

Abstract
This study examined the action of gabapentin (gabapentin,1-(aminomethyl) cyclohexane acetic acid (Neurontin®)) on voltage-gated calcium (Ca2+) channel influx recorded in cultured rat dorsal root ganglion (DRG) neurones.
Voltage-gated Ca2+ influx was monitored using both fura-2 based fluorescence Ca2+ imaging and the whole-cell patch clamp technique.
Imaging of intracellular Ca2+ transients revealed that gabapentin inhibited KCl (30 mM)-evoked voltage-dependent Ca2+ influx. Both the duration for 50% of the maximum response (W50) and total Ca2+ influx were significantly reduced by ∼25-30% in the presence of gabapentin (25 μM).
Gabapentin potently inhibited the peak whole-cell Ca2+ channel current (IBa) in a dose-dependent manner with an estimated IC50 value of 167 nM. Block was incomplete and saturated at a maximal concentration of 25 μM.
Inhibition was significantly decreased in the presence of the neutral amino acid L-isoleucine (25 μM) but unaffected by application of the GABAB antagonist, saclofen (200 μM), suggesting a direct action on the α2δ subunit of the Ca2+ channel.
Gabapentin inhibition was voltage-dependent, producing an ∼7 mV hyperpolarizing shift in current voltage properties and reducing a non-inactivating component of whole-cell current activated at relatively depolarized potentials.
The use of specific Ca2+ channel antagonists revealed a mixed pharmacology of the gabapentin-sensitive current (N-, L- and P/Q-type), which is dominated by N-type current.
The present study is the first to demonstrate that gabapentin directly mediates inhibition of voltage-gated Ca2+ influx in DRG neurones, providing a potential means for gabapentin to effectively mediate spinal anti-nociception.
Keywords: Gabapentin, Ca2+ channels, dorsal root ganglia, patch-clamp, fluorescence calcium imaging, pain, sensory neurones
Introduction
The anti-convulsant drug Gabapentin, 1-(aminomethyl) cyclohexane acetic acid (Neurontin®) has been shown to exhibit clinically effective anti-hyperalgesic activity via an unknown mechanism. Gabapentin was originally developed for the treatment of spasticity and partial epilepsy and has proved effective in a number of different animal seizure models (Taylor et al., 1998; Cesena & Calcutt, 1999; Field et al., 1999; Cheng et al., 2000; Nicholson, 2000). Numerous open label case studies and three large double-blind trials now also provide supporting evidence that gabapentin is successful at treating a wide-ranging number of neuropathic pain conditions, including diabetic neuropathy (Backonja et al., 1998), postherpetic neuralgia (Rowbotham et al., 1998), trigeminal neuralgia, migraine and pain associated with cancer and multiple sclerosis (Di Trapini et al., 2000; Caraceni et al., 1999; Houtchens et al., 1997; see also Magnus, 1999; Laird & Gidal, 2000; Nicholson, 2000). Although gabapentin has now proved an effective treatment for a number of neuropathic pain conditions, numerous studies have investigated its possible mechanisms of action and yet no firm conclusions have been drawn (Taylor et al., 1998).
Originally designed as a GABA mimetic, until very recently, there was no evidence for the ability of gabapentin to influence the functioning of either GABA receptors or GABA re-uptake mechanisms either directly or by inappropriate metabolic substitution (Taylor et al., 1998). However, gabapentin has since been shown to mediate post synaptic membrane excitability through a subtype-specific interaction with a particular GABAB receptor heterodimer (Ng et al., 2001). Gabapentin has also been demonstrated to substitute as a substrate for the system L amino acid transporter (Stewart et al., 1993; Su et al., 1995), providing an effective route of entry for gabapentin into the intracellular environment. Nevertheless, the only specific molecular target for gabapentin identified to date is the α2δ subunit of voltage-dependent Ca2+ channels (Gee et al., 1996). Interaction with this high affinity binding site may provide a means for gabapentin to mediate voltage-dependent Ca2+ influx, by indirectly influencing the functional interaction between the accessory α2δ and the pore-forming α1 subunit of Ca2+ channels (reviewed in Walker & De Waard, 1998).
Previous studies have reported a mixed susceptibility of different Ca2+ channel preparations to the action of gabapentin. Stefani et al. (1998) were the first group to demonstrate that gabapentin inhibited voltage-dependent Ca2+ channel currents recorded from cortical neurons, however the gabapentin-mediated reduction in current varied between different cell types. Nevertheless, despite the inconsistent nature of the gabapentin-sensitive Ca2+ current, a comparative survey of work undertaken by a number of independent groups suggests that, given the right conditions, gabapentin can produce a reduction in Ca2+ channel influx. Gabapentin has been shown to reduce P/Q-type current into synaptosomes and inhibit release of excitatory amino acids from neocortical and trigeminal nucleus slices in a localized and stimulus-dependent manner (Dooley et al., 2000; Fink et al., 2000; Meder & Dooley, 2000; Maneuf & Mcknight, 2000).
In view of these findings, the effects of gabapentin were therefore tested on voltage-dependent Ca2+ influx recorded in cultured rat dorsal root ganglia (DRG) neurones. These neurones are thought to play a crucial role in pain processing (Reichling & Levine, 1999) and have been shown to express a variety of high voltage-activated (HVA) Ca2+ channel α1 subunits (N-, L-, P/Q- and R-type; Mintz et al., 1992; Rusin & Moises, 1995) – all of whose properties have been shown to be affected by co-association with accessory α2δ subunits (Gurnett et al., 1996; Klugbauer et al., 1999; Gao et al., 2000; reviewed in Walker & De Waard, 1998; Felix, 1999). In the present study, the affect of gabapentin on HVA Ca2+ influx was assessed using both Ca2+ imaging and the whole-cell patch clamp technique. Some of the data in this manuscript has previously been published in abstract form (Martin et al., 2000; Mcclelland et al., 2000; Sutton et al., 2000).
Methods
Cell culture
Briefly, dorsal root ganglia were dissected from 1- to 3-day-old Wistar rats that had been killed by decapitation. Ganglia were then incubated in Collagenase/Dispase (1 mg ml−1, Boehringer) at 37°C for 45 min in Ca/Mg-free PBS before being transferred to DMEM-based media (Gibco+10% FBS, 2 mM L-glutamine, penicillin (5000 IU ml−1), streptomycin (5000 mg ml−1)) and mechanically dissociated by trituration through a 23G hyperdermic needle. The cell suspension was strained to remove debris (<40 μm, Falcon), spun and re-suspended in the above culture media containing 100 ng ml−1 7s-NGF (Promega) before being plated onto poly-L-lysine/laminin-coated coverslips (∼5×103/coverslip, 10 mm diameter.) Cells were treated with cytosine arabinoside (2.5 μM) for the first 48 h in culture (37°C in air containing 5% CO2) after which they were transferred to F14-based culture media (Imperial Laboratories +10% HS (Gibco), penicillin (5000 IU ml−1), streptomycin (5000 mg ml−1), 2 mM L-glutamine, 14 mM NaHCO3, 10 ng ml 2.5S NGF (Sigma)). Cells used for Ca2+ imaging experiments were only cultured in F14-based media (20 ng ml−1 NGF). and were not treated with cytosine aribinoside. Cells were used for experiments after an additional 48 h and re-fed with fresh culture media every 2 – 3 days.
Calcium imaging
Cultured DRG neurones were incubated for 1 h in NaCl-based extracellular solution (mM): NaCl, 130; KCl, 3.0; MgCl2, 0.6; CaCl2, 2.0; NaHCO3, 1.0; HEPES, 10.0; glucose, 5.0 and 0.01, fura-2AM (Sigma, 1 mM stock in dimethylformamide), pH 7.4 (NaOH), osmolarity 310 – 320 mOsm (sucrose). The cells were then washed for 10 – 20 min with NaCl-based extracellular solution to remove the extracellular fura-2AM and to allow cytoplasmic de-esterification of the Ca2+ sensitive fluorescent dye. The cells were constantly perfused with NaCl-based extracellular solution (1 – 2 ml/min) and viewed under an inverted Olympus BX50WI microscope with a KAI-1001 S/N 5B7890-4201 Olympus camera attached. The fluorescence ratiometric images from data obtained at excitation wavelengths of 340 nm and 380 nm were viewed and analysed using OraCal pro, Merlin morphometry temporal mode (Life Sciences resources, version 1.20). The neurones were stimulated with NaCl-based extracellular solution containing high K+ (30 mM), which produced depolarization, activation of voltage-gated Ca2+ channels and large transient increases in intracellular Ca2+. Three consistent transient increases of intracellular Ca2+ could be obtained in a single experiment. The actions of gabapentin (25 μM) were investigated on the response to the second stimulus. The actions of gabapentin on the Ca2+ transient amplitude, duration at 50% peak amplitude (W50) and total Ca2+ flux were measured.
Measurement of membrane potentials
The effects of changing extracellular [K+] on membrane potential were evaluated using the whole-cell variant of the patch clamp technique. Culture DRG neurones were bathed in NaCl-based extracellular solution and low resistance patch pipettes were filled with a patch pipette solution containing (mM), KCl 140, EGTA 5, CaCl2 0.1, MgCl2 2, HEPES 10, ATP 2, pH 7.2 (TRIS), 310 – 315 mOsmol/l (sucrose). After entering the whole-cell recording configuration, neurones were allowed to equilibrate for approximately 5 min before stable membrane potentials were made. Extracellular solutions containing 10, 20, 30 or 40 mM K+ were applied by low pressure ejection via a blunt micropipette (tip diameter about 10 μm) positioned approximately 100 μm from the neurone and the new stable membrane potentials recorded.
Electrophysiology and solutions
The sensitivity of HVA calcium channels to gabapentin was assessed utilising the whole-cell mode of the patch clamp technique (2 – 5 mM Ba). Extracellular recording saline comprised of (mM): Choline chloride 130, TTX 0.0025, TEA 25, KCl 3, BaCl2 2/5, MgCl2 0.6, NaHCO3 1, HEPES 10, Glucose 4, pH 7.4 NaOH, 320 mOsml (sucrose). Intracellular patch pipettes were filled with (mM): CsCl2 140, EGTA 10, CaCl2 0.1, MgCl2 2.0, HEPES 10, ATP, 2 pH 7.2. Tris, 310 mOsml (sucrose). GTP (1 mM) was included in the intracellular patch pipette solution for all experiments except the dose response data to increase stability of recordings over time. All experiments were performed at room temperature (20 – 22°C). Whole-cell patch-clamp recordings were performed using an Axopatch 200 amplifier (Axon Instruments Inc., Foster City, CA, U.S.A.) linked to a personal computer equipped with pCLAMP Version 8.0. Patch pipettes (World Precision Instruments 1B12OF-3) were pulled using a Sutter P-87 microelectrode puller and showed typical resistances of 2 – 4 MΩ. Series resistance had typical values of 8 – 12 MΩ and was electronically compensated by at least 90%. Recordings were low-pass filtered at 2 kHz using the built in Bessel filter of the amplifier and digitized at 5 kHz using a Digidata 1200 A/D converter (Axon Instruments). Peak inward currents (IBa) were activated from a Vh of −80 mV using 100 ms steps to Vc of 0 mV every 15 s. To control for full equilibration with the internal patch solution, data was obtained from currents that had been allowed to stabilize for up to 10 min before any measurements were made. Drugs were applied by gravity-fed bath perfusion from an outlet placed close to the recorded cell. Data was analysed using Clampfit (Axon Instruments, Inc.) All curve fitting was carried out in GraphPad Prism Version 3.0. Unless stated otherwise, data are expressed as means±s.e.mean, numbers in parentheses displayed on the figures reflect numbers of individual cells recorded, and P values given reflect unpaired 2-tailed Student's t-tests.
Stocks of gabapentin, L-isoleucine (Sigma), ω-Agatoxin IVA and ω-Conotoxin GVIA (Alamone) were prepared in water and stored at −20°C. Stocks of saclofen and nifedipine (Sigma) were prepared in DMSO and stored at −20°C.
Results
Gabapentin inhibits calcium influx into cultured DRG neurones
Experiments were initially carried out to optimize the recording conditions and to justify the protocols used in this study. Electrophysiological and fura-2-based fluorescence imaging data was obtained for neuronal responses over a range of stimulating KCl concentrations. Increases in intracellular Ca2+ were shown to exhibit a graded linear correlation with changes to membrane potential over increasing concentrations of KCl (Figure 1a,b). Triplicate responses of consistent amplitude and duration which showed complete recovery from the stimulus were obtained using a maximal concentration of 30 mM KCl (Figure 1c – e). Mean values calculated for the duration (width) at 50% of the maximum response (W50) were 84.6±10.8 s, 84.1±7.0 s and 87.5±7.3 s for three consecutive stimuli, respectively (Figure 1e).
Figure 1.
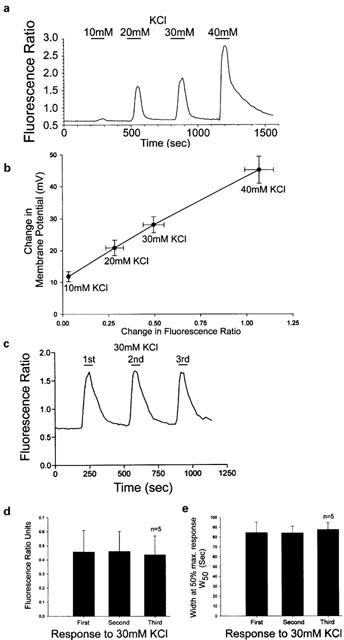
(a) Trace showing changes in fluorescence ratio produced by changes in KCl in imaging experiments. (b) Graph showing linear correlation between change in fluorescence ratio and membrane potential at varying concentrations of K+. The plot was produced from mean data±s.e.mean obtained from imaging experiments (n=19) and measuring membrane potential changes (n=6 – 7); R2=0.9977. (c) Trace from an individual DRG neurone showing that, three pulses of 30 mM KCl gave repeatable responses that fully recovered between stimulation. (d and e) Bar graphs show that consistent control Ca2+ transients were obtained in response to repeated stimuli of 30 mM KCl applied to the same cells. Data plotted are the mean response as a fluorescence ratio or duration at 50% of the transient amplitude.
A 1 – 2 min pre-application of 25 μM gabapentin was not seen to alter the mean peak amplitude of the Ca2+ transient evoked by 30 mM K+. Changes in fluorescence ratio were calculated to be 0.3290±0.029, 0.3230±0.0240 and 0.3030±0.025 for control, gabapentin and recovery responses, respectively (n=27; Figure 2a). Nevertheless, gabapentin (25 μM) did significantly reduce both the duration of the Ca2+ transients and the total Ca2+ influx evoked by 30 mM KCl – measured as the area under the transient curve (Figure 2b,c). The duration for 50% of the maximum response (W50) was significantly and reversibly reduced from control in the presence of gabapentin (calculated values were 88.4±4.9 s, 67.4±3.8 s and 96.2±7.4 s, for control, gabapentin and recovery, respectively, n=27; Figure 2b). Similar results were obtained when analysing the data for the gabapentin effect on total Ca2+ flux normalized with respect to the first control response, where gabapentin reversibly reduced the influx to 79.9±3.8% with the recovery response calculated to be 99.7±3.3% (n=27; Figure 2c).
Figure 2.
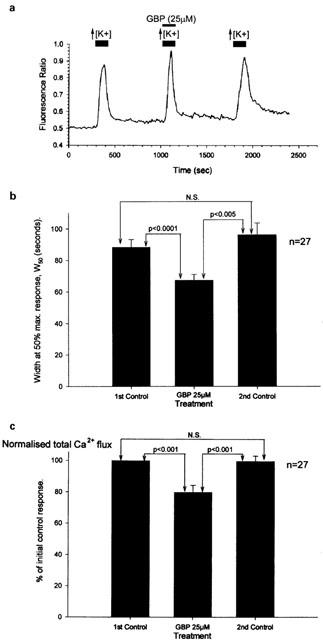
Gabapentin inhibited Ca2+ influx into cultured DRG as measured using fura-2 fluorescence imaging. (a) Trace from a single cultured DRG neurone showing a decrease in response duration and total Ca2+ flux by gabapentin (25 μM), but no significant change in the amplitude of the peak Ca2+ transient. (b and c) Bar charts showing the inhibitory actions of 25 μM gabapentin (GBP) on the duration of the Ca2+ transients evoked by 30 mM KCl measured at 50% of the peak amplitude (W50) and the total Ca2+ flux normalized with respect to the first control Ca2+ transient.
Gabapentin inhibits whole-cell voltage-gated calcium channel current in DRG neurones
Gabapentin-mediated inhibition of HVA Ca2+ channel current was initially assessed using the whole-cell patch clamp technique with barium as the charge carrier. From a holding potential of −80 mV, peak inward currents were activated every 15 s using 100 ms step depolarizations to Vc=0 mV. Using these conditions the total peak recorded current is dominated by HVA channel influx. As shown in Figure 3a, sustained bath perfusion of gabapentin (25 μM) significantly and irreversibly blocked a component of peak HVA current (IBa). Onset of block was slow to develop and completely saturated within 10 min of application.
Figure 3.
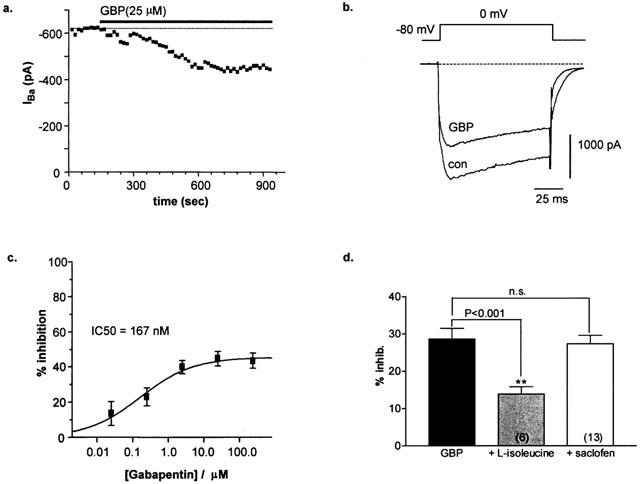
(a) Time-course showing gabapentin inhibition of peak IBa. (b) Whole-cell current trace of peak HVA IBa activated under control conditions (con) and following a 10 min application of gabapentin (GBP). (c) Dose-response of GBP-mediated inhibition of peak HVA IBa. Data points represent mean±s.e.mean (n=3 – 8 determinants) and were fitted with the following sigmoidal equation: Y=max/1+(IC50/[GBP]n) where n is the Hill slope factor=0.55, max is maximum inhibition=46.8% and IC50=167 nM. (d) Bar graph (mean±s.e.mean) showing that inhibition of peak HVA IBa by gabapentin (GBP) is significantly reduced in the presence of L-isoleucine (25 μM) but is unaffected following treatment with saclofen (200 μM, n=10, 6 and 13, respectively).
The action of gabapentin was tested over a broad range of concentrations (25 nM – 250 μM). Inhibition of the HVA current was incomplete, with maximal block saturating at a concentration of 25 μM GBP (44.7±4.1%, n=8; Figure 3c). Block of the peak current was dose-dependent and fit of the mean dose-response data with the Hill equation yielded an estimated IC50 value of 167 nM. This value is close to the Kd value ∼40 nM obtained in binding studies (Suman-Chauhan et al., 1993; Gee et al., 1996; Brown & Gee, 1998) and below estimates of clinically effective plasma concentrations (10 – 100 μM; Goa & Sorkin, 1993). The lower potency of gabapentin in vivo may be due endogenous levels of L-system substrates, such as L-leucine, which have been demonstrated to decrease the efficacy of gabapentin by competing for the α2δ binding site (Gee et al., 1996). Indeed, pre-application of L-isoleucine (25 μM) significantly reduced inhibition by gabapentin to just 13.8±2.0%, n=6 (P<0.001; Figure 3d), sufficient to increase the estimated IC50 value to a value greater than 10 μM, as observed previously for in vivo studies. This data therefore indicates that the Ca2+ channel α2δ subunit is the effective site of functional interaction for gabapentin in this preparation.
A recent report suggested that gabapentin may indirectly influence Ca2+ channels by interacting with a specific splice variant of the GABAB receptor (Ng et al., 2001). However, in agreement with previous observations (Mcclelland et al., 2000), in this study inhibition by gabapentin (25 μM, 28.6±7.1%, n=10) was unaffected by pre-application of the specific GABAB antagonist, saclofen (200 μM), reducing the calcium channel current by 27.2±1.8% (n=13; P=0.66; Figure 3d). This data, coupled with a reduced potency in the presence of neutral amino acids, therefore suggests that in DRG neurones gabapentin does indeed mediate its affect via a direct interaction with the Ca2+ channel.
Voltage-dependence of gabapentin inhibition
In DRG neurones, the voltage-dependence of the HVA whole-cell current represents the combined properties of a number of different channel subtypes (L-, N-, P/Q-, and R-type; Mintz et al., 1992; Rusin & Moises, 1995), each differentially influenced by their associated accessory subunits (Walker & De Waard, 1998). Using 2 mM Ba2+ as the charge carrier, maximal activation of the HVA current occurred within the voltage range of Vc=−5 and 0 mV. Voltage-dependence of gabapentin inhibition was subsequently determined using a ramped step protocol (0.9 ms/mV, Vh=−80 mV). Currents were activated under control conditions or following a 10 min application of gabapentin. Boltzmann fit of the data showed that gabapentin inhibited a proportion of current activated at relatively depolarized potentials (Figure 4a). The decrease in HVA conductance was accompanied by an ∼7 mV hyperpolarizing shift in the voltage-dependence of current activation (V50act=−8.6±1.5, and −15.2±0.8 mV, n=3 and 4, for control and gabapentin-treated cells respectively; P<0.05), with no corresponding change in the estimated reversal (null) potential determined from Boltzmann fit of the data (Erev=29.5±1.3 and 29.6±1.9 mV).
Figure 4.
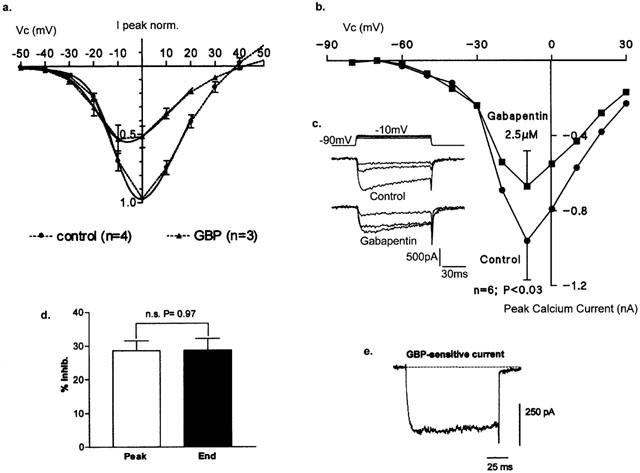
(a) Mean±s.e.mean ramp-activated whole-cell currents (0.9 ms/mV) from −50 mV under control conditions (n=4) and following a 10 min application of gabapentin (25 μM; n=3). Solid line indicates Boltzmann fit to the data. (b) Mean (±s.e.mean; n=6 determinants) I/V relationship for whole-cell voltage-gated Ca2+ currents (2 mM Ca2+) recorded under control conditions and following a 3 – 5 min application of gabapentin by low pressure ejection (2.5 μM; P<0.03). (c) inset records showing Ca2+ currents recorded in the absence or presence of gabapentin (Vc=−30, −20 and 10 mV). (d) Bar graph of mean inhibition (±s.e.mean; n=10 determinants) of HVA IBa measured at the peak vs the end of the current step. (e) Gabapentin-sensitive difference current obtained by subtracting peak HVA current traces recorded under control conditions and following a 10 min application of gabapentin (25 μM).
The effects of gabapentin (2.5 μM) were also investigated on voltage-gated currents using 2 mM Ca2+ as the charge carrier. From a holding potential of −90 mV, current/voltage (I/V) relationships were generated using 100 ms voltage step commands to potentials between −150 to +30 mV. No effects on the mean low threshold current (Vc=−50 to −30 mV) were detected. Nevertheless, as for Ba2+, a 3 – 5 min application of gabapentin produced a significant 33±9% inhibition of the peak current at Vc=−10 mV, although no significant change in the mean voltage-dependence of channel activation was observed (Figure 4b; P<0.03, n=6). However, in two of the six recorded neurones there was a similar tendency for the I/V relationship to shift to the left in the presence of gabapentin as for currents recorded in the presence of Ba2+ (see inset traces, Figure 4c).
Examination of both the Ba2+ and Ca2+ current waveforms following inhibition by gabapentin (25 μM) did not reveal any changes in rate of current inactivation. Both peak and end Ba2+ current amplitudes were equally inhibited by gabapentin (28.6±2.9% vs 28.8±3.4% respectively; P=0.97, n=10; Figure 4d). Current subtraction to obtain the gabapentin-sensitive difference Ba2+ current revealed a square waveform, confirming its non-inactivating properties (Figure 4e).
Pharmacological characterization of gabapentin-sensitive current
The pharmacological profile of the gabapentin-sensitive current was investigated using a variety of specific Ca2+ channel antagonists. A number of the different HVA Ca2+ channels expressed in DRG neurones can be blocked using selective inhibitors (N-, P/Q- and L-type; Mintz et al., 1992; Rusin & Moises, 1995). Isolation of the components of HVA current were determined using 100 nM ω-Agatoxin IVA to inhibit P/Q-type current (13.5±4.4%, n=8), 1 μM ω-Conotoxin GVIA (ω-CgTx GVIA) to inhibit N-type current (57.3±6.6%, n=7) and the L-type blocker, nifedipine (5 μM; 29.8±2.2%, n=6; Figure 5a).
Figure 5.
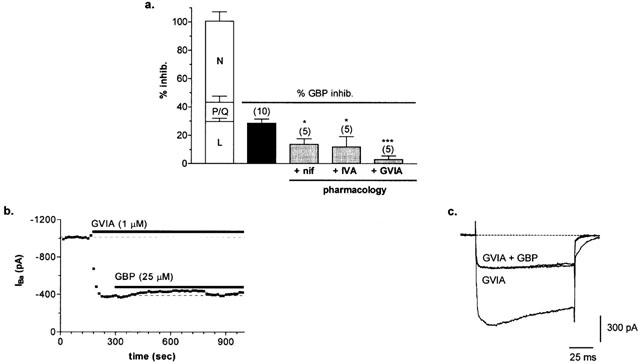
(a) Pharmacology of gabapentin-sensitive peak HVA IBa (mean±s.e.mean). Clear bars represent mean data for inhibition of peak HVA IBa with Ca2+ channel antagonists (L-, P/Q- or N-type; n=6, 8 and 7, respectively). The black bar represents mean total current inhibited by gabapentin. Grey bars represent mean gabapentin-sensitive current remaining in the presence of L- (+nif), P/Q- (+IVA) or N- (+GVIA) type blockers. (b) Time-course showing gabapentin inhibition of peak IBa is virtually eliminated following block of the N-type current with ω-CgTx GVIA. (c) Whole-cell current trace of peak HVA IBa activated under control conditions and following sequential applications of ω-CgTx GVIA (1 μM, GVIA) followed by gabapentin (25 μM) in the presence of GVIA (GVIA+GBP).
As shown in Figure 5, pre-application of each Ca2+ channel antagonist significantly reduced inhibition by gabapentin. Pre-block by ω-CgTx GVIA almost completely eliminated the gabapentin-sensitive current, reducing it from 28.6±2.9% (n=10) to just 3.1±2.5% (n=5; P<0.001), suggesting that gabapentin inhibition is mediated mainly by N-type current (Figure 5b,c). However, prior inhibition of either L- or P/Q-type current also significantly reduced the effectiveness of gabapentin, although to a lesser extent, as gabapentin inhibition was just 13.7±3.9% and 12.0±7.2%, in the presence of nifedipine and ω-Agatoxin IVA, respectively (n=5, 5; P<0.05). The mixed pharmacology of the gabapentin-sensitive current suggests that inhibition may reflect a direct action of gabapentin on a number of different sub-types of calcium channel, dominated by the N-type current in this cell-type.
Discussion
This study is the first to demonstrate a potent inhibition by gabapentin of the Ca2+ influx through voltage-gated Ca2+ channels in DRG neurones. Voltage-gated Ca2+ channels are critical in mediating both the development and maintenance of the neuronal sensitization process associated with neuropathic pain, and provide attractive candidates for the development of antihyperalgesic drugs (reviewed in Vanegas & Schaible, 2000). DRG neurones are thought to play a crucial role in pain processing (Reichling & Levine, 1999) and have been shown to express a variety of HVA Ca2+ channel α1 subunits (Mintz et al., 1992; Rusin & Moises, 1995). Recordings taken from dorsal horn neurons in spinal cord slices from adult hyperalgesic rats or neonatal control slices have confirmed a presynaptic (DRG) site of action for gabapentin (Patel et al., 2000; Shimoyama et al., 2000), and depression of presynaptic Ca2+ currents participating in nociceptive synaptic transmission may well be expected to contribute to the antihyperalgesic action of gabapentin. Nevertheless, previous studies indicated that gabapentin had no effect on freshly dissociated DRG neurones, suggesting that inhibition of Ca2+ channels in this cell-type did not contribute to its mechanism of action (Rock et al., 1993). More recently however, a decrease in Ca2+ channel current has been detected in DRG neurones by a number of different groups using both electrophysiological and Ca2+ imaging techniques (Martin et al., 2000; Mcclelland et al., 2000; Sarantopoulos et al., 2000; Sutton et al., 2000). Under conditions used in the present study, gabapentin potently inhibited voltage-dependent Ca2+ influx in DRG neurones, with effective reductions achieved using nM concentrations of drug.
The mixed pharmacology of the gabapentin-sensitive Ca2+ current in DRG neurones suggests that inhibition may reflect a direct action of gabapentin on a number of different channel sub-types. Of particular interest was the unique sensitivity of the ω-CgTx GVIA-sensitive, N-type current to inhibition by gabapentin. N-type Ca2+ channels are believed to play an important role in mediating neuropathic pain (Vanegas & Schaible, 2000) and the potential for gabapentin-dependent modulation of presynaptic N-type channels in DRG neurones would prove an important means for mediating neuronal excitability and neurotransmitter release (Dunlap et al., 1995). In this study, reduced sensitivity to gabapentin was also demonstrated following inhibition of other subtypes of HVA Ca2+ channel (P/Q- and L-type). Not only did the gabapentin-responsive current exhibit mixed Ca2+ channel pharmacology, there was also a degree of overlap in the sensitivity of gabapentin inhibition to various Ca2+ channel blockers. These findings are supported by a recent paper by Stefani et al. (2001), who also noted an apparent overlap in the Ca2+-dependent pharmacology of gabapentin action in isolated rat cortical neurones. In the current study, the reduced gabapentin sensitivity observed following the use of specific Ca2+ channel antagonists might reflect an additional, more indirect influence of the blockers on the gabapentin-sensitive target. Activation of voltage-dependent Ca2+ influx per se could participate in ‘priming' the cell for gabapentin action, and the addition of Ca2+ channel inhibitors could play a secondary role in altering any Ca2+-dependent modulation of the level of gabapentin responsiveness of the cell.
The interesting observation of the relatively non-inactivating nature of the gabapentin-sensitive current given its mixed pharmacology, can be explained accordingly. The binding site for gabapentin is located on the accessory α2δ subunit and not the major α1 pore-forming unit of the channel. Moreover, DRG neurones have been shown to express a mixed population of voltage-gated Ca2+ channels consisting of a number of different subtypes of α1 subunits. The α2δ subunits (1 – 3) have all been shown to functionally associate with a variety of different α1 subunits (Gurnett et al., 1996; Dolphin et al., 1999; Klugbauer et al., 1999; Gao et al., 2000; reviewed in Walker & De Waard, 1998; Felix, 1999). Through its interaction with α2δ, gabapentin may therefore indirectly affect a heterogenous mix of α1 subunits as no preferential partnership between α1 and α2δ subtypes has yet been demonstrated. Co-expression experiments have shown that functional association with α2δ affects comparable changes in current amplitude and voltage-dependent properties of a variety of different α1 subunits (Walker & De Waard, 1998; Felix, 1999). Likewise in a similar manner, through its interaction with α2δ, gabapentin might also be inferred to produce a similar overall trend of a decrease in current amplitude carried by a variety of different α1 subunits, regardless of the underlying biophysical properties of the α1 subunit. This data therefore suggests that in DRG neurones, gabapentin may modulate a mixture of different α1 subunits producing a reduction of a non-inactivating high-voltage activated component of the whole-cell current. This finding is supported by previous work, whereby gabapentin has been shown to inhibit disparate components of L- vs P/Q- and N-type current in different preparations (Stefani et al., 1998; Fink et al., 2000).
In support of these findings, the mixed ability of gabapentin to inhibit Ca2+ channels has been demonstrated previously for a number of different preparations. Stefani et al. (1998) recorded a differing sensitivity of rat brain neurones to gabapentin and no detectable affects of gabapentin were observed on Ca2+ channel currents recorded from hippocampal neurones taken from patients with temporal lobe epilepsy (Schumacher et al., 1998). Gabapentin inhibition of synaptosomal P/Q-type Ca2+ current and affects on neurotransmitter release have also been shown to be both tissue-specific and dependent upon the stimulus protocol (Dooley et al., 2000; Fink et al., 2000; Meder & Dooley, 2000; Maneuf & Mcknight, 2000). The recent identification of three different subtypes of α2δ provides a means for subunit – specific interactions with gabapentin, which could restrict its affects to more localized sites of action. (Klugbauer et al., 1999; Su et al., 2000). Moreover, sub-cellular distribution and tissue specificity of Ca2+ channel subunit combinations have not yet been determined. Thus, gabapentin may require particular subunit variants (α1, α2δ, and β), coupled with the presence of additional accessory proteins and/or activation of second messenger cascades to be effective (Sutton et al., 2000; Reichling & Levine, 1999). Consequently, the pre-requisite of distinct, permissive conditions may well act to limit the actions of gabapentin, in contrast to other established Ca2+ channel ligands. Indeed, as a drug gabapentin is not effective in all patients (Laird & Gidal, 2000), a finding which could reflect a biovariability in its pharmacokinetic properties and/or the target binding interactions of gabapentin.
As the only ligand identified to-date that interacts with the accessory α2δ subunit, gabapentin provides a unique, alternative candidate for mediating Ca2+ influx to control the development and maintenance of chronic pain. In support of this theory, the differing α2δ binding affinities of stereo-selective analogues of gabapentin correlate well with their structure-activity relationships in animal models of pain (Field et al., 2000). Moreover, α2δ subunits have been shown to be upregulated in dorsal horn tissue obtained from hyperalgesic rats (Philp et al., 1999a,1999b; Luo et al., 2000). Levels of mRNA, protein and [3H]-gabapentin binding sites were all shown to be increased and were accompanied by a corresponding increase in expression of the pore-forming α1B (N-type) subunit (Philp et al., 2000). A finding mirrored in this study, by the observed sensitivity of the N-type current in DRG neurones to inhibition by gabapentin
Nevertheless, the affects of gabapentin are not restricted to a presynaptic α2δ site of action. Postsynaptic affects on spinal synaptic transmission have also been demonstrated, whereby gabapentin has been shown to variably modulate either NMDA- or AMPA-mediated synaptic transmission in freshly dissociated DRG neurones, control slices and in vivo (Rock et al., 1993; Chizh et al., 2000; Shiyoma et al., 2000). Most recently, gabapentin has also been shown to target a specific GABAB receptor heterodimer, promoting activation of a postsynaptic inwardly rectifying K+ (Kir) conductance and altering membrane excitability (Ng et al., 2001). Nevertheless, experiments using saclofen undertaken in this study support previous findings that the actions of gabapentin on Ca2+ channel currents in this preparation are not mediated via a corresponding interaction with the GABAB receptor (Mcclelland et al., 2000). Gabapentin can also gain access to the intracellular cytoplasmic environment by virtue of its ability to substitute as a substrate for the system L amino acid transporter (Stewart et al., 1993; Su et al., 1995). As a consequence, accumulating concentrations of gabapentin could interact with any number of different cytoplasmic proteins and second messenger cascades, indirectly influencing Ca2+ influx and /or other neuronal functions. Indeed, gabapentin has been shown to alter metabolic turnover and promote non-vesicular release of the inhibitory neurotransmitter, GABA (Taylor et al., 1998; Honmou et al., 1995a,1995b; Petroff et al., 1996).
Thus, in summary, the overall mechanism of action for gabapentin may well reflect a combined action mediated by both extra and intracellular targets, which together provide a means for reducing overall neuronal excitability. However, the α2δ subunit of the calcium channel is the only molecular target identified to date that interacts with gabapentin with high affinity. Moreover, as this study has demonstrated, in DRG neurones voltage-gated Ca2+ channel current is sensitive to inhibition by gabapentin, which consequently provides a potential means for gabapentin to effectively mediate spinal anti-nociception.
Acknowledgments
We thank Dr Alison Stacey for tissue culture support and Dr Jason Brown for helpful discussion in the writing of this manuscript. Duncan Martin and Dr Roderick Scott thank Pfizer (formerly Parke-Davis) for financial support.
Abbreviations
- Ca2+
Calcium
- (DRG)
dorsal root ganglia
- (GBP)
gabapentin,1-(aminomethyl) cyclohexane acetic acid (Neurontin®)
- (HVA)
high voltage-activated
- IBa
peak whole-cell calcium channel current carried by barium
- (W50)
duration for 50% of the maximum response
References
- BACKONJA M., BEYDOUN A., EDWARDS K.R., SCHWARTZ S.L., FONESCA V., HES M., LAMOREAUX L., GAROFALO E. Gabapentin for the symptomatic treatment of painful neuropathy in patients with diabetes mellitus. JAMA. 1998;280:1831–1836. doi: 10.1001/jama.280.21.1831. [DOI] [PubMed] [Google Scholar]
- BROWN J.P., GEE N.S. Cloning and deletion mutagenesis of the α2δ calcium channel subunit from porcine cerebral cortex. J. Biol. Chem. 1998;273:25458–25465. doi: 10.1074/jbc.273.39.25458. [DOI] [PubMed] [Google Scholar]
- CARACENI A., ZECCA E., MARTINI C., DE CONNO F. Gabapentin as an adjuvant to opioid analgesia for neuropathic cancer pain. J. Pain. Symp. Manag. 1999;17:441–445. doi: 10.1016/s0885-3924(99)00033-0. [DOI] [PubMed] [Google Scholar]
- CESENA R.M., CALCUTT N.A. Gabapentin prevents hyperalgesia during the formalin test in diabetic rats. Neurosci. Lett. 1999;262:101–104. doi: 10.1016/s0304-3940(99)00057-9. [DOI] [PubMed] [Google Scholar]
- CHENG J-K,. , PAN H-L., EISENACH J.C. Antiallodynic effect of intrathecal gabapentin and its interaction with clonidine in a rat model of postoperative pain. Anesthesiology. 2000;92:1126–1131. doi: 10.1097/00000542-200004000-00031. [DOI] [PubMed] [Google Scholar]
- CHIZH B.A., SCHEEDE M., SCHULTZ H. Antinociception and (R,S)-alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid antagonism by gabapentin in the rat spinal cord in vivo. Naunyn Schmiedebergs Arch. Pharmacol. 2000;362:197–200. doi: 10.1007/s002100000275. [DOI] [PubMed] [Google Scholar]
- DI TRAPANI G., MEI D., MARRA C., MAZZA S., CAPUANO A. Gabapentin in the prophylaxis of migraine: a double-blind randomized placebo-controlled study. Clin. Ter. 2000;151:145–148. [PubMed] [Google Scholar]
- DOLPHIN A.C., WYATT C.N., RICHARDS J., BEATTIE R.E., CRAIG P., LEE J.-H., CRIBBS L.L., VOLSEN S.G., PEREZ-REYES E. The effect of α2-δ and other accessory subunits on expression and properties of the calcium channel α1G. J. Physiol. 1999;519.1:35–45. doi: 10.1111/j.1469-7793.1999.0035o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOOLEY D.J., DONOVAN C.M., PUGSLEY T.A. Stimulus-dependent modulation of [3H]Norepinephrine release from rat neocortical slices by gabapentin and pregabalin. J. Pharmacol. Exp. Ther. 2000;295:1086–1093. [PubMed] [Google Scholar]
- DUNLAP K., LUEBKE J.I., TURNER T.J. Exocytotic Ca2+ channels in mammalian central neurons. TINS. 1995;18:89–98. [PubMed] [Google Scholar]
- FELIX R. Voltage-dependent Ca2+ channel α2δ auxiliary subunit: structure, function and regulation. Receptors and Channels. 1999;6:351–362. [PubMed] [Google Scholar]
- FIELD M.J., HUGHES J., SINGH L. Further evidence for the role of the α2δ subunit of voltage dependent calcium channels in models of neuropathic pain. Br. J. Pharmacol. 2000;131:282–286. doi: 10.1038/sj.bjp.0703604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FIELD M.J., MCCLEARY S., HUGHES J., SINGH L. Gabapentin and pregabalin, but not morphine and amitriptyline, block both static and dynamic components of mechanical allodynia induced by streptozocin in the rat. Pain. 1999;80:391–398. doi: 10.1016/s0304-3959(98)00239-5. [DOI] [PubMed] [Google Scholar]
- FINK K., MEDER W., DOOLEY D.J., GOTHERT M. Inhibition of neuronal Ca2+ influx by gabapentin and subsequent reduction of neurotransmitter release from rat neocortical slices. Br. J. Pharmacol. 2000;130:900–906. doi: 10.1038/sj.bjp.0703380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAO B., SEKIDO Y., MAXMOV A., SAAD M., FORGACS E., LATIF F., WEI M.H., LERMAN M., LEE J.-H., PEREZ-REYES E., BEZPROZVANNY I., MINNA J.D. Functional properties of a new voltage-dependent calcium channel α2δ auxiliary subunit gene (CACNA2D2) J. Biol. Chem. 2000;275:12237–12242. doi: 10.1074/jbc.275.16.12237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GEE N.S., BROWN J.P., DISSANAYAKE V.U. , OFFORD J., THURLOW R., WOODRUFF G.N. The novel anticonvulsant drug, gabapentin (neurontin), binds to the alpha2delta subunit of a calcium channel. J. Biol. Chem. 1996;271:5768–5776. doi: 10.1074/jbc.271.10.5768. [DOI] [PubMed] [Google Scholar]
- GOA K.L., SORKIN E.M. Gabapentin. A review of its pharmacological properties and clinical potential in epilepsy. Drugs. 1993;46:409–427. doi: 10.2165/00003495-199346030-00007. [DOI] [PubMed] [Google Scholar]
- GURNETT C.A., DE WAARD M., CAMPBELL K.P. Dual function of the voltage-dependent Ca2+ channel alpha 2 delta subunit in current stimulation and subunit interaction. Neuron. 1996;16:431–446. doi: 10.1016/s0896-6273(00)80061-6. [DOI] [PubMed] [Google Scholar]
- HONMOU O., KOCSIS D., RICHERSON G.B. Gabapentin potentiates the conductance increase induced by nipecotic acid in CA1 pyramidal neurons in vitro. Epilepsy Res. 1995a;20:193–202. doi: 10.1016/0920-1211(94)00076-9. [DOI] [PubMed] [Google Scholar]
- HONMOU O., OYELESE A.A., KOCSIS D. The anticonvulsant gabapentin enhances promoted release of GABA in hippocampus: a field potential analysis. Brain Res. 1995b;692:273–277. doi: 10.1016/0006-8993(95)00634-3. [DOI] [PubMed] [Google Scholar]
- HOUTCHENS M.K., RICHERT J.R., SAMI A., ROSE J.W. Open label gabapentin treatment for pain in multiple sclerosis. Multiple Sclerosis. 1997;3:250–253. doi: 10.1177/135245859700300407. [DOI] [PubMed] [Google Scholar]
- KLUGBAUER N., LACINOVA L., MARAIS E., HOBOM M., HOFMANN F. Molecular diversity of the calcium channel alpha2delta subunit. J. Neurosci. 1999;19:684–691. doi: 10.1523/JNEUROSCI.19-02-00684.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAIRD M.A., GIDAL B.E. Use of Gabapentin in the treatment of neuropathic pain. Annal. Pharmacother. 2000;34:802–807. doi: 10.1345/aph.19303. [DOI] [PubMed] [Google Scholar]
- LUO Z.D., CHAPLAN S.R., SORKIN L.S., HIGUERA E.S., WEBB M., STAUDERMAN K.A. Retrograde regulation of calcium channel α2δ subunit in dorsal root ganglia of spinal nerve injured rats and its potential roles in neuropathic pain. Soc. Neurosci. 2000;26:352.4. [Google Scholar]
- MAGNUS L. Nonepileptic uses of Gabapentin. Epilepsia. 1999;40 Suppl. 6:S66–S72. doi: 10.1111/j.1528-1157.1999.tb00936.x. [DOI] [PubMed] [Google Scholar]
- MANEUF Y.P., MCKNIGHT A.T. Gabapentin inhibits substance P- and calcitonin gene-related peptide-facilitated K+-evoked release of [3H]-glutamate from rat caudal trigeminal nucleus slices. Soc. Neurosci. 2000;26:722.9. [Google Scholar]
- MARTIN D.J., IBBOTSON T., SCOTT R.H. The inhibitory effects of gabapentin on Ca2+ influx into cultured rat dorsal root ganglion neurones and F11 neuroblastoma cells. J. Physiol. 2000;528P:PC45. [Google Scholar]
- MCCLELLAND D., HERD M.B., MARTIN D.J., SUTTON K.G., LEE K., SCOTT R.H. Comparisons between responses evoked by GABA receptor mechanisms and gabapentin in cultured neonatal rat dorsal root ganglion neurones. J. Physiol. 2000;528P:C53. [Google Scholar]
- MEDER W.P., DOOLEY D.J. Selective modulation of K+-induced synaptosomal calcium influx in discrete mammalian CNS regions by Gabapentin. Soc. Neurosci. 2000;26:234.6. [Google Scholar]
- MINTZ I.M., ADAMS M.E., BEAN B.P. P-type calcium channels in rat central and peripheral neurons. Neuron. 1992;9:85–95. doi: 10.1016/0896-6273(92)90223-z. [DOI] [PubMed] [Google Scholar]
- NG G.Y.K., BERTRAND S., SULLIVAN R., ETHIER N., WANG J., YERGEY J., BELLEY M., TRIMBLE L., BATEMAN K., ALDER L., SMITH A., MCKERNAN R., METTERS K., O NEILL G.P., LACAILLE J-C., HEBERT T.E. γ-aminobutyric acid type B receptors with specific heterodimer composition and postsynaptic actions in hippocampal neurons are targets of anticonvulsant gabapentin action. Mol. Pharmacol. 2001;59:144–152. [PubMed] [Google Scholar]
- NICHOLSON B. Gabapentin use in neuropathic pain syndromes. Acta Neurol. Scand. 2000;101:359–371. doi: 10.1034/j.1600-0404.2000.0006a.x. [DOI] [PubMed] [Google Scholar]
- PATEL M.K., GONZALEZ M.I., BRAMWELL S., PINNOCK R.D., LEE K. Gabapentin inhibits excitatory synaptic transmission in the hyperalgesic spinal cord. Br. J. Pharmacol. 2000;130:1731–1734. doi: 10.1038/sj.bjp.0703530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PETROFF O.A., ROTHMAN D.L., BEHAR K.L., LAMOUREUX D., MATTSON R.H. The effect of gabapentin on brain gamma-aminobutyric acid in patients with epilepsy. Ann. Neurol. 1996;39:95–99. doi: 10.1002/ana.410390114. [DOI] [PubMed] [Google Scholar]
- PHILP L., HOLLOMAN E., FIELD M.J., WILLIAMS R.G.Levels of spinal α2δ protein and mRNA in a rat model of neuropathic pain Soc. Neurosci. Abstr. 1999b25425.7p [Google Scholar]
- PHILP L., HOLLOMAN E., MEECHAM K., BLYTH K., PINNOCK R., HUGHES J., WILLIAMS R. [3H]-Gabapentin binding and α2δ immunoreactivity in the spinal cord of the rat following chronic constriction injury of the sciatic nerve. Br. Neurosci. Assoc. Abstr. 1999a;15:46.08. [Google Scholar]
- PHILP L., HOLLOMAN H., REES H., WILLIAMS R.G. Upregulation of voltage dependent calcium channel subunits in C-fibres in the chronic constriction injury model of neuropathic pain. Soc. Neurosci. 2000;26:351.6. [Google Scholar]
- REICHLING D.B., LEVINE J.D. The primary afferent nociceptor as a pattern generator. Pain. 1999. pp. S103–109. [DOI] [PubMed]
- ROCK D.M., KELLY K.M., MACDONALD R.L. Gabapentin actions on ligand- and voltage-gated responses in cultured rodent neurons. Epilepsy Res. 1993;16:89–98. doi: 10.1016/0920-1211(93)90023-z. [DOI] [PubMed] [Google Scholar]
- ROWBOTHAM M., HARDEN N.N., STACEY B., BERNSTEIN P., MAGNUS-MILLER L. Gabapentin for the treatment of postherpetic neuralgia: a multicentre, double-blind cross-over study. JAMA. 1998;280:1837–1842. doi: 10.1001/jama.280.21.1837. [DOI] [PubMed] [Google Scholar]
- RUSIN K.I., MOISES H.C. μ-opioid receptor activation reduces multiple components of high-threshold calcium current in rat sensory neurons. J. Neurosci. 1995;15:4315–4327. doi: 10.1523/JNEUROSCI.15-06-04315.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SARANTOPOULOS C.D., MCCALLUM J.B., KWOK W.M., CLIFFORD P.S., HOGAN O. Gabapentin decreases membrane voltage-activated calcium currents in injured and intact mammalian DRG neurons. Soc. Neurosci. 2000;26:453.9. [Google Scholar]
- SCHUMACHER T.B., BECK H., STEINHAUSER C., SCHRAMM J., ELGER C.E. Effects of phenytoin, carbamazepine and gabapentin on calcium channels in hippocampal granule cells from patients with temporal lobe epilepsy. Epilepsia. 1998;39:355–363. doi: 10.1111/j.1528-1157.1998.tb01387.x. [DOI] [PubMed] [Google Scholar]
- SHIMOYAMA M., SHIMOYAMA N., HORI Y. Gabapentin affects glutamatergic excitatory neurotransmission in the rat dorsal horn. Pain. 2000;85:405–414. doi: 10.1016/S0304-3959(99)00283-3. [DOI] [PubMed] [Google Scholar]
- STEFANI A., SPADONI F., BERNARDI G. Gabapentin inhibits calcium currents in isolated rat brain neurons. Neuropharmacol. 1998;37:83–91. doi: 10.1016/s0028-3908(97)00189-5. [DOI] [PubMed] [Google Scholar]
- STEFANI A., SPADONI F., GIACOMINI P., LAVARONI F., BERNARDI G. The effects of gabapentin on different ligand- and voltage-gated currents in isolated cortical neurons. Epilepsy Res. 2001;43:239–248. doi: 10.1016/s0920-1211(00)00201-1. [DOI] [PubMed] [Google Scholar]
- STEWART B.H., KUGLER A.R., THOPSON P.R., BOCKBRADER H.N. A saturable transport mechanism in the intestinal absorption of gabapentin is the underlying cause of the lack of proportionality between increasing dose and drug levels in plasma. Pharmaceut. Res. 1993;10:276–281. doi: 10.1023/a:1018951214146. [DOI] [PubMed] [Google Scholar]
- SU T., GONG C.H., HANG J., KOHLER W., DICKERSON M. Human α2δ2 subunit of calcium channel: a novel gabapentin binding protein in brain. Soc. Neurosci. 2000;26:40.20. [Google Scholar]
- SU T., LUNNEY E., CAMPBELL G., OXENDER D.L. Transport of gabapentin, a γ-amino acid drug, by system L α-amino acid transporters: a comparative study in astrocytes, synaptosomes and CHO cells. J. Neurochem. 1995;64:2125–2131. doi: 10.1046/j.1471-4159.1995.64052125.x. [DOI] [PubMed] [Google Scholar]
- SUMAN-CHAUHAN N., WEBDALE L., HILL D.R., WOODRUFF G.N. Characterisation of [3H]-gabapentin binding to a novel site in the rat brain: homogenate binding studies. Eur. J. Pharmacol. 1993;244:293–301. doi: 10.1016/0922-4106(93)90155-3. [DOI] [PubMed] [Google Scholar]
- SUTTON K.G., SCOTT R.H., LEE K., PINNOCK R.D. Gabapentin inhibits high threshold calcium channel currents in cultured rat dorsal root ganglia neurones. Soc. Neurosci. Abstr. 2000;26:234.4. doi: 10.1038/sj.bjp.0704439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAYLOR C.P., GEE N.S., SU T.-Z., KOCSIS J.D., WELTY D.F., BROWN J.P., DOOLEY D.J., BODEN P., SINGH L. A summary of mechanistic hypotheses of gabapentin pharmacology. Epilepsy Res. 1998;29:233–249. doi: 10.1016/s0920-1211(97)00084-3. [DOI] [PubMed] [Google Scholar]
- VANEGAS H., SCHAIBLE H-G. Effects of antagonists to high-threshold calcium channels upon spinal mechanismsm of pain, hyperalgesia and allodynia. Pain. 2000;85:9–18. doi: 10.1016/s0304-3959(99)00241-9. [DOI] [PubMed] [Google Scholar]
- WALKER D., DE WAARD M. Subunit interaction sites in voltage-dependent Ca2+ channels: role in channel function. TINS. 1998;21:148–154. doi: 10.1016/s0166-2236(97)01200-9. [DOI] [PubMed] [Google Scholar]