

Abstract
We identified the ethacrynic-acid derivative DCPIB as a potent inhibitor of ICl,swell, which blocks native ICl,swell of calf bovine pulmonary artery endothelial (CPAE) cells with an IC50 of 4.1 μM. Similarly, 10 μM DCPIB almost completely inhibited the swelling-induced chloride conductance in Xenopus oocytes and in guinea-pig atrial cardiomyocytes. Block of ICl,swell by DCPIB was fully reversible and voltage independent.
DCPIB (10 μM) showed selectivity for ICl,swell and had no significant inhibitory effects on ICl,Ca in CPAE cells, on chloride currents elicited by several members of the CLC-chloride channel family or on the human cystic fibrosis transmembrane conductance regulator (hCFTR) after heterologous expression in Xenopus oocytes. DCPIB (10 μM) also showed no significant inhibition of several native anion and cation currents of guinea pig heart like ICl,PKA, IKr, IKs, IK1, INa and ICa.
In all atrial cardiomyocytes (n=7), osmotic swelling produced an increase in chloride current and a strong shortening of the action potential duration (APD). Both swelling-induced chloride conductance and AP shortening were inhibited by treatment of swollen cells with DCPIB (10 μM). In agreement with the selectivity for ICl,swell, DCPIB did not affect atrial APD under isoosmotic conditions.
Preincubation of atrial cardiomyocytes with DCPIB (10 μM) completely prevented both the swelling-induced chloride currents and the AP shortening but not the hypotonic cell swelling.
We conclude that swelling-induced AP shortening in isolated atrial cells is mainly caused by activation of ICl,swell. DCPIB therefore is a valuable pharmacological tool to study the role of ICl,swell in cardiac excitability under pathophysiological conditions leading to cell swelling.
Keywords: DCPIB; ICl,swell; swelling-activated chloride channel; atrium; action potential
Introduction
The swelling-induced chloride current ICl,swell is present in most cells (Nilius et al., 1994; Okada, 1997) including that of the mammalian heart (Sorota, 1992; Sakai et al., 1995) and it might play an important role in cell volume regulation. The molecular correlate of ICl,swell is not yet known.
In isolated cardiac cells, activation of ICl,swell either by positive pressure inflation (Hagiwara et al., 1992) or osmotic swelling causes a slight depolarization of the resting membrane potential (Vrest) (Akiyama & Fozzard, 1975; Ehara & Hasegawa, 1983; Zhang et al., 1993; Du & Sorota, 1997) and a shortening of the action potential (Kawata et al., 1974; Ehara & Hasegawa, 1983; Vandenberg et al., 1997). Previous studies on ventricular tissue indicate that during ischaemia and reperfusion cell swelling occurs (Tranum-Jensen et al., 1981; Jennings et al., 1986; Garcia-Dorado & Oliveras, 1993) which, with such a magnitude in isolated cells (Sorota, 1992; Hagiwara et al., 1992), could likely activate ICl,swell (Sorota, 1994; Hiraoka et al., 1998). Ischaemia and reperfusion are often associated with cardiac arrhythmias (Janse & Wit, 1989), suggesting that activation of ICl,swell and its linked APD shortening might participate in the genesis of arrhythmias (Hiraoka et al., 1998; Mulvaney et al., 2000). Cardiac ICl,swell has been described in ventricular and atrial cardiomyocytes of several mammalian species including humans, but seems to be predominantly expressed in atrial and sino-atrial node cells (Hagiwara et al., 1992; Sorota, 1992; Vandenberg et al., 1994; Sakai et al., 1995; Li et al., 1996). Thus, the activity of ICl,swell may have a greater impact on atrial, rather than ventricular, electrical activity and it might play a role in controlling the electrical activity of atrial tissue and possibly in pacemaking (Hiraoka et al., 1998; Mulvaney et al., 2000).
Whereas several studies investigated the contribution of ICl,swell to swelling-induced electrical changes in ventricular cells, to our knowledge, no study has analysed the function of ICl,swell in atrial action potential duration.
Here we investigated the effects of osmotic swelling on the APD of atrial cardiomyocytes isolated from guinea-pig hearts. We show for the first time that the non-diuretic ethacrynic-acid derivative DCPIB is a potent blocker of native ICl,swell currents present in different cell types like Xenopus oocytes, calf bovine pulmonary artery endothelial (CPAE) cells and guinea-pig atrial myocytes. At 10 μM, DCPIB is selective for ICl,swell since it has no effect on the activity of ICl,Ca in CPAE cells, on various cloned chloride channels after heterologous expression in Xenopus oocytes (CLC-1, -2, -4, -5, CLC-K1 and hCFTR) or on native currents in guinea-pig cardiomyoctes (IKs, IKr, IK1, INa, ICa and ICl,PKA). In addition, DCPIB did not alter the currents elicited by hKv1.5, hKv4.3, hminK and HERG, which encode the most important repolarizing currents in the human heart (IKur, Ito1, IKs and IKr,), after their expression in Xenopus oocytes. Consistently, DCPIB (10 μM) had no effects on atrial APD under isoosmotic conditions.
This novel selective blocker of ICl,swell allowed us to specifically determine the contribution of ICl,swell to swelling-induced changes in atrial APD without interfering with other cardiac currents critically involved in AP generation and shaping. We show that swelling of atrial myocytes by hypotonic perfusion activated ICl,swell and concomitantly caused a drastic shortening of APD, which was reversed by addition of DCPIB after cell swelling occurred. Moreover, this strong APD shortening was completely prevented by incubating cells with DCPIB prior to hypotonic perfusion, which blocked ICl,swell but not cell swelling.
Methods
Cell preparation and cell culture
Single cardiac myocytes were obtained from guinea-pig hearts using standard enzymatic techniques. Briefly, guinea-pigs (350 – 450 g) were killed by cervical dislocation after stunning and the hearts were quickly removed. The aorta was cannulated and the heart was retrogradely perfused for 5 min with a nominally Ca2+-free Tyrode solution at 37.5°C. The Ca2+-free Tyrode solution contained (in mM): NaCl 143, KCl 5.4, MgCl2 0.5, NaH2PO4 0.25, glucose 10, HEPES 5; and pH was adjusted to 7.2 with NaOH (290±5 mOsmol). The solution was aerated with O2 for 20 – 30 min. Subsequently the heart was perfused with the same solution which in addition contained 0.33 mg ml−1 collagenase (Type CLS II, 270 U mg−1, Biochrom KG) and 15 μM CaCl2. Atrium and ventricle were removed and further dissected into small pieces, and cell dissociation was achieved by gentle mechanical agitation with a pipette in the storage solution. The storage solution contained (in mM): L-glutamine acid 50, KCl 40, taurine 20, KH2PO4 20, MgCl2 1, glucose 10, HEPES 10, EGTA 2; and the pH was adjusted to 7.2 with KOH (260±5 mOsmol). The suspension was filtered through a nylon mesh and stored at room temperature.
Xenopus laevis oocytes were obtained from tricaine anaesthetized animals. Ovaries were surgically removed, cut into pieces and collagenase treated (1 mg ml−1, Worthington, type II) in OR 2 solution containing (in mM): NaCl 82.5, KCl 2 , MgCl2 1, HEPES 5, pH 7.4 with NaOH for 120 min. Oocytes were stored in recording ND 96 solution containing (in mM): NaCl 96 , KCl 2, CaCl2 1.8, MgCl2 1, HEPES 5, pH 7.4, additional containing Na+ pyruvate (275 mg l−1), theophylline (90 mg l−1) and gentamycin (50 mg l−1) at 18°C. Oocytes were injected with approximately 10 ng cRNA. Macroscopic currents were recorded 2 – 4 days after injection. For measurements of ICl,swell, oocytes were manually defolliculated and stored at 18°C in ND 96.
Calf pulmonary artery endothelial cells were grown in DMEM with 2 mM L-glutamine, 100 μg ml−1 streptomycin, 100 U ml−1 penicillin and 20% foetal bovine serum. Cultures were maintained at 37°C in 5% CO2. Cells were detached by exposure to 0.05% trypsin in a Ca2+- and Mg2+-free solution, reseeded, and kept in culture for 2 – 4 days before use. Only non-confluent cells were used.
Solutions
ICl,swell in CPAE cells
The standard external solution was a modified Krebs solution containing (in mM): NaCl 150, KCl 6, MgCl2 1, CaCl2 1.5, glucose 10, HEPES 10, adjusted to pH 7.4 with NaOH. The osmolarity was adjusted to 320±5 mOsmol using a vapour pressure osmometer (Vogel, OM 801). At the beginning of the recording the modified Krebs solution was replaced by an isotonic Cs+ solution (320±5 mOsmol) containing (in mM): NaCl 105, CsCl 6, MgCl2 1, CaCl2 1,5, D-mannitol 90, glucose 10, HEPES 10, adjusted to pH 7.4 with NaOH. ICl,swell was activated by exposing the cells to a 25% hypotonic solution (240±5mOsmol) by omitting 90 mM D-mannitol. The standard pipette solution contained (in mM): CsCl 40, Cs-aspartate 100, MgCl2 1, CaCl2 1.93, EGTA 5, Na2ATP 4, HEPES 10, adjusted to pH 7.2 with CsOH (290±5 mOsmol).
ICl,Ca in CPAE cells
The modified Krebs solution was replaced by a slightly hypertonic Krebs-Cs+ solution (345±5 mOsmol) containing (in mM): NaCl 150, CsCl 6, MgCl2 1, CaCl2 1.5, glucose 10, D-mannitol 25, HEPES 10, adjusted to pH 7.4 with NaOH, to prevent co-activation of ICl,swell. ICl,Ca was activated by loading the CPAE cells via the patch pipette with 1 μM free Ca2+ as described previously (Nilius et al., 1997a,1997b). The standard pipette solution contained (in mM): CsCl 40, Cs-aspartate 100, MgCl2 1, CaCl2 4.33, EGTA 5, Na2ATP 4, HEPES 10, adjusted to pH 7.2 with CsOH (290±5 mOsmol).
ICl,swell in Xenopus oocytes
The isoosmotic solution (220±5 mOsmol) was a mannitol containing ND 48 of the following composition (in mM): NaCl 48 , KCl 2, CaCl2 1.8, MgCl2 1, mannitol 110, and HEPES 5; pH was adjusted to 7.4 with NaOH. The hypotonic solution to evoke ICl,swell was ND 48 without mannitol (110±5 mOsmol).
ICl,swell in gp cardiomyocytes
The isoosmotic external solution (290±5 mOsmol) contained (in mM): NaCl 70, sucrose 140, MgCl2 2, BaCl2 2, HEPES 5, and the pH was adjusted to 7.5 with CsOH. 2 μM nicardipine was added to block Ca2+-channels and 20 μM ouabain to block swelling-induced Na+/K+ pump currents (Whalley et al., 1993). The standard hypoosmotic solution (160±5 mOsmol) was as above except that sucrose was omitted. The internal pipette solution (285±5 mOsmol) contained (in mM): CsCl 58, Cs-aspartate 52, tetraethylammonium (TEA) chloride 20, EGTA 10, Mg2ATP 5, Na3GTP 0.2, HEPES 5; pH was adjusted to 7.3 with CsOH.
Action potential recordings
The standard isoosmotic external solution (290±5 mOsmol) was (in mM): NaCl 100, sucrose 80, KCl 5.4, MgCl2 0.5, CaCl2 1.8, NaH2PO4 0.33, glucose 11, HEPES 5, pH was adjusted to 7.4 with NaOH. The hypoosmotic solution was similar to the isoosmotic solution except that sucrose was omitted (205±5 mOsmol). The internal pipette solution contained (in mM): K+-aspartate 100, KCl 20, EGTA 0.2, Na2-phosphocreatine 5, MgATP 5, MgCl2 1, Na3GTP 0.2, and HEPES 5, pH was adjusted to 7.3 with NaOH.
IKs, IKr, IK1 in gp cardiomyocytes
The standard solution contained (in mM): NaCl 140, KCl 4.7, CaCl2 1.3, MgCl2 1, glucose 10, and HEPES 10; pH was adjusted to 7.4 with NaOH. The solution for IKr additionally contained 5 μM nifedipine to block Ca2+-channels. The internal pipette solution contained (in mM): KCl 140, NaCl 10, MgCl2 1.1, K2ATP 1, EGTA 1, and HEPES 10; pH was adjusted to 7.2 with KOH.
IKs in gp cardiomyocytes during cell swelling
Recordings of IKs during cell swelling were performed as previously described (Rees et al., 1995). Myocytes were perfused with an isoosmotic solution containing (in mM): NaCl 100, sucrose 80, KCl 5.4, MgCl2 0.5, NaH2PO4 0.33, glucose 5.5, and HEPES 5.0; pH was adjusted to 7.4 with NaOH (290±5 mOsmol). In the hypotonic solution (200±5mOsmol) the sucrose was omitted. The solutions contained 0.2 μM dofetilide to block IKr. The internal pipette solution contained (in mM): K-aspartate 140, MgCl2 5, K2ATP 5, EGTA 10, and HEPES 5; pH was adjusted to 7.4 with KOH (310±5 mOsmol).
INa in gp cardiomyocytes
The external solution contained (in mM): NaCl 20, CsCl 30, tetramethylammonium chloride 20, CoCl2 2, MgCl2 1, 4-aminopyridine 5, sucrose 80, glucose 5, and HEPES 5, pH was adjusted to 7.4 with HCl. The internal solution contained (in mM): NaCl 16, CsCl 115, MgCl2 1, CaCl2 0.3, EGTA 10 and HEPES 5; pH was adjusted to 7.2 with CsOH.
ICa in gp cardiomyocytes
The bath solution contained (in mM): NaCl 132, CsCl 4.8, MgCl2 1.2, CaCl2 1, glucose 5, and HEPES 10; pH was adjusted to 7.3 with CsOH. The internal pipette solution had the following composition (in mM): CsCl 140, MgCl2 2, CaCl2 1 EGTA, 11, and HEPES 10; pH was adjusted to 7.3 with CsOH.
ICl,PKA in gp cardiomyocytes
The external solution was the same as the isotonic solution for ICl,swell recordings, except that it contained NaCl 140 mM and sucrose was omitted. To evoke ICl,PKA isoprenaline, prepared as 1 mM stock solution in water containing 100 mM ascorbic acid, was added to superfusion solutions to a final concentration of 1 μM, or alternatively 3-isobutyl-1-methylxanthine (IBMX), prepared as 1 M stock solution in DMSO, was added to a final concentration of 500 μM. The internal pipette solution was the same as for ICl,swell.
CLCs and CFTR expressed in Xenopus oocytes
The standard solution was ND 96. For activation of hCFTR the solution contained 1 mM IBMX and 10 μM forskolin; prepared from 1 and 0.1 M stock solutions in DMSO, respectively.
Voltage clamp protocols
During two-electrode voltage clamp experiments, the holding potential in Xenopus oocytes experiments was −30 mV. Inhibition values are given at +40 mV, except for rCLC-2 with −100 mV and hCLC-4 and hCLC-5 with +60 mV. For hCLC-1 a voltage step protocol from −120 to +80 mV with 40 mV increments for 500 ms was used. In rCLC-2 recordings voltage steps from −160 to +40 mV in 20 mV increments for 5 s were performed and inhibition data were obtained by stepping to −120 mV and subsequent to −100 mV for 10 s, using a sweep interval of 60 s. hCLC-4 and hCLC-5 were measured by a protocol stepping from −140 to +100 mV in 20 mV increments for 500 ms. CLC-K1 and hCFTR were analysed with a protocol that ranged from −100 to +60 mV in 20 mV steps. hCFTR was recorded for 500 ms and CLC-K1 for 2 s.
At the beginning of each measurement in guinea-pig cardiomyocytes an I-V relationship was obtained by a triangular ramp protocol; first hyperpolarizing to −140 mV within 40 ms, subsequent depolarizing to +80 mV in 500 ms and within 30 ms back to the holding potential. Inhibition data of ICl,swell and ICl,PKA are given at +40 mV. Voltage clamp protocols are illustrated within the figures.
Current measurements and data analysis
Standard two-electrode voltage clamp recordings in Xenopus oocytes were performed with a Turbo Tec 10CD (NPI) amplifier and an ITC-16 interface. Patch clamp recordings were amplified with an EPC 9 Series D (HEKA). Both recording methods were combined with Pulse software (HEKA) and Igor for data acquisition on Pentium II PC. Patch clamp recordings were performed in whole-cell mode at 35±1°C via a thermocouple feedback circuit. Patches of CPAE cells and Na+ currents were recorded at room temperature (21±1°C). In Na+ current experiments linear capacitative and leakage currents were subtracted by the P-P/n (P, test voltage; n=4) method (Bezanilla & Armstrong, 1977) and series resistance compensation was used at 40 – 60%. Cells were visualized with an inverted microscope (Zeiss, Axiovert 35) at a magnification of ×400. Changes in cell size were monitored by a camera (Panasonic, WV-CD 22) combined with a VHS video recorder (Panasonic, AG-7355). Photos were obtained by connecting a camera to the microscope (Contax, 167). Volume changes in ventricular myocytes are proportional to changes in cell width (Roos, 1986; Drewnowska & Baumgarten, 1991). In some experiments the cell width was monitored as an indicator of cell volume, as previously reported (Sorota, 1992). Changes in cell width were analysed using a reticle in the occular and the photographs, where the average width at three different locations was calculated. In experiments with cardiomyocytes tip resistances of 2.0 – 3.5 MΩ were used. For ICl,swell recordings tip resistances were 1.5 – 2.5 MΩ and after membrane rupture the suction port of the electrode holder was opened to the atmosphere to ensure that no pressure was applied to the back of the pipette. For action potential recordings electrodes with tip resistances of 3.0 – 4.5 MΩ were used. Action potentials were elicited by a 0.8 – 1 nA current pulse applied for 1 – 3 ms at a frequency of 0.2 Hz operating in current clamp mode. In all experiments the patch pipette current was zeroed before seal formation. All results are reported as means±s.e.mean. Statistical differences of resting membrane potential and action potential duration were evaluated by a two population Student's paired t-test using Origin software. IC50 for block of ICl,swell was estimated by a Hill equation using Igor software. Data were calculated for significance and indicated by asterisks, with * for P<0.05 and ** for P<0.01, compared to isotonic conditions unless stated otherwise.
Results
DCPIB is a potent blocker of native ICl,swell
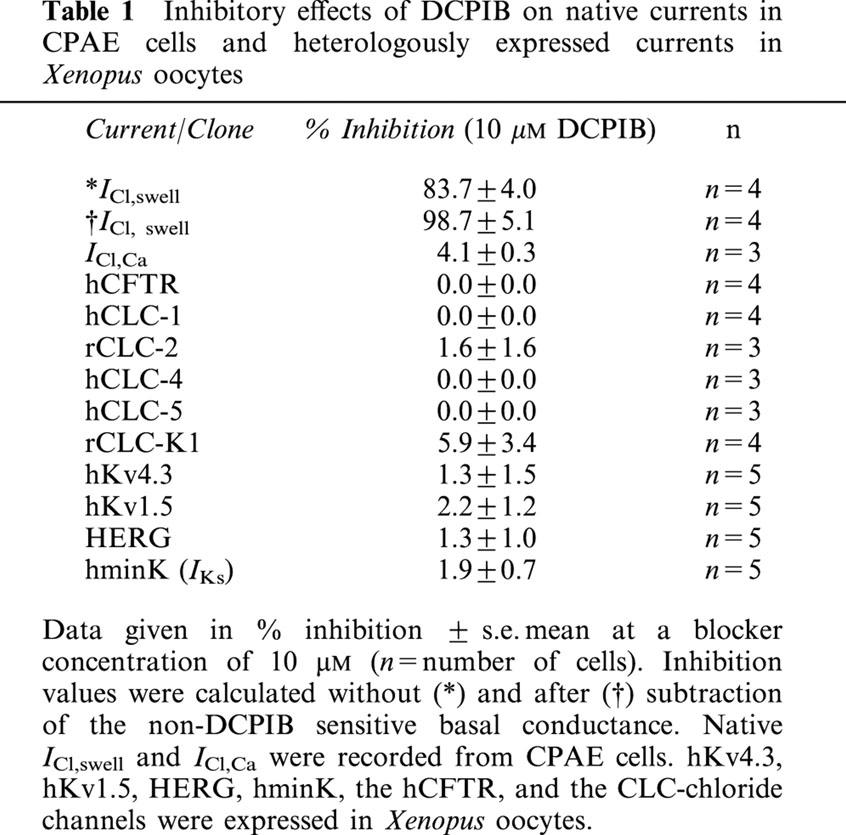
The non-diuretic acylaryloxyacid derivative DCPIB (Figure 1) has been shown to inhibit astroglial swelling (Bourke et al., 1981). The related compound IAA-94 has been shown to block epithelial chloride currents (Landry et al., 1989) and ICl,swell (Sorota, 1994). This prompted us to analyse whether DCPIB is able to block ICl,swell. Three different types of cells were used, CPAE cells, Xenopus oocytes and guinea-pig atrial cells, which all possess a native swelling-sensitive chloride current. In CPAE cells a large chloride current representing ICl,swell could be activated by osmotic swelling (Nilius et al., 1994). Maximal activation of ICl,swell occurred within 3 – 5 min of hypotonic perfusion. Treatment of swollen cells with 10 μM DCPIB inhibited ICl,swell by 83.7±4% (n=4) (Figure 2a – c and Table 1), reflecting an almost complete block of the swelling-induced changes in Cl−-conductance. Maximal block was achieved within 3 – 5 min. The IC50 for block of ICl,swell by DCPIB was 4.1 μM (Figure 1a). The block was voltage independent (Figure 1b) and fully reversible after washout of DCPIB by hypotonic solution. In Xenopus oocytes endogenous ICl,swell can only be measured if oocytes are follicle enclosed (Arellano & Miledi, 1995; Arellano et al., 1996) or if they are manually defolliculated (Ackerman et al., 1994). Oocytes were stored in theophyllin-free medium because otherwise the amplitude of the swelling-activated chloride current was reduced (see cyclic AMP dependence: Nagasaki et al., 2000). DCPIB also potently inhibited ICl,swell of Xenopus oocytes. The block of ICl,swell was 53.3±3.0% (n=4) at 10 μM.
Figure 1.
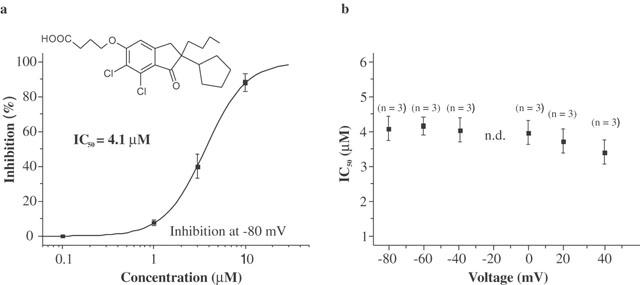
Chemical structure and dose response curve of DCPIB on ICl,swell in CPAE cells. (a) DCPIB blocked ICl,swell in CPAE cells with a half-maximal concentration (IC50) of 4.1 μM at −80 mV. Inhibition values were calculated without subtraction of the basal current. If the basal non-DCPIB sensitive current is subtracted the resulting IC50 is 2.5 μM at −80 mV. (b) IC50 values of DCPIB determined at different voltages without subtraction of the basal current (n.d.=not determined).
Figure 2.
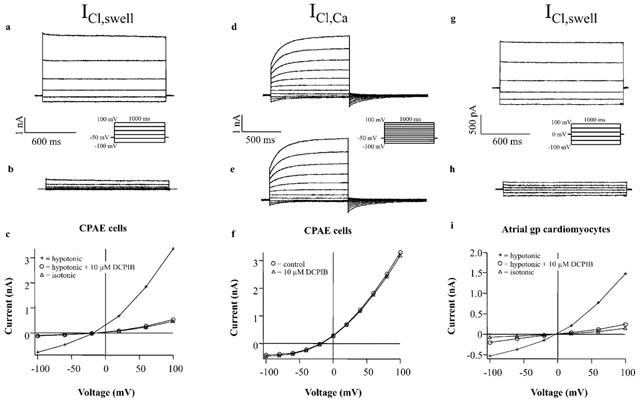
Effects of DCPIB on native ICl,swell and ICl,Ca currents. (a) ICl,swell current traces from CPAE cells after hypotonic solution was applied for 3 – 5 min in the absence and (b) in the presence of 10 μM DCPIB. (c) I – V curves of ICl,swell obtained with the illustrated step pulse protocol, in isotonic medium (isotonic), after cell swelling in hypotonic medium (hypotonic) and following the treatment of swollen cells with 10 μM DCPIB (hypotonic+10 μM DCPIB). (d) ICl,Ca currents recorded from CPAE cells in response to voltage steps applied every 3 s from a holding potential of −50 mV. (e) ICl,Ca currents in the presence of 10 μM DCPIB. (f) I – V relationships from currents in (d) and (e). Currents were measured at the end of the voltage steps. (g) ICl,swell currents of guinea-pig atrial cardiomyocytes. Hypotonic solution was applied for 5 – 10 min. Atrial currents were measured starting from a holding potential of 0 mV using the illustrated step pulse protocol. (h) ICl,swell currents in presence of 10 μM DCPIB in the hypotonic bath solution. (i) Corresponding I – V curves.
Table 1.
Inhibitory effects of DCPIB on native currents in CPAE cells and heterologously expressed currents in Xenopus oocytes
Next we tested the effect of DCPIB on atrial ICl,swell using previously reported methods (Vandenberg et al., 1994). DCPIB effectively blocked atrial ICl,swell resulting in an inhibition of 72.9±7.4% (n=4) at 10 μM (Figure 2g – i and Table 2). Thus, in all cells examined 10 μM DCPIB was able to potently block native ICl,swell. All cells displayed a basal conductance under isoosmotic conditions. In CPAE cells, the addition of 50 μM DCPIB led to a reduction of the basal current by 15.8±4.2% (n=4), whereas the basal current of atrial cardiomyocytes was only reduced by 8.5±3.0% (n=4). This is consistent with a low basal activity of ICl,swell in isoosmotic environment. Therefore, after subtraction of the non-DCPIB sensitive basal conductance, inhibition of the swelling-induced changes in Cl−-conductance of CPAE and atrial cells was almost complete (Figure 2c,i).
Table 2.
Inhibitory effects of DCPIB on native currents of guinea-pigs

DCPIB does not affect cell swelling
Volume changes in isolated cardiomyocytes are proportional to changes in cell width (Roos, 1986; Drewnowska & Baumgarten, 1991). In some experiments the cell width was monitored as an indicator of cell volume. Representative experiments are illustrated in Figure 3. In all atrial cells (n=6) a detectable increase in cell width was observed during hypotonic perfusion. The average cell width increased by a factor of 1.31±0.04 (n=6) (Figure 3b) after 10 min of hypotonic perfusion. Chloride currents increased over the time course of the experiments. Addition of DCPIB to swollen cells in the hypotonic medium did not reverse swelling of atrial cardiomyocytes. After 5 min of perfusion with a 10 μM DCPIB containing hypotonic solution (Figure 3c) the average increase in cell width was 1.41±0.07 fold (n=7) compared to the original isotonic value. Since DCPIB did not decrease cell size, an inhibition of ICl,swell through attenuation of the hypotonicity-induced cell swelling is very unlikely. Moreover, preincubation of cardiomyocytes with DCPIB did not attenuate subsequent swelling in hypotonic medium. Average cell width after 5 min of preincubation with 10 μM DCPIB was 1.01±0.02% (n=4) (Figure 3e) and subsequent cell swelling for 10 min was of the same magnitude as for the swelling in the absence of DCPIB. In DCPIB containing hypotonic solution the cell size increased by a factor of 1.35±0.07 (Figure 3f).
Figure 3.
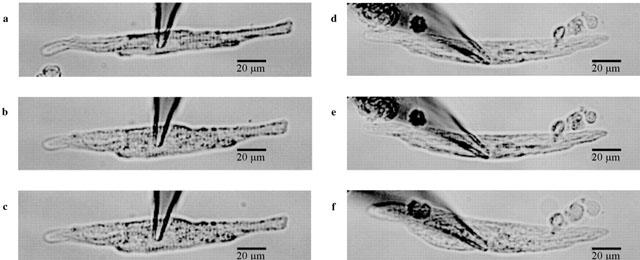
Photographs of guinea-pig atrial cardiomyocytes during whole cell patch clamp recording. Cell width was analysed by averaging the cell width at three different places of the myocyte. (a) In isotonic solution. (b) Following 10 min of hypotonic perfusion and (c) after perfusion (5 min) with 10 μM DCPIB of swollen cells from (b). The figure illustrates that DCPIB did not decrease the cell swelling during hypotonicity. (d) Atrial cardiomyocyte in isotonic solution and (e) after 5 min of preincubation with 10 μM DCPIB and (f) after perfusion with hypotonic medium containing 10 μM DCPIB. Preincubation with DCPIB did not inhibit subsequent cell swelling in hypotonic medium.
Specificity of DCPIB
To test the specificity for ICl,swell, we first analysed the effects of DCPIB on several Cl− currents, namely on native ICl,Ca in CPAE cells, on several members of the CLC-chloride channel family and on human CFTR after heterologous expression in Xenopus oocytes. While we could not functionally express hCLC-3 in Xenopus oocytes, as has been previously reported (Borsani et al., 1995), injection of cRNA encoding the other chloride channels generated currents as previously reported (Bear et al., 1991; Drumm et al., 1991; Thiemann et al., 1992; Uchida et al., 1994; Steinmeyer et al., 1994; 1995; Friedrich et al., 1999). As illustrated in Figure 2d – f, DCPIB (10 μM) did not alter ICl,Ca in CPAE cells. DCPIB also had no effects on CLC-1, -2, -4, -5, -K1 and hCFTR expressed in Xenopus oocytes (Table 1 and Figure 4).
Figure 4.
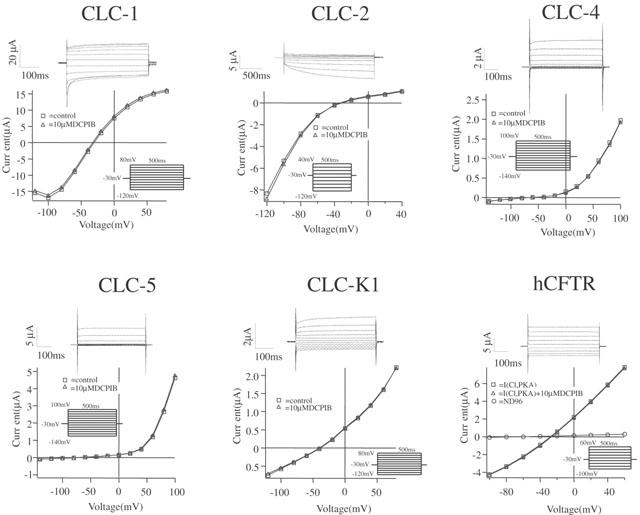
Effects of 10 μM DCPIB on CLC-chloride channels and on hCFTR expressed in Xenopus oocytes. hCFTR was activated by 1 mM IBMX and 10 μM forskolin. Currents were elicited starting from a holding potential of −30 mV according to the illustrated step protocols. Original current traces show control currents. I – V relationships were determined from control currents and currents in the presence of DCPIB.
To determine the specificity of DCPIB for cardiac ion currents involved in AP generation and shaping, isolated guinea-pig cardiomyocytes of the atrium and ventricle were treated with 10 μM DCPIB (Figure 5). DCPIB did not affect amplitudes of the following cationic currents: IKr, IKs, IK1, INa and ICa (Table 2). In addition, we did not observe a shift in I – V relationships of these currents which would also alter APD (data not shown). Similar to hCFTR expressed in Xenopus oocytes (Figure 4), ICl,PKA, which is thought to represent the native cardiac CFTR chloride current, was not inhibited by 10 μM DCPIB (Table 2 and Figure 5). To confirm the data obtained with native currents of guinea-pig cardiomyocytes, we tested the effects of DCPIB (10 μM) on the currents elicited by hKv1.5, hKv4.3, hminK and HERG, which underly the most important repolarizing currents in the human heart (IKur, Ito1, IKs and IKr). Again, DCPIB did not alter these currents after expression in Xenopus oocytes (Figure 6).
Figure 5.
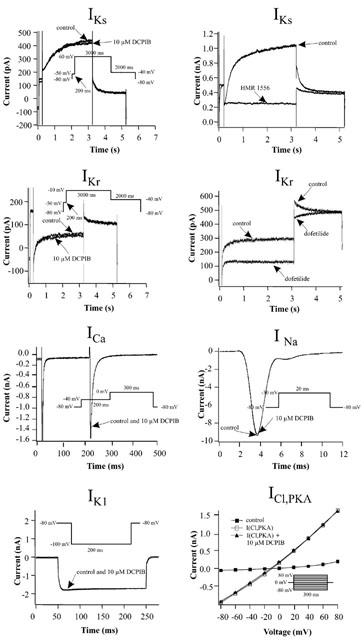
Influence of 10 μM DCPIB on currents of guinea-pig cardiomyocytes. Holding potential in whole cell patches was routinely −80 mV, but 0 mV for measurement of ICl,PKA. Inhibition of IKr was analysed from the tail currents at −40 mV. IKs inhibition data were obtained from the steady state current at +60 mV. To show that the used voltage protocols (Bosch et al., 1998) can separate IKr and IKs we tested their inhibition by 0.2 μM dofetilide and 1.0 μM HMR 1556, respectively. IK1 inhibition values were obtained at −100 mV. INa inward currents were analysed by stepping from the holding potential to −30 mV for 20 ms, with a frequency of 1 Hz. ICa was studied by a step protocol to analyse its peak inward current. The pulses were applied every 15 s to minimize rundown. ICl,PKA was activated by isoprenaline (1 μM) or alternatively IBMX (500 μM).
Figure 6.
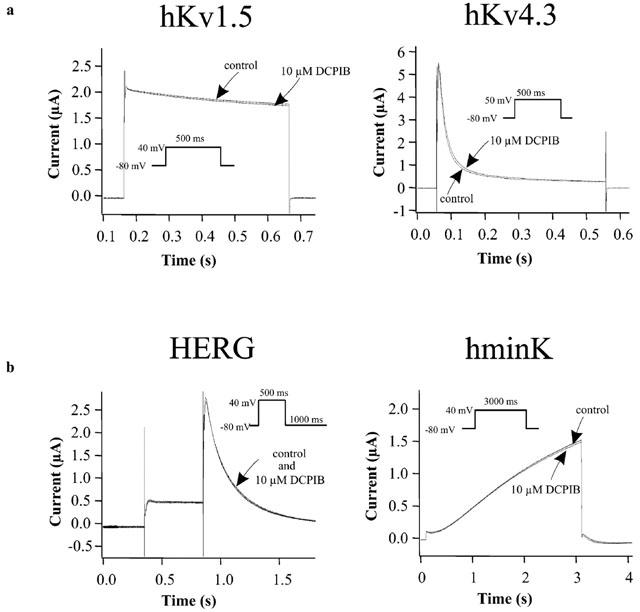
Effects of 10 μM DCPIB on human Kv and delayed rectifier channels, which encode the most important repolarizing K+ currents in the human heart. The channels were expressed in Xenopus oocytes. Holding potential was −80 mV. (a) Current traces of hKv1.5 (IKur) and hKv4.3 (Ito1) before (control) and subsequent to the addition of 10 μM DCPIB. (b) Original current traces of HERG (IKr) and hminK (IKs) injected Xenopus oocytes in ND96 (control) and in the presence of 10 μM DCPIB.
These results demonstrated that DCPIB, at a concentration sufficient to almost completely block ICl,swell, does not affect the major currents involved in the cardiac action potential. Therefore, DCPIB at this concentration is selective for ICl,swell and is a valuable pharmacological tool to study the role of Cl− currents in swelling-induced action potential changes.
Effects of cell swelling on ICl,swell, Vrest and APD
Atrial cells are more sensitive to cell swelling than cardiomyocytes isolated from the ventricle (Vandenberg et al., 1994). In agreement with this, ICl,swell could be activated in 100% (n=7) of atrial cells when perfused with 29% hypotonic solution for 5 – 10 min maximum. In contrast, only 25% (n=8) of ventricular cells in hypotonic solution elicited an ICl,swell. Within this time range activation of ICl,swell was fully reversible on return to isotonic conditions and sensitive to DCPIB (Table 2). Under isotonic conditions the resting membrane potential of atrial cardiomyocytes was −62.9±0.8 mV (n=13). APD90 was 138.3±13.3 ms (n=13) and APD50 was 97.8±14.7 ms (n=13). Consistent with its ineffectiveness on all tested cardiac currents, DCPIB showed no significant effect on isotonic atrial APD (APD90 105.5±3.3% (n=6), APD50 96.5±1.9% (n=6)), (Table 3b and Figure 7b). Also, the resting membrane potential was not significantly different when 10 μM DCPIB was present in isoosmotic medium (Table 3b).
Table 3.
Action potential duration and membrane potential

Figure 7.
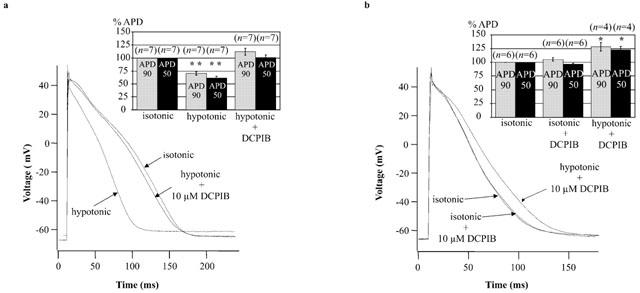
Influence of hypotonicity and 10 μM DCPIB on atrial action potentials. (a) Individual action potential recordings of control cells (isotonic) were compared with AP traces from swollen cells (hypotonic), obtained by perfusion of cells with hypotonic medium for 5 – 10 min. Hypotonic solution caused a marked reduction in APD. Subsequent addition of 10 μM DCPIB reversed APD shortening (hypotonic+10 μM DCPIB). Attenuation of the peak voltage under hypotonic medium was only observed in two out of seven cells. Inset: The histogram illustrates the results displayed in Table 3a. Action potential durations were normalized and expressed as percentage of the isotonic APD. **P<0.01 compared with isotonic values. (b) Prevention of swelling-induced atrial action potential shortening by 10 μM DCPIB. Individual action potentials were recorded from atrial myocytes after perfusion with 10 μM DCPIB in isotonic solution for 5 min. DCPIB did not affect APD under isotonic conditions (compare isotonic vs isotonic+10 μM DCPIB). Subsequent treatment with hypotonic medium containing 10 μM DCPIB for 5 – 10 min did not result in APD shortening (hypotonic+10 μM DCPIB). Inset: The histogram illustrates the results displayed in Table 3b. Action potential durations were normalized to isotonic conditions and expressed as percentage of the isotonic values. *P<0.05 compared with isotonic values.
Hypotonic perfusion, lasting for the same time period required for maximal activation of ICl,swell, significantly shortened atrial APD (Figure 7a). APD90 was shortened to 70.2±3.4% (n=7; P<0.01) and APD50 to 61.3±3.5% (n=7; P<0.01) of the isotonic value (taken as 100%). Addition of 10 μM DCPIB to hypotonic medium abolished APD shortening (Figure 7a) and APD90, as well as APD50, were not statistically different from the original isotonic values (Table 3a). In some experiments APD50 tended to remain slightly shortened, whereas APD90 appeared to be increased. However, the overall resulting mean values of the APD90 and APD50 did not differ statistically from the original values (Table 3a). Thus, in all atrial myocytes (n=7), APD shortening occurred during cell swelling and was fully recovered by treatment with DCPIB. The resting membrane potential depolarized only slightly from −62.9±1.2 (n=7) to −60.9±1.2 mV (n=7) (P<0.05) during cell swelling. Addition of DCPIB to swollen cells did not result in a significant repolarization and Vrest remained at −61.8±1.1 mV (P>0.05). In addition to APD shortening, in some cells (two out of seven) the peak voltage of action potentials decreased during cell swelling. This reduction was almost completely antagonised by 10 μM DCPIB (Figure 7a). To exclude that the reduction of the action potential duration was due to the opening of KATP channels, we applied a triangular ramp pulse protocol to record KATP currents after cell swelling. However, I – V relationships of resulting currents did not provide any evidence that KATP channels opened during osmotic cell swelling (data not shown).
In recordings from ventricular cells, initial prolongation of APD was routinely observed during hypoosmotic challenge (Vandenberg et al., 1997). This effect was not seen in atrial cells.
Perfusion of atrial myocytes with 10 μM DCPIB in isotonic solution for 5 min completely prevented activation of ICl,swell (data not shown) and action potential shortening following the switch to hypotonic DCPIB-containing solution (Figure 7b). Even 10 min after the shift to hypotonic solution, a time sufficient to strongly activate ICl,swell, APD was not shortened. In contrast, a slight prolongation was observed and APD90 was 128.3±7.5% (n=4) and APD50 was 122.8±6.5% (n=4) as compared to isotonic conditions (P=0.03 for APD90, P=0.04 for APD50) (Table 3b). However, in the prolonged presence of DCPIB, the resting membrane potential was not significantly changed during osmotic swelling of cardiomyocytes.
Effects of DCPIB on IKs during cell swelling
To rule out that DCPIB blocks general mechanisms of ion channel activation triggered by cell swelling, we tested whether DCPIB inhibits the swelling-enhanced IKs currents (Rees et al., 1995). We also tested whether DCPIB affected IKs under hypotonic conditions, since a stronger inhibition of IKs during cell swelling could also have contributed to the observed APD lengthening. The IKs measurements during cell swelling were performed as reported previously (Rees et al., 1995). Cells were swollen under Ca2+-free conditions to shift activation of IKr to more negative potentials and activation of IKs to more positive values (Sanguinetti & Jurkiewicz, 1992; Jurkiewicz & Sanguinetti, 1993). In addition, 0.2 μM dofetilide was added to block remaining IKr. Control IKs currents measured at 4.9 s of the voltage step were 0.71±0.08 nA (n=6) and cell swelling caused an increase by 137.1±35.6% (n=3) to 1.68±0.31 nA (Figure 8), similar to previous findings (Rees et al., 1995). DCPIB (10 μM) was not able to block IKs after cell swelling occurred (Figure 8a).
Figure 8.
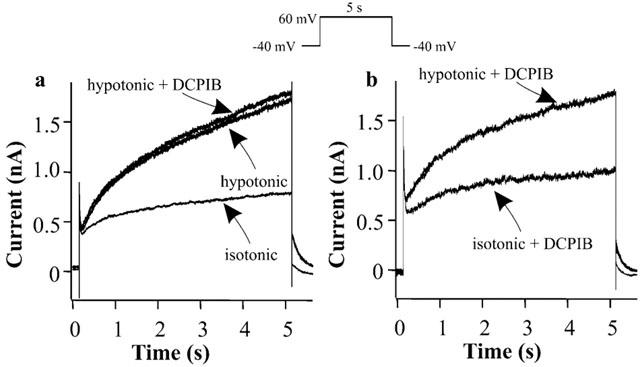
Effects of DCPIB on swelling-enhanced IKs currents. IKs was evoked by stepping from a holding potential of −40 to +60 mV for 5 s under Ca2+-free conditions. (a) Representative IKs current traces in isotonic solution (isotonic), after cell swelling caused by hypotonic perfusion (hypotonic) and after addition of 10 μM DCPIB to the hypotonic solution (hypotonic+DCPIB). (b) Representative IKs current traces in the presence of 10 μM DCPIB in the isotonic solution (isotonic+DCPIB) and after cell swelling in the presence of DCPIB (hypotonic+DCPIB).
In the presence of DCPIB, IKs also increased during swelling by 140.3±31.2% (n=3) (control current of 0.68±0.12 to 1.63±0.35 nA). Thus, DCPIB was not able to inhibit the swelling-induced increase in IKs current amplitude (Figure 8b). The exact mechanism of ICl,swell block by DCPIB is not yet known. These results, together with the cell size measurements, speak in favour of a blockade of ICl,swell itself and against inhibition of a general swelling-induced mechanism of ion channel activation.
Discussion
In almost every cell, swelling is linked to the activation of an outwardly rectifying chloride current, ICl,swell, that is possibly involved in subsequent regulatory volume decrease (RVD). In excitable cells activation of this chloride conductance may additionally interfere with their electrical activity. Since chloride equilibrium potential is close to Erest and ICl,swell is an outwardly rectifying current, opening of ICl,swell should enhance repolarization of the action potential and thereby lead to a shortened action potential duration. The resulting decrease in effective refractory period might favour reentrant arrhythmias (Hiraoka et al., 1998; Mulvaney et al., 2000). In particular, ischaemia and subsequent reperfusion of affected cardiac tissue cause swelling of cardiomyocytes and therefore will most likely activate ICl,swell, which in turn will result in differences in action potential duration and excitability of ischaemic compared to non ischaemic tissue (Hiraoka et al., 1998). The increased dispersion of refractoriness associated with this pathophysiological state might render the heart arrhythmic.
Several studies have been dedicated to the possible pathophysiological role of ICl,swell in the heart. However, conclusions drawn from these studies are hampered by the use of rather unspecific and less potent drugs. Here, we investigated the possibility that DCPIB is a potent and selective blocker of ICl,swell. It is structurally related to indanyloxyacetic acid, a known blocker of chloride channels in epithelial cells (Landry et al., 1989). Here we demonstrate for the first time that DCPIB is a potent blocker of ICl,swell in various cells. DCPIB was very effective in preventing adenosine-stimulated astroglial cell swelling (Bourke et al., 1981) and while a possible inhibition of a coupled Cl−-cation transport pathway has been discussed, to our knowledge, the precise target of DCPIB in this experimental setting has never been identified. It also remains unclear whether the corresponding transport protein is present in atrial cardiomyocytes and if so, whether it is able to modulate APD. The finding that DCPIB did not alter isotonic APD does not strictly rule out its presence in cardiomyocytes, since it is possibly only activated during cell swelling. However, in contrast to its effects on astroglial cells, DCPIB did not inhibit swelling of cardiomyocytes, making it rather unlikely that this Cl−-cation transport pathway is present in atrial cardiomyocytes. Moreover, our finding that DCPIB inhibits ICl,swell suggests that blocking of ICl,swell might somehow have contributed to the beneficial effects of DCPIB treatment in astroglial cell swelling.
The IC50 for block of ICl,swell in endothelial cells was 4.1 μM. Thus, DCPIB is one of the most potent known inhibitors for this current. More importantly and in contrast to classical chloride channel blockers, like DIDS, NPPB, niflumic acid and 9-AC, DCPIB seems to be selective for ICl,swell at 10 μM. Even the structurally related compound IAA-94, also a known blocker of ICl,swell, is not suitable as a pharmacological tool because of its extremely slow time course of inhibition (up to 20 min) and its nonspecific effects on action potential configuration in the absence of cell swelling (Sorota, 1994). Since block by DCPIB is voltage independent, observed changes in APD at different membrane potentials reflect the genuine influence of ICl,swell on APD, rather than the different effects caused by voltage dependent blockers.
In contrast to known chloride channel blockers, we did not observe effects of DCPIB (10 μM) on various other chloride channels. Since it also showed no effects on several cardiac currents that essentially generate and shape the action potential, we regarded it as a useful tool to analyse action potential alterations during osmotic cell swelling. A further prerequisite was that DCPIB did not affect the cardiac action potential under isotonic conditions. Although we did not individually test all cardiac ion currents involved in action potential shaping, or transporters and exchangers which, under certain conditions, may also modulate the action potential, this result is consistent with the proposed specificity of DCPIB. It also supports the observation that ICl,swell is only weakly active under isoosmotic conditions in isolated cardiac cells.
Previous studies have already demonstrated that swelling causes slight changes in resting membrane potential and a reduction of APD in ventricular cells (Vandenberg et al., 1997). Here we studied the role of ICl,swell in the AP of atrial cells since ICl,swell seems to be preferentially and possibly more strongly expressed in atrial cells and atrial swelling was always accompanied by strong action potential shortening. Shortening of APD50 was generally stronger than that of APD90, which can be explained in part by the outward rectification of ICl,swell. Furthermore, the APD50 is evaluated at a potential positive to the chloride equilibrium potential (ECl) in cardiomyocytes, whereas APD90 emerges at a potential very close to ECl. Since estimates of the Cl− activity, aiCl, in cardiac muscle place ECl in the range of −71 to −45 mV under normal physiological conditions (Walker, 1986; Harvey & Hume, 1989), driving force of both inward and outward Cl− currents near ECl is very low. This results in an APD shortening over the whole voltage range but to a lesser extent during the late phase of repolarization. The narrow distance of the reported value of ECl to the recorded Vrest of the atrial cardiomyocytes in our experiments partly explains the weak depolarization observed during cell swelling. Harvey & Hume (1989) suggested that chloride currents in cardiomyocytes, which are dialysed with 22 mM Cl− in a 5.4 mM external K+ solution, contribute little to the resting membrane conductance because it is dominated by the inward rectifier K+ current IK1. Consistent with the results by Yamawake et al. (1992), the observed effects on the atrial membrane potential might have been more pronounced with a lower [K+]o present in our study, since this is expected to reduce the conductance of IK1 channels (Sakmann & Trube, 1984). Vandenberg et al. (1997) observed a depolarization of ventricular cardiomyocytes during hypotonic perfusion of 3.5 mV (Group A), a value which is very close to the 2.0 mV depolarization we measured for atrial cells. However, the depolarization in ventricular cardiomyocytes (Vandenberg et al., 1997) occurred also in cells which lacked a DIDS-sensitive component of APD shortening during cell swelling.
Atrial APD shortening was highly sensitive to DCPIB and inhibition of ICl,swell by DCPIB always caused a reversal of APD shortening, though the achieved effectivity varied slightly from cell to cell. In some cells, a slight shortening of the APD50 persisted and a weakly increased APD90 remained, in comparison to the original isotonic values, while in other cells, both APD50 and APD90 were not fully reversed to isotonic APD. These weak differences in the resulting APD might be caused by changes in currents other than ICl,swell during osmotic swelling. Also, alterations in voltage trajectory or dilutional changes of intracellular K+ and Cl− concentrations might influence repolarizing K+ currents or the ECl. However, the observed differences in APD were not statistically significant (Table 3a).
In addition to these variations, cell swelling also causes changes in other ionic currents, that also could influence APD: enhancement of IKs (Rees et al., 1995) would lead to decreased APD, while inhibition of IKr (Rees et al., 1995) or ICa augmentation would lead to prolonged APD. The sum of these opposing changes during cell swelling could explain why action potential shortening in isolated atrial guinea-pig cardiomyocytes due to cell swelling is primarily caused by activation of ICl,swell and can be fully reversed by DCPIB.
In contrast to our results from atrial cells, full block of ICl,swell in ventricular cells by DIDS was only capable of reversing 50% of swelling-induced APD shortening (Vandenberg et al., 1997), suggesting that in ventricular myocytes activation of ICl,swell is not the only mechanism contributing to APD shortening during osmotic cell swelling (Vandenberg et al., 1997). However, several points must be considered that could explain these different conclusions: (1) DIDS has been shown to activate currents like IKs and ICl,PKA (Harvey, 1993; Busch et al., 1994), which would also cause APD shortening; (2) Hypotonic perfusion of ventricular cells caused an initial lengthening of the action potential (Vandenberg et al., 1997), which was not observed in atrial cardiomyocytes; (3) In these experiments cells were only swollen for 3 min and only showed an APD shortening of 27% of the isotonic value. Considering the slow time course of ICl,swell activation, its contribution to APD shortening should further increase with time.
However, the contribution of ICl,swell to APD shortening might differ between atrium and ventricle and also depend on the experimental conditions. Particularly, IKATP has been shown to be the main current causing swelling-induced APD shortening in ventricular cardiomyocytes in the presence of low intracellular ATP (Priebe & Beuckelmann, 1998).
It is known that several currents change during cell swelling (Vandenberg et al., 1996), whereas the time course of the changes for each current is not yet known and may also differ from species to species and between atrium and ventricle. The observed increase in atrial APD caused by cell swelling in sustained presence of ICl,swell blockers (Figure 7b), is similar to that previously observed in ventricular cells (Vandenberg et al., 1997). It might in part be due to swelling-induced inhibition of the fast delayed rectifier K+ current (Rees et al., 1995) and an increase in the L-type Ca2+ current (Matsuda et al., 1996). This increase in APD might only become visible during the block of ICl,swell. In addition, as mentioned above, dilutional changes will also influence many currents and ion transport pathways by altering the reversal potentials of the individual ions. Furthermore, sustained inhibition of ICl,swell during continued cell swelling might interact with RVD and lead to the activation of unknown compensatory mechanisms. Therefore, several mechanisms could explain the observed deviation of action potential length during prolonged cell swelling and simultaneous ICl,swell blockage.
We show that DCPIB is a potent, selective blocker of ICl,swell in various tissues. Moreover, we provide the first analysis concerning the influence of ICl,swell in the atrial action potential, from which we conclude that, under the used experimental conditions, swelling-induced atrial AP shortening is primarily caused by activation of ICl,swell. Therefore, DCPIB is a valuable tool for comparative studies of ICl,swell effects on atrial and ventricular APD and to further our understanding of the role of ICl,swell in cardiac physiology and pathophysiology.
Acknowledgments
We thank Karin Kopp, Astrid Weyermann, Stefan Müller and Birgit Herzog for technical support and Ilona Gutcher for editorial help.
Abbreviations
- CPAE
calf pulmonary artery endothelial cells
- DCPIB
(4-(2-Butyl-6,7-dichlor-2-cyclopentyl-indan-1-on-5-yl) oxybutyric acid)
- ECl
chloride equilibrium potential
- EGTA
ethylene glycol-O,O′-bis(2-aminoethyl)-N,N,N′,N′-tetraacetic acid
- HEPES
N-(hydroxyethyl)piperazine-N′-(2-ethanesulphonic acid)
- ICl,Ca
calcium-activated chloride current
- ICl,swell
swelling-activated chloride current
- [K+]o
external K+ concentration
- RVD
regulatory volume decrease
- Vrest
resting membrane potential
References
- ACKERMAN M.J., WICKMAN K.D., CLAPHAM D.E. Hypotonicity activates a native chloride current in Xenopus oocytes. J. Gen. Physiol. 1994;103:153–179. doi: 10.1085/jgp.103.2.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AKIYAMA T., FOZZARD H.A. Influence of potassium ions and osmolality on the resting membrane potential of rabbit ventricular papillary muscle with estimation of the activity and the activity coefficient of internal potassium. Circ. Res. 1975;37:621–629. doi: 10.1161/01.res.37.5.621. [DOI] [PubMed] [Google Scholar]
- ARELLANO R.O., MILEDI R. Functional role of follicular cells in the generation of osmolarity-dependent Cl− currents in Xenopus follicles. J. Physiol. 1995;488:351–357. doi: 10.1113/jphysiol.1995.sp020971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARELLANO R.O., WOODWARD R.M., MILEDI R. Ion channels and membrane receptors in follicle-enclosed Xenopus oocytes. New York: Plenum Press; Ion Channels. 1996;vol. 4:203–259. doi: 10.1007/978-1-4899-1775-1_6. [DOI] [PubMed] [Google Scholar]
- BEAR C.E., DUGUAY F., NAISMITH A.L., KARTNER N., HANRAHAN J.W., RIORDAN J.R. Cl− channel activity in Xenopus oocytes expressing the cystic fibrosis gene. J. Biol. Chem. 1991;266:19142–19145. [PubMed] [Google Scholar]
- BEZANILLA F., ARMSTRONG C.M. Inactivation of the sodium channel. I. Sodium current experiments. J. Gen. Physiol. 1977;70:549–566. doi: 10.1085/jgp.70.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BORSANI G., RUGARLI E.I., TAGLIALATELA M., WONG C., BALLABIO A. Characterization of a human and murine gene (CLCN3) sharing similarities to voltage-gated chloride channels and to a yeast integral membrane protein. Genomics. 1995;27:131–141. doi: 10.1006/geno.1995.1015. [DOI] [PubMed] [Google Scholar]
- BOSCH R.F., GASPO R., BUSCH A.E., LANG H.J., LI G.R., NATTEL S. Effects of the chromanol 293B, a selective blocker of the slow, component of the delayed rectifier K+ current, on repolarization in human and guinea pig ventricular myocytes. Cardiovasc. Res. 1998;38:441–450. doi: 10.1016/s0008-6363(98)00021-2. [DOI] [PubMed] [Google Scholar]
- BOURKE R.S., WALDMAN J.B., KIMELBERG H.K., BARRON K.D., SAN FILIPPO B.D., POPP A.J., NELSON L.R. Adenosine-stimulated astroglial swelling in cat cerebral cortex in vivo with total inhibition by a non-diuretic acylaryloxyacid derivative. J. Neurosurg. 1981;55:364–370. doi: 10.3171/jns.1981.55.3.0364. [DOI] [PubMed] [Google Scholar]
- BUSCH A.E., HERZER T., WAGNER C.A., SCHMIDT F., RABER G., WALDEGGER S., LANG F. Positive regulation by chloride channel blockers of IsK channels expressed in Xenopus oocytes. Mol. Pharmacol. 1994;46:750–753. [PubMed] [Google Scholar]
- DRUMM M.L., WILKINSON D.J., SMITH L.S., WORRELL R.T., STRONG T.V., FRIZZELL R.A., DAWSON D.C., COLLINS F.S. Chloride conductance expressed by delta F508 and other mutant CFTRs in Xenopus oocytes. Science. 1991;254:1797–1799. doi: 10.1126/science.1722350. [DOI] [PubMed] [Google Scholar]
- DREWNOWSKA K., BAUMGARTEN C.M. Regulation of cellular volume in rabbit ventricular myocytes: bumetanide, chlorothiazide, and ouabain. Am. J. Physiol. 1991;260:C122–C131. doi: 10.1152/ajpcell.1991.260.1.C122. [DOI] [PubMed] [Google Scholar]
- DU X.Y., SOROTA S. Cardiac swelling-induced chloride current depolarizes canine atrial myocytes. Am. J. Physiol. 1997;272:H1904–H1916. doi: 10.1152/ajpheart.1997.272.4.H1904. [DOI] [PubMed] [Google Scholar]
- EHARA T., HASEGAWA J. Effects of hypertonic solution on action potential and input resistance in the guinea-pig ventricular muscle. Jpn. J. Physiol. 1983;33:151–167. doi: 10.2170/jjphysiol.33.151. [DOI] [PubMed] [Google Scholar]
- FRIEDRICH T., BREIDERHOFF T., JENTSCH T.J. Mutational analysis demonstrates that CLC-4 and CLC-5 directly mediate plasma membrane currents. J. Biol. Chem. 1999;274:896–902. doi: 10.1074/jbc.274.2.896. [DOI] [PubMed] [Google Scholar]
- GARCIA-DORADO D., OLIVERAS J. Myocardial oedema: a preventable cause of reperfusion injury. Cardiovasc. Res. 1993;27:1555–1563. doi: 10.1093/cvr/27.9.1555. [DOI] [PubMed] [Google Scholar]
- HAGIWARA N., MASUDA H., SHODA M., IRISAWA H. Stretch-activated anion currents of rabbit cardiac myocytes. J. Physiol. (Lond.) 1992;456:285–302. doi: 10.1113/jphysiol.1992.sp019337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARVEY R.D. Effects of stilbenedisulfonic acid derivatives on the cAMP-regulated chloride current in cardiac myocytes. Pflügers Arch. 1993;422:436–442. doi: 10.1007/BF00375068. [DOI] [PubMed] [Google Scholar]
- HARVEY R.D., HUME J.R. Autonomic regulation of a chloride current in heart. Science. 1989;244:983–985. doi: 10.1126/science.2543073. [DOI] [PubMed] [Google Scholar]
- HIRAOKA M., KAWANO S., HIRANO Y., FURUKAWA T. Role of cardiac chloride currents in changes in action potential characteristics and arrhythmias. Cardiovasc. Res. 1998;40:23–33. doi: 10.1016/s0008-6363(98)00173-4. [DOI] [PubMed] [Google Scholar]
- JANSE M.J., WIT A.L. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol. Rev. 1989;69:1049–1169. doi: 10.1152/physrev.1989.69.4.1049. [DOI] [PubMed] [Google Scholar]
- JENNINGS R.B., REIMER K.A., STEENBERGEN C. Myocardial ischemia revisited. The osmolar load, membrane damage, and reperfusion. J. Mol. Cell. Cardiol. 1986;18:769–780. doi: 10.1016/s0022-2828(86)80952-x. [DOI] [PubMed] [Google Scholar]
- JURKIEWICZ N.K., SANGUINETTI M.C. Rate-dependent prolongation of cardiac action potentials by a methanesulfonanilide class III antiarrhythmic agent. Specific block of rapidly activating delayed rectifier K+ current by dofetilide. Circ. Res. 1993;72:75–83. doi: 10.1161/01.res.72.1.75. [DOI] [PubMed] [Google Scholar]
- KAWATA H., KAWAGOE K., TATEYAMA I. Effects of osmolarity change on the excitation-contraction coupling of bullfrog ventricle. Jpn. J. Physiol. 1974;24:587–603. doi: 10.2170/jjphysiol.24.587. [DOI] [PubMed] [Google Scholar]
- LANDRY D.W., AKABAS M.H., REDHEAD C., EDELMAN A., CRAGOE E.J., JR, AL-AWQATI Q. Purification and reconstitution of chloride channels from kidney and trachea. Science. 1989;244:1469–1472. doi: 10.1126/science.2472007. [DOI] [PubMed] [Google Scholar]
- LI G.R., FENG J., WANG Z., NATTEL S. Transmembrane chloride currents in human atrial myocytes. Am. J. Physiol. 1996;270:C500–C507. doi: 10.1152/ajpcell.1996.270.2.C500. [DOI] [PubMed] [Google Scholar]
- MATSUDA N., HAGIWARA N., SHODA M., KASANUKI H., HOSODA S. Enhancement of the L-type Ca2+ current by mechanical stimulation in single rabbit cardiac myocytes. Circ. Res. 1996;78:650–659. doi: 10.1161/01.res.78.4.650. [DOI] [PubMed] [Google Scholar]
- MULVANEY A.W., SPENCER C.I., CULLIFORD S., BORG J.J., DAVIES S.G., KOZLOWSKI R.Z. Cardiac chloride channels: physiology, pharmacology and approaches for cidentifying novel modulators of activity. Drug Discov. Today. 2000;5:492–505. doi: 10.1016/s1359-6446(00)01561-0. [DOI] [PubMed] [Google Scholar]
- NAGASAKI M., YE L., DUAN D., HOROWITZ B., HUME J.R. Intracellular cyclic AMP inhibits native and recombinant volume-regulated chloride channels from mammalian heart. J. Physiol. (Lond.) 2000;523:705–717. doi: 10.1111/j.1469-7793.2000.00705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NILIUS B., OIKE M., ZAHRADNIK I., DROOGMANS G. Activation of a Cl- current by hypotonic volume increase in human endothelial cells. J. Gen. Physiol. 1994;103:787–805. doi: 10.1085/jgp.103.5.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NILIUS B., PRENEN J., SZUCS G., WEI L., TANZI F., VOETS T., DROOGMANS G. Calcium-activated chloride channels in bovine pulmonary artery endothelial cells. J. Physiol. 1997a;498:381–396. doi: 10.1113/jphysiol.1997.sp021865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NILIUS B., PRENEN J., VOETS T., VAN DEN BREMT K., EGGERMONT J., DROOGMANS G. Kinetic and pharmacological properties of the calcium-activated chloride current in macrovascular endothelial cells. Cell Calcium. 1997b;22:53–63. doi: 10.1016/s0143-4160(97)90089-0. [DOI] [PubMed] [Google Scholar]
- OKADA Y. Volume expansion-sensing outward-rectifier Cl− channel: fresh start to the molecular identity and volume sensor. Am. J. Physiol. 1997;273:C755–C789. doi: 10.1152/ajpcell.1997.273.3.C755. [DOI] [PubMed] [Google Scholar]
- PRIEBE L., BEUCKELMANN D.J. Cell swelling causes the action potential duration to shorten in guinea-pig ventricular myocytes by activating IKATP. Pflügers Arch. 1998;436:894–898. doi: 10.1007/pl00008087. [DOI] [PubMed] [Google Scholar]
- REES S.A., VANDENBERG J.I., WRIGHT A.R., YOSHIDA A., POWELL T. Cell swelling has differential effects on the rapid and slow components of delayed rectifier potassium current in guinea pig cardiac myocytes. J. Gen. Physiol. 1995;106:1151–1170. doi: 10.1085/jgp.106.6.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROOS K.P. Length, width, and volume changes in osmotically stressed myocytes. Am. J. Physiol. 1986;251:H1373–H1378. doi: 10.1152/ajpheart.1986.251.6.H1373. [DOI] [PubMed] [Google Scholar]
- SAKAI R., HAGIWARA N., KASANUKI H., HOSODA S. Chloride conductance in human atrial cells. J. Mol. Cell. Cardiol. 1995;27:2403–2408. doi: 10.1016/s0022-2828(95)92199-0. [DOI] [PubMed] [Google Scholar]
- SAKMANN B., TRUBE G. Conductance properties of single inwardly rectifying potassium channels in ventricular cells from guinea-pig heart. J. Physiol. 1984;347:641–657. doi: 10.1113/jphysiol.1984.sp015088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SANGUINETTI M.C., JURKIEWICZ N.K. Role of external Ca2+ and K+ in gating of cardiac delayed rectifier K+ currents. Pflügers Arch. 1992;420:180–186. doi: 10.1007/BF00374988. [DOI] [PubMed] [Google Scholar]
- SOROTA S. Swelling-induced chloride-sensitive current in canine atrial cells revealed by whole-cell patch-clamp method. Circ. Res. 1992;70:679–687. doi: 10.1161/01.res.70.4.679. [DOI] [PubMed] [Google Scholar]
- SOROTA S. Pharmacologic properties of the swelling-induced chloride current of dog atrial myocytes. J. Cardiovasc. Electrophysiol. 1994;5:1006–1016. doi: 10.1111/j.1540-8167.1994.tb01143.x. [DOI] [PubMed] [Google Scholar]
- STEINMEYER K., LORENZ C., PUSCH M., KOCH M.C., JENTSCH T.J. Multimeric structure of ClC-1 chloride channel revealed by mutations in dominant myotonia congenita (Thomsen) EMBO J. 1994;13:737–743. doi: 10.1002/j.1460-2075.1994.tb06315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEINMEYER K., SCHWAPPACH B., BENS M., VANDEWALLE A., JENTSCH T.J. Cloning and functional expression of rat CLC-5, a chloride channel related to kidney disease. J. Biol. Chem. 1995;270:31172–31177. doi: 10.1074/jbc.270.52.31172. [DOI] [PubMed] [Google Scholar]
- THIEMANN A., GRÜNDER S., PUSCH M., JENTSCH T.J. A chloride channel widely expressed in epithelial and non-epithelial cells. Nature. 1992;356:57–60. doi: 10.1038/356057a0. [DOI] [PubMed] [Google Scholar]
- TRANUM-JENSEN J., JANSE M.J., FIOLET W.T., KRIEGER W.J., D'ALNONCOURT C.N., DURRER D. Tissue osmolality, cell swelling, and reperfusion in acute regional myocardial ischemia in the isolated porcine heart. Circ. Res. 1981;49:364–381. doi: 10.1161/01.res.49.2.364. [DOI] [PubMed] [Google Scholar]
- UCHIDA S., SASAKI S., FURUKAWA T., HIRAOKA M., IMAI T., HIRATA Y., MARUMO F. Molecular cloning of a chloride channel that is regulated by dehydration and expressed predominantly in kidney medulla. J. Biol. Chem. 1994;268:3821–3824. [PubMed] [Google Scholar]
- VANDENBERG J.I., BETT G.C., POWELL T. Contribution of a swelling-activated chloride current to changes in the cardiac action potential. Am. J. Physiol. 1997;273:C541–C547. doi: 10.1152/ajpcell.1997.273.2.C541. [DOI] [PubMed] [Google Scholar]
- VANDENBERG J.I., REES S.A., WRIGHT A.R., POWELL T. Cell swelling and ion transport pathways in cardiac myocytes. Cardiovasc. Res. 1996;32:85–97. [PubMed] [Google Scholar]
- VANDENBERG J.I., YOSHIDA A., KIRK K., POWELL T. Swelling-activated and isoprenaline-activated chloride currents in guinea pig cardiac myocytes have distinct electrophysiology and pharmacology. J. Gen. Physiol. 1994;104:997–1017. doi: 10.1085/jgp.104.6.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALKER J.L.Intracellular inorganic ions in cardiac tissue The Heart and Cardiovascular System 1986New York: Raven Press; 561–572.ed. Fozzard H.A., pp [Google Scholar]
- WHALLEY D.W., HOOL L.C., TEN EICK R.E., RASMUSSEN H.H. Effect of osmotic swelling and shrinkage on Na+/K+ pump activity in mammalian cardiac myocytes. Am. J. Physiol. 1993;265:C1201–C1210. doi: 10.1152/ajpcell.1993.265.5.C1201. [DOI] [PubMed] [Google Scholar]
- YAMAWAKE N., HIRANO Y., SAWANOBORI T., HIRAOKA M. Arrhythmogenic effects of isoproterenol-activated Cl− current in guinea-pig ventricular myocytes. J. Mol. Cell. Cardiol. 1992;24:1047–1058. doi: 10.1016/0022-2828(92)91871-2. [DOI] [PubMed] [Google Scholar]
- ZHANG J., RASMUSSON R.L., HALL S.K., LIEBERMAN M. A chloride current associated with swelling of cultured chick heart cells. J. Physiol. (Lond.) 1993;472:801–820. doi: 10.1113/jphysiol.1993.sp019974. [DOI] [PMC free article] [PubMed] [Google Scholar]