ZD6169 versus 0.3 μM, levcromakalim). The potency of levcromakalim was clearly greater than ZD6169, as has been observed in detrusor smooth muscles (guinea-pig, Gopalakrishnan et al., 1999; pig, Buckner et al., 2000). However, the potencies of each KATP channel opener in pig urethra were slightly less than in urinary bladder. In cell-attached configuration, at the same concentration (10 μM), ZD6169 was again less potent than levcromakalim in inducing channel activity in pig urethra (Teramoto et al., 1999; 2001a). This coincided well with the present observations on urethral tension measurements. However, in guinea-pig detrusor, Li et al. (1995) reported that there was no significant difference between the peak amplitude of the 10 μM levcromakalim-induced currents and that of the 10 μM ZD6169-induced currents, suggesting that the potency of ZD6169 is the same as that of levcromakalim. We are not certain whether this significant difference of the potency of these KATP channel openers may be due to different experimental conditions or to species difference.
In patch-clamp experiments, we have recently reported that ZD6169 possesses a dual effect (i.e. agonistic and antagonistic action) on the activity of the KATP channel in pig urethra (Teramoto et al., 2001a). We have also reported that continuous application of ZD6169 (⩾30 μM) suppressed the activity of KATP channels after their initial activation (Teramoto et al., 2001a). However, in the present tension experiments, application of ZD6169 caused a stable and concentration-dependent urethral relaxation between 100 nM – 100 μM. Furthermore, the ZD6169 (⩾30 μM)- induced urethral relaxation was partially but not completely inhibited by application of 1 μM glibenclamide. These results strongly suggest that other relaxing mechanisms may be involved in the ZD6169-induced urethral relaxation in addition to the activation of KATP channels. Indeed, in the present experiments, although the ability of the levcromakalim-induced relaxation was not significantly different in the presence and absence of 1 μM Bay K8644, the ZD6169-induced relaxation was significantly reduced in the presence of Bay K8644 in comparison with the control. These results suggest that the glibenclamide-insensitive relaxation induced by ZD6169 may involve voltage-dependent Ca2+ channels in pig urethra.
Physiological roles of voltage-dependent Ca2+ channels in the smooth muscle cells of pig urethra
In tension measurement, when extracellular Ca2+ was removed and 2 mM EGTA was added, the resting urethral tone was reversibly reduced, suggesting that Ca2+ influx may play an important role in regulating the resting tone. Although application of 1 μM nifedipine also relaxed the smooth muscle tone, 1 μM BAY K8644 caused a dramatic increase in resting tone. These results suggest that Ca2+ entry through voltage-dependent Ca2+ channels may play a major role in generating the resting tone.
Voltage-dependent Ca2+ currents have been demonstrated in urethral myocytes by use of whole-cell recordings (sheep, Cotton et al., 1997; pig, Teramoto & Brading, 1998). The inward Ba2+ currents studied in the present study in pig urethra probably flow through L-type voltage-dependent Ca2+ channels due to the following electrophysiological and molecular properties; (1) There was no hump or second peak at less positive membrane potential level in the current-voltage relationships. (2) When the holding membrane potential was changed from −40 to −90 mV, the threshold potential to produce inward Ba2+ current did not shift. (3) There was no difference in the decay of inward currents recorded at two different holding membrane potentials (−40 and −90 mV). (4) Application of nifedipine, one of the most common dihydropyridine (DHP) Ca2+ channel antagonists, suppressed the amplitude of the inward Ba2+ current at a holding potential of −50 mV (Ki=31 nM; Teramoto & Brading, 1998). (5) Calciseptine suppressed the inward Ba2+ currents. (6) RT – PCR studies show that α1C subunit mRNA was expressed in pig urethra with the expected fragment size for rabbit α1C subunit. (7) By use of cell-attached mode, only one class of the voltage-dependent Ba2+ channel (27 pS voltage-dependent Ba2+ channel, 90 mM Ba2+ solution) which was suppressed by subsequent application of 10 μM nifedipine was recorded from −40 to +20 mV. These results suggest that the L-type Ca2+ channel is probably the only type of voltage-dependent Ca2+ channel present in pig urethra. Other smooth muscles may also possess only a single type of voltage-dependent Ca2+ channel (reviewed by Bolton et al., 1999).
Inhibitory effects of ZD6169 on the voltage-dependent Ba2+ currents in pig urethra
The same amplitude of voltage-dependent Ba2+ currents was produced when holding at −80 mV or more negatively, suggesting that all of the voltage-dependent Ca2+ channels at these potentials may be in the resting state. The ability of 100 μM ZD6169 to suppress the peak amplitude of the Ba2+ currents evoked by a depolarizing pulse from three different holding potentials (−90, −100 and −120 mV) were not significantly different, suggesting that at these negative holding potentials ZD6169 may inhibit the voltage-dependent Ba2+ currents in a voltage-independent manner (resting state block). When the holding potential was elevated to −50 from −90 mV, voltage-dependent inhibition by ZD6169 was seen and the concentration response curve was shifted to the left. The voltage-dependent inactivation curve was also shifted to the left after application of 100 μM ZD6169. These results suggest the voltage-dependent inhibitory actions of ZD6169 occur at the inactivated state of voltage-dependent Ba2+ channels in pig urethra (voltage-dependent block). The dissociation constant for drug binding to the channel to the inactivated state could be estimated from the shift of the voltage-dependent inactivation curve and the concentration response curve obtained at the resting state by use of the following equation (Uehara & Hume, 1985),
where ΔVhalf is the amplitude of the shift of the voltage-dependent inactivation curve, k is a slope factor for the inactivation curve and [D] is the concentration of drug applied. Kinact and Krest are dissociation constants of ZD6169 for the inactivated and the resting states of voltage-dependent Ba2+ channels, respectively. In the present experiments, the Krest value was estimated to be 122 μM from the concentration response curve at a holding potential of −90 mV. When ΔVhalf value was obtained from the results using 8 s conditioning pulses, the estimated Kinact value was 9 μM. Given this, we suggest that ZD6169 may bind to the inactivated state with approximately 14 times higher affinity than to the resting state in pig urethra. In single-channel recordings, we have demonstrated that ZD6169 irreversibly inhibited the activity of the unitary Ba2+ current which was also suppressed by 10 μM nifedipine. From these results, we suggest that ZD6169 may inhibit the voltage-dependent Ba2+ currents through the blocking of the unitary 27 pS Ba2+ channel in pig urethra.
Multiple effects of ZD6169 on the membrane currents in pig urethra
Although various types of KATP channel openers have been reported to possess multiple effects on cellular functions in smooth muscle (such as Ca2+ antagonist-like effects, internal Ca2+ reuptake actions, NO releaser etc., reviewed by Quayle et al., 1997), we have been able to demonstrate at least two different effects of ZD6169 on two significantly different ion channels by use of single-channel recordings in an independent manner. Namely, at lower concentrations (⩽10 μM), ZD6169 seems to cause an urethral relaxation mainly through activation of KATP channels. In contrast, at higher concentrations (30 μM ⩾), ZD6169 causes a significant and stable relaxation, by inhibiting the voltage-dependent Ca2+ channels (after initially activating KATP channels). ZD6169 may thus be termed not only a KATP channel opener but also a voltage-dependent Ca2+ channel blocker, depending on its concentration. In the present experiments, 100 μM ZD6169 was able to induce a similar level of urethral relaxation in the presence and absence of 1 μM glibenclamide, although the maximum level of relaxation was achieved much more slowly in the presence of glibenclamide.
In conclusion, we have been able to demonstrate an inhibitory effect of ZD6169 on the voltage-dependent L-type Ca2+ channels in pig urethra, causing a significant ZD6169-induced urethral relaxation. ZD6169 is not selective for KATP channel in urethral smooth muscle.
Acknowledgments
We are grateful to AstraZeneca Pharmaceuticals for the generous gift of ZD6169. We thank Prof Alison F. Brading (University, Department of Pharmacology, Oxford, U.K.) for her helpful discussion and critical reading of the manuscript. We also thank Dr Y. Mori (Department of Information Physiology, National Institute for Physiological Sciences, Japan) for generously providing rabbit α1C cDNA clone. We thank SmithKline Beecham Pharmaceuticals for the generous gift of levcromakalim. This work was supported in part by the grants-in-aid from the Clinical Research Foundation (Fukuoka, Japan) and the Kaibara Morikazu Medical Science Promotion Foundation (Fukuoka, Japan), awarded to Noriyoshi Teramoto and from the Japan Society for the Promotion of Science (Grant Number 12671543).
Abbreviations
- Bay K 8644
methyl 1,4-dihydro-2,6-dimethyl-3-nitro-4-(2-trifluoromethylphenyl)-pyridine-5-carboxylate
- DHP
dihydropyridine
- DMSO
dimethylsulphoxide
- k
slope factor for the inactivation curve
- KATP channels
ATP-sensitive K+ channels
- Kinact
dissociation constant for the inactivated state of voltage-dependent Ba2+ channels
- Krest
dissociation constant for the resting state of voltage-dependent Ba2+ channels
- NO
nitric oxide
- PSS
physiological salt solution
- RT – PCR
reverse transcriptase-polymerase chain reaction
- TEA+
tetraethylammonium
References
- BOLTON T.B., PRESTWICH S.A., ZHOLOS A.V., GORDIENKO D.V. Excitation-contraction coupling in gastrointestinal and other smooth muscles. Annu. Rev. Physiol. 1999;61:85–115. doi: 10.1146/annurev.physiol.61.1.85. [DOI] [PubMed] [Google Scholar]
- BUCKNER S.A., MILICIC I., DAZA A., DAVIS-TABER R., SCOTT V.E., SULLIVAN J.P., BRIONI J.D. Pharmacological and molecular analysis of ATP-sensitive K+ channels in the pig and human detrusor. Eur. J. Pharmacol. 2000;400:287–295. doi: 10.1016/s0014-2999(00)00388-5. [DOI] [PubMed] [Google Scholar]
- COTTON K.D., HOLLYWOOD M.A., MCHALE N.G., THORNBURY K.D. Ca2+ current and Ca2+-activated chloride current in isolated smooth muscle cells of the sheep urethra. J. Physiol. 1997;505:121–131. doi: 10.1111/j.1469-7793.1997.121bc.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOPALAKRISHNAN M., WHITEAKER K.L., MOLINARI E.J., DAVIS-TABER R., SCOTT V.E., SHIEH C.C., BUCKNER S.A., MILICIC I., CAIN J.C., POSTL S., SULLIVAN J.P., BRIONI J.D. Characterization of the ATP-sensitive potassium channels (KATP) expressed in guinea pig bladder smooth muscle cells. J. Pharmacol. Exp. Ther. 1999;289:551–558. [PubMed] [Google Scholar]
- HOWE B.B., HALTERMAN T.J., YOCHIM C.L., DO M.L., PETTINGER S.J., STOW R.B., OHNMACHT C.J., RUSSELL K., EMPFIELD J.R., TRAINOR D.A., BROWN F.J., KAU S.T. ZENECA ZD6169: A novel KATP channel opener with in vivo selectivity for urinary bladder. J. Pharmacol. Exp. Ther. 1995;274:884–890. [PubMed] [Google Scholar]
- LI J.H., YASAY G.D., ZOGRAFOS P., KAU S.T., OHNMACHT C.J., RUSSELL K., EMPFIELD J.R., BROWN F.J., TRAINOR D.A., BONEV A.D., HEPPNER T.J., NELSON M.T. Zeneca ZD6169 and its analogs from a novel series of anilide tertiary carbinols: in vitro KATP channel opening activity in bladder detrusor. Pharmacology. 1995;51:33–42. doi: 10.1159/000139314. [DOI] [PubMed] [Google Scholar]
- MIKAMI A., IMOTO K., TANABE T., NIIDOME T., MORI Y., TAKESHIMA H., NARUMIYA S., NUMA S. Primary structure and functional expression of the cardiac dihydropyridine-sensitive calcium channel. Nature. 1989;340:230–233. doi: 10.1038/340230a0. [DOI] [PubMed] [Google Scholar]
- QUAYLE J.M., NELSON M.T., STANDEN N.B. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol. Rev. 1997;77:1165–1232. doi: 10.1152/physrev.1997.77.4.1165. [DOI] [PubMed] [Google Scholar]
- TERAMOTO N., BRADING A.F. The effects of nifedipine and other calcium antagonists on the glibenclamide-sensitive K+ currents in smooth muscle cells from pig urethra. Br. J. Pharmacol. 1998;123:1601–1608. doi: 10.1038/sj.bjp.0701777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TERAMOTO N., BRADING A.F., ITO Y. Glibenclamide-sensitive K+ channels underlying levcromakalim-induced relaxation in pig urethra. Eur J. Pharmacol. 1999;365:291–300. doi: 10.1016/s0014-2999(98)00885-1. [DOI] [PubMed] [Google Scholar]
- TERAMOTO N., ITO Y. Comparative studies on the relaxing action of several adenosine 5′-triphosphate-sensitive K+ channel openers in pig urethra. J. Smooth Muscle Res. 1999;35:11–22. doi: 10.1540/jsmr.35.11. [DOI] [PubMed] [Google Scholar]
- TERAMOTO N., OGATA R., OKABE K., KAMEYAMA A., KAMEYAMA M., WATANABE T.X., KURIYAMA H., KITAMURA K. Effects of calciseptine on unitary barium channel currents in the guinea-pig portal vein. Pflügers Arch. 1996;432:462–470. doi: 10.1007/s004240050158. [DOI] [PubMed] [Google Scholar]
- TERAMOTO N., YUNOKI T., TAKANO M., YONEMITSU Y., MASAKI I., SUEISHI K., BRADING A.F., ITO Y. Dual action of ZD6169, a novel K+ channel opener, on ATP-sensitive K+ channels in pig urethral myocytes. Br. J. Pharmacol. 2001a;133:154–164. doi: 10.1038/sj.bjp.0704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TERAMOTO N., YUNOKI T., TANAKA K., ITO Y. Actions of a novel ATP-sensitive K+ channel opener ZD6169 on pig urethral smooth muscle. Jpn. J. Pharmacol. 2001b;146P:P-346. doi: 10.1038/sj.bjp.0704408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRIVEDI S., STETZ S.L., POTTER-LEE L., MCCONVILLE M., LI J.H., EMPFIELD J., OHNMACHT C.J., RUSSELL K., BROWN F.J., TRAINOR D.A., KAU S.T. K-channel opening activity of ZD6169 and its analogs: effect on 86Rb efflux and 3H-P1075 binding in bladder smooth muscle. Pharmacology. 1995;50:388–397. doi: 10.1159/000139308. [DOI] [PubMed] [Google Scholar]
- UEHARA A., HUME J.R. Interactions of organic calcium channel antagonists with calcium channels in single frog atrial cells. J. Gen. Physiol. 1985;85:621–647. doi: 10.1085/jgp.85.5.621. [DOI] [PMC free article] [PubMed] [Google Scholar]