

Abstract
In the present experiments, we investigated the effects of methylecgonidine (MEG) on nitric oxide (NO) production in cultured neonatal rat cardiomyocytes. Incubation of cultured cardiomyocytes with carbachol or MEG for 48 h significantly enhanced NO production. No release was increased from 1.48±0.13 μM (mg protein)−1 for control to 5.73±0.19 μM (mg protein)−1 for 1 μM carbachol treated cells (P<0.001). In addition, incubation with 1 μM MEG enhanced NO production to 5.55±0.28 μM (mg protein)−1. The effects of MEG on NO production were concentration-dependent. The muscarinic antagonist atropine prevented the enhancement of NO production induced by carbachol or MEG. Compared to MEG-induced NO production, cocaine was much less potent.
The enhancement of NO production by carbachol or MEG was even greater in cultured cardiomyocytes transfected with the M2 cDNA. After 48-h incubation with 1 μM carbachol or 1 μM MEG, NO production was increased by 6.5 and 6.7 fold, respectively, in cardiomyocytes overexpressing M2 receptors. Coincubation with atropine or NG-nitro-L-arginine methyl ester abolished the enhancement of NO production. In contrast, NO production enhanced by carbachol or MEG in M1- or M3-transfected cardiomyocytes was similar to the level in non-transfected cells.
Western blot analysis showed that the protein levels of M1, M2, and M3 were significantly increased in cardiomyocytes transfected with the receptor cDNAs, but MEG had no effect on the expressions. It is interesting that both carbachol and MEG caused a significant increase in constitutive endothelial NO synthase (eNOS) only in M2-transfected cardiomyocytes, not in non-transfected, M1- or M3-transfected cells. Again, atropine blocked the MEG-produced induction of eNOS.
Our data demonstrate that MEG significantly enhanced NO production in cultured cardiomyocytes and that the enhancement of NO production may result from MEG stimulation of muscarinic M2 receptors.
Keywords: Methylecgonidine, carbachol, muscarinic receptor, nitric oxide, cardiomyocyte
Introduction
Nitric oxide (NO) is formed by a family of three known NO synthases (NOSs) during the catalytic conversion of L-arginine to L-citrulline in the presence of oxygen and NADPH. Evidence has shown that cardiac myocytes have the capacity to express both the constitutive endothelial (eNOS or NOS3) and the inducible (iNOS or NOS2) NO synthase (Balligand et al., 1995a, 1995b). It is known that eNOS in the heart is responsible for NO production during a receptor-dependent signalling process. However, iNOS is expressed in cardiomyocytes in response to specific cytokines. NO is an important signalling molecule for both muscarinic and β-adrenergic stimulation, and has effects on inotropic and chronotropic function in cultured neonatal or adult ventricular myocytes (Balligand et al., 1993).
Muscarinic cholinergic innervation in mammalian hearts plays an important role in the regulation of heart rate and myocardial contractility. Five different molecular forms (M1 – 5) of muscarinic receptors have been described. The M2 subtype is the predominant one expressed in mammalian hearts (Peralta et al., 1987). Experimental data show that NO production during muscarinic receptor stimulation is due to activation of eNOS in adult rat ventricular myocytes (Balligand et al., 1995a). A link between muscarinic cholinergic stimulation and NOS activation in cardiomyocytes has been evidenced by an elevation of NO production and by an increase in cyclic GMP content which is due to the NO-induced activation of guanylyl cyclase (Balligand et al., 1993). In addition, a recent study in cardiac myocytes shows that the enhancement of NO production by the muscarinic agonist carbachol results from stimulation of muscarinic M2 receptors (Feron et al., 1999). Therefore, NO and cyclic GMP are the possible mediators responsible for the action of muscarinic cholinergic agonists on heart rate and on contractile function, presumably via stimulation of muscarinic M2 receptors in the heart.
Drug abuse is a serious social and health problem in many countries. Cocaine users have a high incidence of cardiovascular complications, including acute myocardial infarction (Isner et al., 1986; Cregler & Mark, 1985), sudden death (Benchimol et al., 1978), myocarditis (Virmani et al., 1988), and hypertension (Resnick et al., 1977). Cocaine and the principal pyrolysis product of crack cocaine base, methylecgonidine (MEG), at relatively low concentrations produced a negative inotropic effect in ferret myocardium and isolated ventricular myocytes (Miao et al., 1996; Huang et al., 1997; Woolf et al., 1997). The muscarinic antagonist atropine and the selective M2 receptor blocker methoctramine abolished the negative inotropic effect of cocaine and MEG, but M1 (pirenzepine) and M3 (4-diphenylacetoxy-N-methylpiperidine methiodide) blocking agents did not (Huang et al., 1997). These results suggest that the negative inotropic effect of cocaine and MEG result from, at least in part, stimulation of muscarinic receptors. We recently demonstrated that cocaine and MEG increased the production of cyclic GMP and decreased intracellular cyclic AMP content in cultured human embryonic lung cells by stimulation of muscarinic M2 receptors (Yang et al., 2001). To examine whether the increase in cyclic GMP production caused by MEG resulted from an increase of NO production, we investigated the effects of MEG on eNOS expression and NO production in cultured neonatal rat cardiomyocytes with or without overexpression of muscarinic receptor subtypes. In addition, we evaluated whether the effects of MEG on NO production resulted from an alteration of M2 receptor expression. The effects of MEG on other muscarinic receptor subtypes, M1 and M3, were also determined in cultured neonatal rat cardiomyocytes.
Methods
Culture of neonatal rat cardiomyocytes
Primary cultures of neonatal cardiac myocytes were prepared from the hearts of 1-day-old neonatal rats with a commercial isolation kit (Worthington Biochem. Corp., NJ, U.S.A.) This kit utilizes purified enzyme preparations to produce healthy beating cells which were seeded into 100 mm Petri culture dishes. Cells were incubated at 37°C in air with 5% CO2 added and 98% relative humidity (model 3123, Forma Scientific, Marietta, OH, U.S.A.) The culture medium was changed every other day. Cardiac myocytes used for NO measurement or protein expression analysis were cultured for 5 to 10 days. MEG, cocaine, atropine, carbachol, and L-NAME were obtained from Sigma (Sigma, St. Louis, MO, U.S.A.) Different concentrations of compound were added into culture medium and cells were incubated for 48 h before measurement of NO production and protein levels. Atropine or NG-nitro-L-arginine methyl ester (L-NAME) was added to culture dishes 30 min ahead of the treatment of cocaine, MEG or carbachol to block muscarinic receptors.
Assay for nitrite (NO−2) production
Nitrite (NO−2), the stable metabolic end product of NO synthesis, was measured in cultured cardiomyocytes as described previously (Oddis et al., 1995). NO production collected from two culture dishes with the same treatment was measured as a single run (n=1). Equal volumes of cell culture medium and Griess Reagent (2.9 mM sulphanilic acid and 0.2 mM N-[1-naphthyl]ethylenediamine-HCl in 5% phosphoric acid) were mixed and incubated at room temperature for 1 h. The absorbance at 570 nm was determined with a microplate reader (Molecular Devices Co., Menlo Park, CA, U.S.A.) The concentration of NO−2 in medium samples was developed by using a standard curve of NaNO2 (sodium nitrite).
Transfection of muscarinic receptor cDNAs, M1, M2, and M3
Human muscarinic receptor, M1, M2, and M3, cDNAs (Hm1pCD, Hm2pCD, Hm3pCD) were generous gifts provided by Dr Bonner TI (NIH, MD, U.S.A.) In these plasmids, human muscarinic receptor, M1, M2, and M3, gene complete sequences were cloned in the Okayama-Berg pcD mammalian expression vector with an SV40 early promoter (Okayama et al., 1983). The cultures were 60 – 90% confluent in 100 mm dishes at the time of transfection. Neonatal rat cardiomyocytes were transfected with 8 μg plasmid DNA per dish by using a calcium phosphate precipitation method (Chen et al., 1987). Cells were treated with different reagents 48 h after transfection.
Western blot analysis
For detection of muscarinic receptor and NOS proteins, cells were lysed in ice-cold lysis buffer (1% Triton X-100, 150 mM NaCl, 10 mM Tris, pH 7.4, 1 mM EDTA, 1 mM EGTA, pH 8.0, 0.2 mM sodium orthovanadate, 0.2 mM PMSF, 0.5% NP-40). After sonication for 45 s on ice, the resulting lysates were centrifuged at 10,000×g for 10 min at 4°C to remove cell debris. Protein content was determined using a standard protein assay kit (Bio-Rad Labs., Hercules, CA, U.S.A.) Two hundred μg protein samples were fractionated by 10% SDS-polyacrylamide gels electrophoresis, blotted onto a nitrocellulose membrane (Millipore Cor., Bedford, MA, U.S.A.) Membranes were blocked for 1 h at room temperature with 5% nonfat milk in Tris-buffered saline (25 mM Tris and 150 mM NaCl) containing 0.05% Tween-20 and incubated with antibodies specific to M1, M2, M3 (goat, 0.5 μg (ml)−1, Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) or eNOS (mouse, 1 μg (ml)−1, Transduction Lab., Lexington, KY, U.S.A.) overnight at 4°C. After washing, the blots were probed with 1 : 8000 dilution of horseradish peroxidase (HRP)-conjugated anti-goat IgG (for M1, M2, and M3) or HRP-conjugated anti-mouse (for eNOS) IgG (Sigma, St Louis, MO, U.S.A.) and visualized using the ECL system (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) Blots were exposed to autoradiographic X-ray film and bands were quantified with ImageQuant software (Molecular Dynamic, Sunnyvale, CA, U.S.A.) To reduce variability of Western blot analyses caused by sample loading and processing, cyclophilin A expressed in cardiomyocytes was used as an internal standard (Ito et al., 2000).
Data analysis
To correct variation of cell masses among different cultured samples, the data for NO measurement were normalized to total protein content. Blots of eNOS, M1, M2, and M3 receptor proteins with or without cDNA transfection were normalized to their corresponding protein levels of cyclophilin A during blotting measurement. The number of measurements of NO and proteins is the sum of single replicates. Each sample collected from two culture dishes with the same treatment was measured as a single run (n=1). Data were presented as mean±s.e.mean unless otherwise noted. Statistical differences between two groups were determined by the unpaired Student's t-test. Data derived from more than two experimental groups were tested by one-way analysis of variance (ANOVA). Statistical significance was considered at the level of P<0.05.
Results
Increases in NO production by carbachol and MEG
Carbachol is a potent and nonselective cholinergic agonist which enhances NO production in rat cardiomyocytes (Balligand et al., 1995a; Yamamoto et al., 1998). To determine whether neonatal cardiomyocytes express muscarinic receptors and what the effect of carbachol on NO production was, cultured neonatal rat cardiomyocytes were incubated with 1 μM carbachol with or without 1 μM atropine for 48 h. Figure 1A shows that NO production was increased from 1.48±0.13 μM (mg protein)−1 for the control to 5.73±0.19 μM (mg protein)−1 for 1 μM carbachol treated cells (n=8, P<0.001). The delta increase in NO production was 4.25 μM (mg protein)−1. To test whether the effect of carbachol on NO production was via stimulation of muscarinic receptors, the nonselective muscarinic antagonist atropine (1 μM) was applied to block muscarinic receptors. While atropine alone had no significant effect on NO production, coincubation with carbachol and atropine abolished the increase in NO production in carbachol-treated neonatal rat cardiomyocytes (Figure 1A). These results demonstrate that cultured neonatal rat cardiomyocytes express ACh muscarinic receptors and carbachol increases their NO production.
Figure 1.
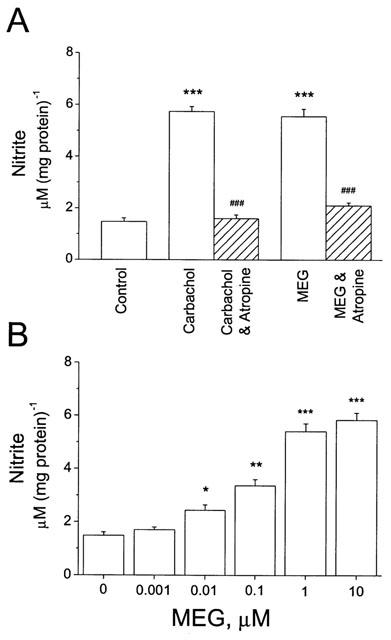
Increase in NO production by carbachol and MEG in cultured neonatal rat cardiomyocytes. (A) Myocytes were incubated with 1 μM carbachol or 1 μM MEG for 48 h. Nitrite in cell medium was measured in the absence (n=8) and presence of carbachol (n=8) or MEG (n=8). The enhancement of NO production by carbachol or MEG was abolished or significantly attenuated after coincubation with 1 μM atropine (Carbachol & Atropine and MEG & Atropine). ***P<0.001, vs control; ###P<0.001, vs carbachol or MEG. (B) MEG produced concentration-dependent increase in NO production in cultured neonatal cardiomyocytes. Each concentration had at least five individual runs (n⩾5). *P<0.05; **P<0.01; ***P<0.001; vs control.
The major pyrolysis product formed from heating cocaine base is MEG (Meisels & Loke, 1993), which has structure similarity to the muscarinic agonists arecoline and anatoxin A (Ettinger & Albin, 1989). A muscarinic stimulatory effect of MEG on airway smooth muscles (El-Fawal & Wood, 1995) and on cardiomyocytes (Huang et al., 1997) has been reported. To compare the effect of MEG on NO production with that of carbachol, cultured neonatal rat cardiomyocytes were incubated with 1 μM MEG for 48 h. Figure 1A shows that 1 μM MEG caused an increase in NO production similar to that of carbachol. Pre-incubation of cardiomyocytes with 1 μM atropine prevented the MEG-induced increase in NO production (Figure 1A). The levels of NO production were 1.48±0.13 μM (mg protein)−1 for the control, 5.55±0.28 μM (mg protein)−1 for 1 μM MEG (n=8, P<0.001, Figure 1A), and 2.16±0.12 μM (mg protein)−1 for 1 μM MEG plus 1 μM atropine (n=6). The delta increase in NO production by MEG was 4.07 μM (mg protein)−1. These results indicate that MEG enhanced NO production by stimulation of muscarinic receptors. In addition, MEG-induced NO production was concentration-dependent (Figure 1B). It is surprising that MEG at 0.01 μM was able to cause a significant increase in NO production in cultured neonatal rat cardiomyocytes (P<0.05, n=6, Figure 1B). Compared to MEG, cocaine induced NO production only at much higher concentrations. Incubation of cultured neonatal cardiomyocytes with 1 μM cocaine (n=6) did not significantly alter the basal NO production. However, incubation of cardiomyocytes with higher concentrations of cocaine markedly enhanced NO production, 3.10±0.54 μM (mg protein)−1 for 10 μM cocaine (n=8, P<0.05) and 3.68±0.28 μM (mg protein)−1 for 50 μM cocaine (n=6, P<0.001). These results demonstrate that MEG is more potent than cocaine in regard to enhancing NO production in cultured neonatal cardiomyocytes.
NO production in cardiomyocytes transfected with muscarinic receptors
Evidence has shown that the enhancement of NO production in rat cardiomyocytes by carbachol is via activation of muscarinic M2 receptors (Yamamoto et al., 1998; Feron et al., 1999). Extracellular application of MEG resulted in a negative inotropic effect on ferret cardiomyocytes probably by a stimulation of muscarinic M2 receptors, because methoctramine (a M2 blocker) abolished the MEG's effect (Huang et al., 1997). To test whether carbachol- and MEG-enhanced NO production was by activation of M2 receptors, cultured neonatal rat cardiomyocytes were transfected with the cDNAs of M1, M2, and M3 receptor subtypes. NO production was assessed and compared in non-transfected and transfected cardiomyocytes. Carbachol or MEG caused a significantly greater increase in NO production in M2 cDNA-transfected cardiomyocytes than in non-transfected, or M1- and M2-transfected heart cells. The increase in NO production was 6.5 fold for 1 μM carbachol stimulation, from 1.71±0.16 μM (mg protein)−1 to 11.19±0.61 μM (mg protein)−1 (n=8, P<0.01, Figure 2A), respectively. MEG at 1 μM caused an increase in NO production by 6.7 fold, from 1.71±0.16 μM (mg protein)−1 for the control to 11.52±0.78 μM (mg protein)−1 for MEG (n=6, P<0.01, Figure 2A). The delta increase in NO production was 9.48 μM (mg protein)−1 for carbachol and 9.81 μM (mg protein)−1 for MEG. The effects of carbachol and MEG on NO production were more than 2 fold greater in M2-transfected cardiomyocytes than in non-transfected cells. Again, the carbachol- or MEG-induced NO production was abolished or significantly attenuated in the presence of 1 μM atropine (Figure 2A). In addition, the NO synthase inhibitor NG-nitro-L-arginine methyl ester (L-NAME) at 500 μM also abolished the increase in NO production caused by carbachol or MEG in cultured cardiomyocytes overexpressing muscarinic M2 receptors (n=6, P<0.001, Figure 2A). These results indicate that both carbachol and MEG produced greater NO production in M2-transfected neonatal rat cardiomyocytes, probably via stimulation of muscarinic M2 receptors.
Figure 2.
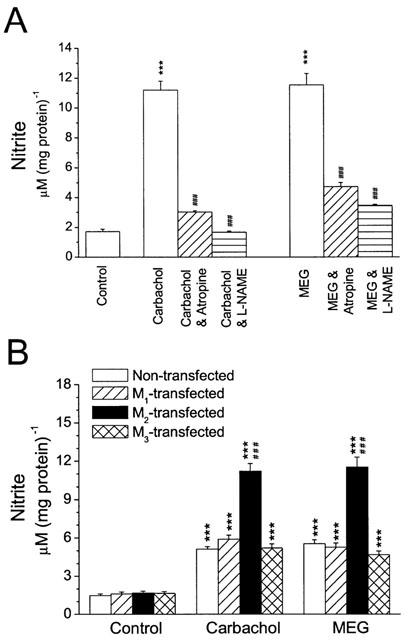
NO production in the presence of carbachol or MEG in cardiomyocytes transfected with muscarinic receptors. (A) Nitrite production caused by 1 μM carbachol or 1 μM MEG was significantly greater in M2-transfected cardiomyocytes than in non-transfected cells. The increase in NO production was 6.5 fold for carbachol (n=8) and 6.7 fold for MEG (n=6), in cardiomyocytes overexpressing M2 subtypes. The enhancement of NO production by carbachol or MEG at 1 μM was significantly attenuated in M2-transfected cardiomyocytes after coincubation with 1 μM atropine (n=8) or 500 μM L-NAME (n=6) for 48 h. ***P<0.001, vs control; ###P<0.001, vs carbachol or MEG alone. (B) Under control conditions (Control), NO production was similar in cardiomyocytes with or without transfection of muscarinic receptors. However, incubation with carbachol (Carbachol) or MEG (MEG) at 1 μM enhanced NO production in cardiomyocytes with or without M1, M2, or M3 transfection (n=8 in each group). The enhancement of NO production caused by carbachol or MEG was not significantly different among non-transfected and M1- or M3-transfected cells, but significantly greater in M2-transfected cardiomyocytes. ***P<0.001, vs control; ###P<0.001, vs non-transfected, M1- and M3-transfected groups.
It is interesting that the basal levels of NO production were not significantly altered in cardiomyocytes after transfection with M1, M2, or M3 receptor subtype (Figure 2B, Control). However, NO production caused by carbachol or MEG was significantly greater in M2-transfected cardiomyocytes than in cardiomyocytes transfected with muscarinic M1 or M3 receptors. Both carbachol and MEG caused an increase in NO production in M1- or M3-transfected cardiomyocytes, but the increase was similar to that in non-transfected cells (Figure 2B). The delta increase in NO production caused by carbachol or MEG in M2-transfected myocytes was more than 2 fold greater than in non-transfected or M1- and M3-transfected cardiomyocytes (Figure 2B). The data further support the hypothesis that the carbachol- or MEG-induced increase in NO production may result from stimulation of muscarinic M2 receptors in cultured neonatal rat cardiomyocytes.
Expression of muscarinic receptors
In mammalian cardiomyocytes, M2 seems to be the predominant subtype of muscarinic receptors (Peralta et al., 1987). Our results from Western blot analysis show that the protein level of muscarinic M2 receptors was 1.5 fold greater than that of M1, and 2.5 fold greater than that of M3, respectively (data not shown). To examine whether the increase in NO production resulted from an induction of muscarinic receptor expression, the protein content of M1, M2, and M3 was assessed in non-transfected cardiomyocytes incubated with carbachol or MEG. In fact, the expression of M1, M2, or M3 was not significantly altered in non-transfected cardiomyocytes incubated with carbachol or MEG at 1 μM for 48 h (Figure 3). The results indicate that the increase in NO production caused by carbachol or MEG is due to stimulation of muscarinic receptors, not to an increase in muscarinic receptor expression.
Figure 3.
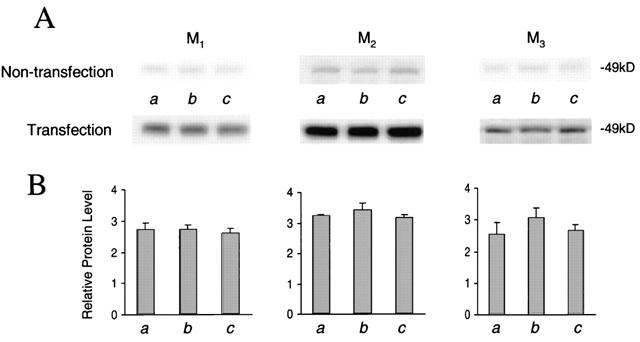
Expression of M1, M2, and M3 receptor subtypes in cultured cardiomyocytes. (A) Representative Western blots of M1, M2, and M3 in cardiomyocytes with or without transfection of muscarinic M1 – 3 receptors are shown. Different groups (n=3 in each group) of cultured cardiomyocytes were treated with control (a), 1 μM carbachol (b), and 1 μM MEG (c). (B) Blots of M1, M2, and M3 receptor proteins as shown in (A) with or without cDNA transfection were quantified with their internal standard cyclophilin A. Relative protein levels were presented as dividing transfection by non-transfection for each own receptor subtype. The expression efficiency was similar for these three muscarinic subtypes.
The greater enhancement of NO production by carbachol or MEG in M2-transfected cells than in M1- or M3-transfected cardiomyocytes was probably due to a difference of receptor expression levels. To test this possibility, Western blot analysis of the protein levels of M1, M2, and M3 was performed in cultured neonatal rat cardiomyocytes with or without transfection of those cDNAs. Figure 3 shows that expression of muscarinic receptors was normalized by transfection over non-transfection for all three receptor subtypes. After cardiomyocytes were transfected with M1, M2, and M3, protein levels increased by 2.33, 3.42, and 2.93 fold for the receptor subtypes, respectively. The expression efficiency was similar for these three muscarinic subtypes. However, compared to M1- or M3-transfected cells, treatment with carbachol or MEG at 1 μM for 48 h further increased NO production only in M2-transfected cells (Figure 2B). In addition, it is obvious that both carbachol and MEG did not significantly alter M1, M2, and M3 protein expression in non-transfected cardiomyocytes and in cells transfected with the cDNAs, M1, M2 or M3 (Figure 3). These results suggest that the greater enhancement of NO production in M2-transfected cardiomyocytes results from carbachol- or MEG-induced stimulation of muscarinic M2 receptors.
Induction of eNOS by MEG
Several studies have shown that in cardiac myocytes the enhancement of NO production by carbachol results from activation and induction of eNOS via stimulation of muscarinic receptors, presumably M2 (Balligand et al., 1995a; Han et al., 1995; Feron et al., 1999). To evaluate the effects of carbachol and MEG on the expression of eNOS, Western blot analysis of eNOS was carried out in carbachol- or MEG-incubated neonatal rat cardiomyocytes. Incubation with carbachol or MEG at 1 μM for 48 h did not significantly alter the protein level of eNOS in cultured neonatal rat cardiomyocytes (Figure 4). However, incubation with carbachol or MEG significantly increased eNOS expression in cardiomyocytes with overexpression of muscarinic M2 receptors (Figure 5). The increase in eNOS expression by carbachol or MEG in M2-transfected cardiomyocytes was abolished when the cells were coincubated with the muscarinic receptor antagonist atropine at 1 μM (Figure 5). In addition, coincubation with L-NAME also prevented the increase in eNOS expression in M2-transfected cardiomyocytes (data not shown). Atropine or L-NAME alone had no effect on eNOS expression in non-transfected or receptor-transfected cardiomyocytes. The results demonstrate that the greater enhancement of NO production caused by carbachol or MEG in M2-transfected cells than in M1- or M3-transfected cardiomyocytes probably results from both activation and induction of eNOS.
Figure 4.
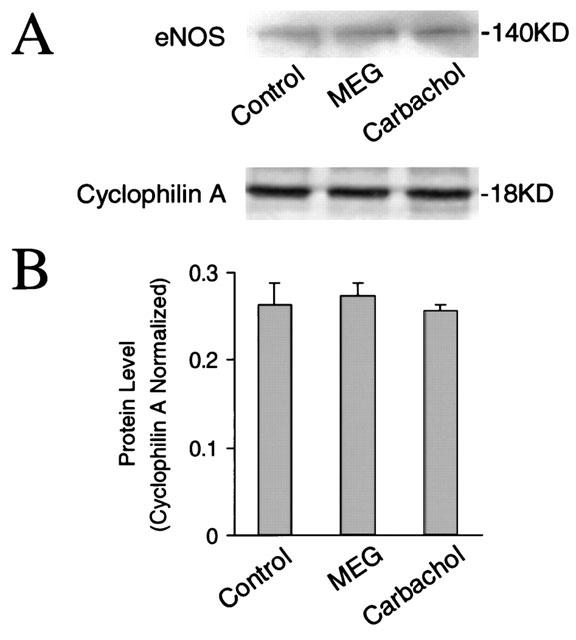
Expression of eNOS in cultured cardiomyocytes without transfection of muscarinic receptors. (A) Representative Western blots of eNOS in cardiomyocytes without transfection of muscarinic receptors are shown. Cultured cardiomyocytes were treated for 48 h with control, 1 μM MEG, and 1 μM carbachol, respectively. (B) Blots of eNOS as shown in (A) were quantified with their corresponding internal standard cyclophilin A. Cardiomyocytes did not show an alteration of eNOS expression by carbachol or EMG (n=3 in each group).
Figure 5.
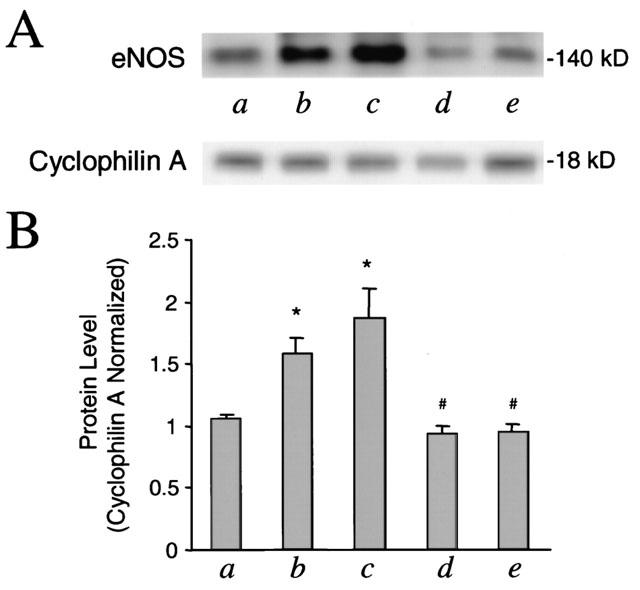
Enhancement of eNOS expression by carbachol or MEG in cultured cardiomyocytes transfected with muscarinic M2 receptors. (A) Representative Western blots of eNOS in cardiomyocytes transfected with muscarinic receptors are shown. Cultured cardiomyocytes were incubated for 48 h with control (a), 1 μM carbachol (b), 1 μM MEG (c), 1 μM carbachol plus 1 μM atropine (d), 1 μM MEG plus 1 μM atropine (e), respectively. (B) Blots of eNOS as shown in (A) were quantified with their internal standard cyclophilin A. Note the induction of eNOS in the presence of carbachol or MEG and the abolishment of the induction by addition of atropine (n=3 in each group). *P<0.005, vs control; #P<0.05, vs carbachol or MEG alone.
Discussion
The results presented in this study show that MEG significantly enhanced NO production in cultured neonatal rat cardiomyocytes. The MEG-enhanced NO production was abolished by addition of the muscarinic receptor antagonist atropine. The data suggest that the effects of MEG on NO production probably resulted from stimulation of muscarinic receptors in cultured neonatal rat cardiac myocytes. Stimulation of muscarinic receptors by MEG has been found in ferret myocardium and isolated ventricular myocytes (Miao et al., 1996; Huang et al., 1997; Woolf et al., 1997), as well as in human ventricular trabeculae (Woolf et al., 1997) and in cultured human embryonic lung cells (Yang et al., 2001). NO is an important signalling molecule for the negative inotropic and chronotropic effects of muscarinic receptor agonists (Calver et al., 1993; Buxton et al., 1993). Several other studies in vivo and in vitro have shown that stimulation of muscarinic cholinergic receptors activates eNOS to increase cardiac NO release (Balligand et al., 1995a; Sterin-Borda et al., 1995; George et al., 1973). Five muscarinic receptor subtypes have been identified in mammalian hearts and the M2 receptor subtype is the predominant one (Peralta et al., 1987; Goyal, 1989; Pappano & Mubagwa, 1992). In this study, we examined the three muscarinic receptor subtypes, M1, M2, and M3, and detected their expression in cultured neonatal rat cardiomyocytes. The M2 subtype was expressed the most among these three subtypes. It has been shown that carbachol-induced NO production results from activation of muscarinic M2 receptors in rat cardiac myocytes (Yamamoto et al., 1998; Feron et al., 1999). Therefore, the MEG-enhanced NO production found in the present study is also possible due to stimulation of muscarinic M2 receptors, even though M1 and M3 were also found in cultured neonatal rat cardiomyocytes.
Our experimental data in muscarinic receptor-transfected cardiomyocytes further support the hypothesis that the MEG-enhanced NO production may result from stimulation of muscarinic M2 receptors. Compared to non-transfected cells, the enhancement of NO production by MEG or carbachol was significantly greater only in M2-transfected cardiomyocytes, but not in M1- or M3-transfected cells. The enhancement of NO production by MEG in M2-transfected cardiomyocytes was not only blocked by atropine, but also by L-NAME which is a well-known NO synthase inhibitor. However, by using the receptor-binding method, Buxton et al. (1993) found that L-NAME was a competitive antagonist of muscarinic M2 receptors in peripheral and central nervous tissues. Therefore, the MEG-enhanced NO production is probably due to stimulation of muscarinic M2 receptors in non-transfected and also in muscarinic cDNA-transfected cardiomyocytes, because the further enhancement of NO production by MEG was only found in M2-transfected cells and blocked by the muscarinic antagonist. Recent studies have shown that cardiomyocytes express eNOS and that NO-dependent parasympathetic signalling results from activation of eNOS in cardiac myocytes (Balligand et al., 1995a; Feron et al., 1999). Based on our present results and others, we speculate that the MEG-enhanced NO production in non-transfected or muscarinic receptor-transfected cardiomyocytes may result from the activation of eNOS by stimulation of muscarinic M2 receptors.
It is interesting that a significant induction of eNOS expression by both carbachol and MEG was found only in M2-transfected neonatal rat cardiomyocytes, not in non-transfected and M1- or M3-transfected cells. The induction of eNOS expression was prevented by addition of atropine. A possible explanation for the MEG-induced eNOS expression found only in M2-transfected cardiomyocytes is that the induction requires a stronger input from stimulation of muscarinic M2 receptors, which is not enough in non-transfected, or in M1- and M3-transfected cells. However, stimulation of overexpressed M2 receptors by MEG or carbachol in M2-transfected cells caused an induction of eNOS. To elucidate the precise mechanism of the induction of eNOS expression by carbachol and MEG in M2-transfected cardiomyocytes requires further experiments.
Crack cocaine users appear to be at high risk of suffering a heart attack (Isaacs et al., 1987), cardiac irritability, and hypertension. The mechanism for drug-related cardiopulmonary complications is not fully delineated (Cregler & Mark, 1986). Cholinergic modulation of the cardiovascular system by cocaine has been demonstrated in conscious dogs (Stambler et al., 1993). This effect was eliminated by combined ganglionic and post-synaptic muscarinic receptor blockers. Our previous data indicated that MEG decreased the responsiveness of the myofilament to Ca2+ (Huang et al., 1997). Muscarinic cholinergic receptors regulate many cardiac activities, such as heart rate and myocardial contractility. MEG-enhanced NO production found in the present study may provide new information for the understanding of drug-induced medical complications at the cellular and molecular level. Diversity of cholinergic receptors in structure, signalling, and regulation was recognized in the last decade (Goyal, 1989; Pappano & Mubagwa, 1992; Sharma et al., 1997). However, the heart is predominated by the M2 receptor subtype (Peralta et al., 1987; Sharkey et al., 1988; Goyal, 1989; Pappano & Mubagwa, 1992). Western blotting analysis in our present experiments confirms that neonatal rat cardiomyocytes express muscarinic M1, M2, and M3 receptors, but M2 predominates. Application of MEG and carbachol did not alter the protein levels of expression of M1, M2, and M3 receptors in both non-transfected and muscarinic cDNA-transfected cardiomyocytes. Therefore, the increase in NO production caused by MEG was due to the functional stimulation of M2 receptors. This stimulation in M2-transfected cardiomyocytes could cause an induction of eNOS expression. In addition, as atropine alone had no effect on M2 or eNOS protein expression, the antagonistic action of atropine on the MEG-enhanced NO production further supports the hypothesis of MEG muscarinic stimulation. The NO effects on cardiac contractility are likely mediated by activation of the soluble guanylyl cyclase which causes an increase in cyclic GMP production in the heart (Watanabe et al., 1975; Yamamoto et al., 1998). In fact, our previous findings showed that MEG and cocaine significantly increased cyclic GMP production in cultured human embryonic lung cells specifically expressing muscarinic M2 receptors (Yang et al., 2001). In the present study, MEG at a concentration as low as 0.01 μM significantly stimulated NO production in cultured neonatal cardiomyocytes. MEG has been detected in human blood and other organs. By gas chromatographic-mass spectrometric detection, the concentrations of MEG were in a range of 3 to 34 ng/ml (equivalent to 0.01 to 0.12 μM) in 13 serum samples from living subjects (Toennes et al., 1999). In another recent study, the concentrations of MEG in blood specimens obtained from 15 postmortem cases were 0 to 42 μg/L, about 0 to 0.15 μM (Shimomura et al., 2001). Different concentrations of MEG were also observed in postmortem liver, brain, and urine. MEG was detected in 12 of 15 postmortem cases and urine had the highest concentration of MEG.
It has been found that excessive NO production causes myocardial depression and beta-adrenergic hypo-responsiveness (Hare et al., 1995). The toxicity of NO depends on O2 concentration (Ioannidis et al., 1998). Cell death was 46% at 10% O2 and reached 95% at 95% O2 in endothelial cells cultured with 2 mM spermineNONOate, an NO donor. The authors attributed the cytotoxic effect mainly to the oxidation of NO. Reaction of NO with O2− produces peroxynitrite (ONOO−) which can cause toxicity independent of NO or O2− (Beckman et al., 1990). NO and its species can inhibit mitochondrial respiration by binding to cytochrome c oxidase (Szabo & Salzman, 1995). Xie & Wolin (1996) reported that in intact cardiac muscle, NO itself had a completely reversible inhibitory effect on respiration, whereas ONOO− enhanced suppression of respiration that was no longer rapidly reversed. Therefore, in the present study MEG-enhanced NO production may cause cardiac toxicity and damage. In addition, we found that incubation of M2-transfected cardiomyocytes with MEG or carbachol increased expression of eNOS, and this induction was abolished by addition of atropine or L-NAME. It has been shown that eNOS can produce O2− and H2O2 which can cause oxidant stress (Pou et al., 1992; Heinzel et al., 1992; Vasquez-Vivar et al., 1998). Oxidant stress is believed to be involved in the pathogenesis of many cardiovascular diseases, including heart failure (Cai & Harrison, 2000). Excessive NO in the heart may contribute to cardiac toxicity of MEG, especially in the co-presence of cocaine and other metabolites of cocaine in vivo. Our data presented here may provide new information for understanding drug cardiotoxicity.
In summary, MEG caused an increase in NO production in cultured neonatal rat cardiomyocytes. The muscarinic receptor antagonist atropine or the NO synthase inhibitor L-NAME (possible M2 antagonist, Buxton et al., 1993) abolished or significantly attenuated the effect. Since a significant further enhancement of NO production by MEG was found only in M2-transfected cells and since muscarinic protein level was not altered in the presence of MEG, the increase in NO production in cardiomyocytes incubated with MEG mainly resulted from functional stimulation of M2 receptors. It is interesting that the expression of eNOS was increased by incubation with MEG only in M2-transfected cells. Our data demonstrate that MEG probably stimulates M2 receptors, and then sequentially activates eNOS to enhance NO production.
Acknowledgments
We are grateful to Dr Alexander Leaf for his generous supply of cultured neonatal rat cardiomyocytes. This study was supported in part by research grants DA11762 and DA12774 (J.P. Morgan) from the National Institute of Drug Abuse, and 9930254N (Y.-F. Xiao) from the American Heart Association.
Abbreviations
- ACh
acetylcholine
- eNOS
constitutive endothelial NO synthase
- L-NAME
NG-nitro-L-arginine methyl ester
- MEG
methylecgonidine
- NO
nitric oxide
References
- BALLIGAND J., KELLY R.A., MARSDEN P.A., SMITH T.W., MICHEL T. Control of cardiac muscle cell function by an endogenous nitric oxide signaling system. Proc. Natl. Acad. Sci. U.S.A. 1993;90:347–351. doi: 10.1073/pnas.90.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BALLIGAND J.-L., KOBZIK L., HAN X., KAYE D.M., BELHASSEN L., O'HARA D.S., KELLY R.A., SMITH T.W., MICHEL T. Nitric oxide-dependent parasympathetic signaling is due to activation of constitutive endothelial (type III) nitric oxide synthase in cardiac myocytes. J. Biol. Chem. 1995a;270:14582–14586. doi: 10.1074/jbc.270.24.14582. [DOI] [PubMed] [Google Scholar]
- BALLIGAND J.-L., UNGUREANU-LONGOIS D., SIMMONS W.W., KOBZIK L., LOWENSTEIN C.J., LAMAS S., KELLY R.A., SMITH T.W., MICHEL T. Induction of NO synthase in rat cardiac microvascular endothelial cells by IL-1β and IFN-γ. Am. J. Physiol. 1995b;268:H1293–H1303. doi: 10.1152/ajpheart.1995.268.3.H1293. [DOI] [PubMed] [Google Scholar]
- BECKMAN J.S., BECKMAN T.W., CHEN J., MARSHALL P.A., FREEMAN B.A. Apparent hydroxyl radical production by peroxynitrite: implication for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U.S.A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENCHIMOL A., BARTALL H., DESSER K.B. Accelerated ventricular Rhythm and cocaine abuse. Ann. Intern. Med. 1978;88:519–520. doi: 10.7326/0003-4819-88-4-519. [DOI] [PubMed] [Google Scholar]
- BUXTON I.L.O., CHEEK D.J., ECKMAN D., WESTFALL D.P., SANDERS K.M., KEEF K.D. N-nitro L-arginine methyl ester and other alkyl esters of arginine are muscarinic receptor antagonists. Circ. Res. 1993;72:387–395. doi: 10.1161/01.res.72.2.387. [DOI] [PubMed] [Google Scholar]
- CAI H., HARRISON D.G. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ. Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- CALVER A., COLLIER J., VALLANCE P. Nitric oxide and cardiovascular control. Exp. Physiol. 1993;78:303–326. doi: 10.1113/expphysiol.1993.sp003687. [DOI] [PubMed] [Google Scholar]
- CHEN C., OKAYAMA H. High efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell. Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CREGLER L.L., MARK H. Medical complications of cocaine abuse. N. Engl. J. Med. 1986;315:1495–1500. doi: 10.1056/NEJM198612043152327. [DOI] [PubMed] [Google Scholar]
- CREGLER L.L., MARK H. Relation of acute myocardial infarction to cocaine abuse. Am. J. Cardiol. 1985;56:794. doi: 10.1016/0002-9149(85)91140-3. [DOI] [PubMed] [Google Scholar]
- EL-FAWAL H.A., WOOD R.W. Airway smooth muscle relaxant effects of the cocaine pyrolysis product, Methylecgonidine. J. Pharmacol. Exp. Ther. 1995;272:991–996. [PubMed] [Google Scholar]
- ETTINGER N.A., ALBIN R.J. A review of the respiratory effects of smoking cocaine. Am. J. Med. 1989;87:664–668. doi: 10.1016/s0002-9343(89)80401-2. [DOI] [PubMed] [Google Scholar]
- FERON O., HAN X., KELLY R.A. Muscarinic cholinergic signaling in cardiac myocytes: Dynamic targeting of M2AChR to sarcolemmal caveolae and eNOS activation. Life Sci. 1999;64:471–477. doi: 10.1016/s0024-3205(98)00590-6. [DOI] [PubMed] [Google Scholar]
- GEORGE W.J., WIKERSON R.D., KADOWITZ P.J. Influence of acetylcholine on contractile force and cyclic nucleotide levels in the isolated perfused rat heart. J. Pharmacol. Exp. Ther. 1973;184:228–235. [PubMed] [Google Scholar]
- GOYAL R.K. Muscarinic receptor subtypes. New Engl. J. Med. 1989;321:1022–1028. doi: 10.1056/NEJM198910123211506. [DOI] [PubMed] [Google Scholar]
- HAN X., SHIMONI Y., GILES W.R. A cellular mechanism for nitric oxide-mediated cholinergic control of mammalian heart rate. J. Gen. Physiol. 1995;106:45–65. doi: 10.1085/jgp.106.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARE J.M., KEANEY J.F., BALLIGAND J.-L., Jr, LOSCALZO J., SMITH T.W., COLUCCI W.S. Role of nitric oxide in parasympathetic modulation of β-adrenergic myocardial contractility in normal dogs. J. Clin. Invest. 1995;95:360–366. doi: 10.1172/JCI117664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEINZEL B., JOHN M., KLATT P., BOHME E., MAYER B. Ca2+/calmodulin-dependent formation of hydrogen peroxide by brain nitric oxide synthase. Biochem. J. 1992;281:627–630. doi: 10.1042/bj2810627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG L., WOOLF J.H., ISHIGURO Y., MORGAN J.P. Effect of cocaine and methylecgonidine on intracellular Ca2+ and myocardial contraction in cardiac myocytes. Am. J. Physiol. 1997;273:H893–H901. doi: 10.1152/ajpheart.1997.273.2.H893. [DOI] [PubMed] [Google Scholar]
- IOANNIDIS I., BATZ M., KIRSCH M., KORTH H.G., SUSTMANN R., DE GROOT H. Low toxicity of nitric oxide against endothelial cells under physiological oxygen partial pressures. Biochem. J. 1998;329:425–430. doi: 10.1042/bj3290425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ISAACS S.O., MARTIN P., WILLOUGHBY J.H. ‘Crack' (an extra potent form of cocaine) abuse: a problem of the eighties. Oral Surg. Oral Med. Oral Pathol. 1987;63:12–16. doi: 10.1016/0030-4220(87)90332-x. [DOI] [PubMed] [Google Scholar]
- ISNER J.M., ESTES M., THOMPSON P.D., COSTANZO-NORDIN M.R., SUBRAMANIAN R., MILLER G., KATSAS G., SWEENEY K., STURNER W.Q. Acute cardiac events temporally related to cocaine abuse. N. Engl. J. Med. 1986;315:1438–1443. doi: 10.1056/NEJM198612043152302. [DOI] [PubMed] [Google Scholar]
- ITO K., YAN X., TAJIMA M., SU Z., BARRY W.H., LORELL B.H. Contractile reserve and intracellular calcium regulation in mouse myocytes from normal and hypertrophied failing hearts. Circ. Res. 2000;87:588–595. doi: 10.1161/01.res.87.7.588. [DOI] [PubMed] [Google Scholar]
- MEISELS I., LOKE J. The pulmonary effects of free-base cocaine. A review. Cleveland Clin. J. Med. 1993;60:325–329. doi: 10.3949/ccjm.60.4.325. [DOI] [PubMed] [Google Scholar]
- MIAO L., QIU Z., MORGAN J.P. Cholinergic stimulation modulates the negative inotropic effect of cocaine on ferret ventricular myocardium. Am. J. Physiol. 1996;270:H678–H684. doi: 10.1152/ajpheart.1996.270.2.H678. [DOI] [PubMed] [Google Scholar]
- ODDIS C.V., SIMMONS R.L., HATTLER B.G., FINKEL M.S. cAMP enhances inducible nitric oxide synthase mRNA stability in cardiac myocytes. Am. J. Physiol. 1995;269:H2044–H2050. doi: 10.1152/ajpheart.1995.269.6.H2044. [DOI] [PubMed] [Google Scholar]
- OKAYAMA H., BERG P. A cDNA cloning vector that permits expression of cDNA inserts in mammalian cells. Mol. Cell. Biol. 1983;3:280–289. doi: 10.1128/mcb.3.2.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAPPANO A.J., MUBAGWA K.Actions of muscarinic agents and adenosine on the heart The Heart and Cardiovascular System 1992New York: Raven Press; 1765–1776.ed. Fozzard, H.A., et al [Google Scholar]
- PERALTA E.G., ASHKENAZI A., WINSLOW J.W., SMITH D.H., RAMACHANDRAN J., CAPON D.J. Distinct primary structures, ligand-binding properties and tissue-specific expression of four human muscarinic acetylcholine receptors. EMBO J. 1987;6:3923–3929. doi: 10.1002/j.1460-2075.1987.tb02733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POU S., POU W.S., BREDT D.D., SNYDER S.H., ROSEN G.M. Generation of superoxide by purified brain nitric oxide synthase. J. Biol. Chem. 1992;267:24173–24176. [PubMed] [Google Scholar]
- RESNICK R.B., KESTENBAUM R.S., SCHWARTZ L.K. Acute systemic effects of cocaine in man. A controlled study by intranasal and intravenous routes. Science. 1977;195:696–698. doi: 10.1126/science.841307. [DOI] [PubMed] [Google Scholar]
- SHARKEY J., RITZ M.C., SCHENDEN J.A., HANSON R.C., KUHAR M.J. Cocaine inhibits muscarinic cholinergic receptors in heart and brain. J. Pharmacol. Exp. Ther. 1988;246:1048–1052. [PubMed] [Google Scholar]
- SHARMA V.K., COLECRAFT H.M., RUBIN L.E., SHEU S.S. Does mammalian heart contain only the M2 muscarinic receptor subtype. Life Sci. 1997;60:1023–1029. doi: 10.1016/s0024-3205(97)00043-x. [DOI] [PubMed] [Google Scholar]
- SHIMONURA E.T., HODGE G.D., PAUL B.D. Examination of postmortem fluids and tissues for the presence of methylecgonidine, ecgonidine, cocaine, and benzoylecgonine using solid-phase extraction and gas chromatography-mass spectrometry. Clin. Chem. 2001;47:1040–1047. [PubMed] [Google Scholar]
- STAMBLER B.S., KOMAMURA K., IHARA T., SHANNON R.P. Acute intravenous cocaine causes transient depression followed by enhanced left ventricular function in conscious dogs. Circulation. 1993;87:1687–1697. doi: 10.1161/01.cir.87.5.1687. [DOI] [PubMed] [Google Scholar]
- STERIN-BORDA L., ECHAGUE A.V., LEIROS A., GENARO C.P., BORDA E. Endogenous nitric oxide signaling system and the cardiac muscarinic acetylcholine receptor-inotropic response. Br. J. Pharmacol. 1995;115:1525–1531. doi: 10.1111/j.1476-5381.1995.tb16646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SZABO C., SALZMAN A.L. Endogenous peroxynitrite is involved in the inhibition of mitochondrial respiration in immuno-stimulated J774.2 macrophages. Biochem. Biophys. Res. Commun. 1995;209:739–743. doi: 10.1006/bbrc.1995.1561. [DOI] [PubMed] [Google Scholar]
- TOENNES S.W., FANDINO A.S., KAUERT G. Gas chromatographic-mass spectrometric detection of anhydroecgonine methyl ester (methylecgonidine) in human serum as evidence of recent smoking of crack. J. Chromatogr. B. Biomed. Sci. Appl. 1999;735:127–132. doi: 10.1016/s0378-4347(99)00412-0. [DOI] [PubMed] [Google Scholar]
- VASQUEZ-VIVAR J., KALYANARAMAN B., MARTASEK P., HOGG N., MASTER B.S., KAROUI H., TORDO P., PRITCHARD K.A., Jr Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc. Natl. Acad. Sci. U.S.A. 1998;95:9920–9925. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VIRMANI R., ROBINOWITZ M., SMIALEK J.E., SMYTH D.F. Cardiovascular effects of cocaine. An autopsy study of 40 patients. Am. Heart J. 1988;115:1068–1075. doi: 10.1016/0002-8703(88)90078-6. [DOI] [PubMed] [Google Scholar]
- WATANABE A.M., BESCH H.R., Jr Interaction between cyclic adenosine monophosphate and cyclic guanosine monophosphate in guinea pig ventricular myocardium. Circ. Res. 1975;37:309–317. doi: 10.1161/01.res.37.3.309. [DOI] [PubMed] [Google Scholar]
- WOOLF J.H., HUANG L., ISHIGURO Y., MORGAN J.P. Negative inotropic effect of methylecgonidine, a major product of cocaine base pyrolysis, on ferret and human myocardium. J. Cardiovasc. Pharmacol. 1997;30:352–359. doi: 10.1097/00005344-199709000-00013. [DOI] [PubMed] [Google Scholar]
- XIE Y.W., WOLIN M.S. Role of nitric oxide and its interaction with superoxide in the suppression of cardiac muscle mitochondrial respiration: involvement in response to hypoxia/reoxygenation. Circulation. 1996;94:2580–2586. doi: 10.1161/01.cir.94.10.2580. [DOI] [PubMed] [Google Scholar]
- YAMAMOTO S., MIYAMOTO A., KAWANA S., NAMIKI A., OHSHIKA H. Role of nitric oxide production through M2-cholinergic receptors in cultured rat ventricular myocytes. Biochem. Biophys. Res. Commun. 1998;251:791–795. doi: 10.1006/bbrc.1998.9547. [DOI] [PubMed] [Google Scholar]
- YANG Y.K., KE Q.E., CAI J.B., XIAO Y.-F., MORGAN J.P. Evidence for cocaine and methylecgonidine stimulation of M2 muscarinic receptors in cultured human embryonic lung cells. Br. J. Pharmacol. 2001;132:451–460. doi: 10.1038/sj.bjp.0703819. [DOI] [PMC free article] [PubMed] [Google Scholar]