

Abstract
The role of smooth muscle-derived lipoprotein lipase (LPL) that translocates to the endothelium surface on vascular dysfunction during atherogenesis is unclear. Thus, the role of vascular LPL on blood vessel reactivity was assessed in transgenic mice that specifically express human LPL in the circulatory system.
Aortic free fatty acids (FFAs) were increased by 69% in the transgenic mice expressing human LPL in aortic smooth muscle cells (L2LPL) compared with their non-transgenic littermates (L2).
Contractility to KCl was increased by 33% in aortae of L2LPL mice. Maximal contraction to phenylephrine (PE) was comparable in L2 and L2LPL animals, while the frequency of tonus oscillation to PE increased by 104% in L2LPL mice.
In L2LPL animals, •NO mediated relaxation to acetylcholine (ACh) and ATP was reduced by 47 and 32%, respectively. In contrast, endothelium-independent relaxation to sodium nitroprusside (SNP) was not different in both groups tested.
ATP-initiated Ca2+ elevation that triggers •NO formation was increased by 41% in single aortic endothelial cells freshly isolated from L2LPL animals.
In aortae from L2LPL mice an increased •O2− release occurred that was normalized by removing the endothelium and by the NAD(P)H oxidase inhibitor DPI and the PKC inhibitor GF109203X.
The reduced ACh-induced relaxation in L2LPL animals was normalized in the presence of SOD, indicating that the reduced relaxation is due, at least in part, to enhanced •NO scavenging by •O2−.
These data suggest that despite normal lipoprotein levels increased LPL-mediated FFAs loading initiates vascular dysfunction via PKC-mediated activation of endothelial NAD(P)H oxidase. Thus, vascular LPL activity might represent a primary risk factor for atherosclerosis independently from cholesterol/LDL levels.
Keywords: Atherosclerosis, contraction, endothelium-dependent relaxation, free fatty acids, NADH/NADPH oxidase, protein kinase, endothelial Ca2+, superoxide anions
Introduction
Lipoprotein lipase (LPL) is the major enzyme responsible for the hydrolysis of triglycerides present in circulating lipoproteins and critical for the generation of HDL particles. In the vascular wall LPL is produced in smooth muscle cells (Ylä-herttuala et al., 1989). Following synthesis the enzyme translocates to the endothelial surface (Goldberg, 1996; Olivecrona & Olivecrona, 1995). Although LPL is generally viewed as an antiatherogenic enzyme due to its effect to improve the plasma lipid profile, convincing experimental evidence points to a role of LPL in atherogenesis. The view of a pro-atherogenic role of the LPL was initiated by the early findings of Zilversmit who reported that large amount of LPL activity was found in atherosclerotic vessels (Zilversmit, 1979). In line with these findings, expression of LPL was also found in macrophages of atherosclerotic lesions (Mattsson et al., 1993; Ylä-herttuala et al., 1989) which suggest that the enzyme might be responsible for lipoprotein uptake into macrophages (Ylä-herttuala et al., 1991) that leads to foam cell formation, an important step in the atherosclerotic plaque formation (Olivecrona & Olivecrona, 1995; Zechner, 1997). Recently, experiments with genetically modified macrophages have clearly demonstrated the proatherogenic potential of macrophage LPL (Babaev et al., 1999; Mead et al., 1999).
In contrast to the large number of reports on the role of macrophage LPL in atherogenesis, the contribution of smooth muscle-derived LPL to the development of atherosclerosis remains unclear. Along this line it is important to note that the response-to-injury hypothesis originally proposed by Ross (1986) suggests that the initial step in atherogenesis involves the development of an endothelial/vascular dysfunction that, in turn, facilitates lipid oxidation and trans-endothelial macrophage movements (Ross, 1986). Since hyperlipoproteinaemia is one of the main causes of atherosclerosis one might expect smooth muscle-derived LPL to play a central role in the initial phase of atherogenesis by facilitating an increased uptake of free fatty acids (FFAs) by the hydrolysis of lipoprotein associated triglycerides. In line with this hypothesis, FFAs were found to impair endothelial cell function and endothelium-dependent relaxation (Endresen et al., 1994; Niu et al., 1995; Vossen et al., 1991). This effect on endothelium-dependent relaxation is independent of the length of the FFA (de kreutzenberg et al., 2000). On the other hand, the ω-3 fatty acids docosahexaenoate, eicosapentaenoate and oleic acid were shown to possess anti-atherogenic properties, due to their inhibitory action on cytokine-/lipopolysaccharide-initiated endothelial cell activation (Carluccio et al., 1999; De Caterina et al., 1994; 1995). In light of the oppositional effects of FFAs on the endothelial cell function, it is important to investigate how an increased FFA uptake into the vascular wall that is mediated by smooth muscle-derived LPL affects vascular function in vivo. Thus, in this study, the role of vascular smooth muscle-derived LPL on blood vessel reactivity as an indicator for early vascular changes that facilitates atherogenesis was explored in transgenic mice that specifically express human LPL in the heart and in vascular smooth muscle cells (Levak-Frank et al., 1999).
Methods
Animals and preparation of mouse aortae
In this study, L2LPL mice that were recently developed by Levak-Frank et al. (1999) were compared with their non-transgenic littermates (L2 mice). Briefly, by fusing a human LPL minigene to 8 kb of the 5′-regulatory sequences of the mouse LPL gene, animals were obtained that express human LPL in cardiac muscle and vascular smooth muscle cells but not in adipose tissue or skeletal muscle (Levak-Frank et al., 1999). If not otherwise stated male and female mice aged 32 to 52 weeks were used for this study. Animals weighing between 20 to 35 g were sacrificed by cervical dislocation. The aortae were immediately removed, carefully freed from adherent tissue and placed into Dulbecco's minimal essential medium (DMEM) and were used for experiments within 30 min of isolation. While wall thickness in each group did not differ, nor vessels with visible plaques were used, in L2LPL mice there was considerably more adipose tissue around the abdominal part of the aorta compared with the non-transgenic littermates.
Reverse-transcriptase-polymerase chain reaction
Total RNAs from mouse aortae were isolated by using RNeasy kit (Qiagen, Vienna, Austria). One microgram of total RNA was treated with RNase-free DNase I (Promega, Mannheim, Germany) and subsequently used as a template for first strand cDNA synthesis in a 30 μl reaction as described in detail elsewhere (Barth et al., 2000). Briefly, the reaction mix contained 0.5 mM dNTPs (Pharmacia Biotech Inc., Vienna, Austria), 15 units of RNAguard (Pharmacia Biotech Inc.), 3.3 μM random hexamer primers (Pharmacia Biotech Inc.), 10 mM DTT (Life Technologies), 1× First Strand Buffer (Life Technologies Inc., Vienna, Austria) and 200 units Moloney murine leukaemia virus reverse transcriptase (Life Technologies Inc.) Reactions were incubated for 1 h at 37°C, heated to 75°C for 10 min and 2.5 μl of the completed reactions were used as template for PCR.
For mouse LPL (mLPL), 50-μl PCR reactions contained 0.2 mM dNTPs, appropriate oligonucleotide primers at 10 μM, 1× PCR buffer (Finnzymes Oy, Vienna, Austria) and 1 unit of DyNAzyme II DNA polymerase (Finnzymes Oy). Reaction mix was heated at 94°C for 4 min, and subsequently amplification was carried out for 35 cycles (30 s at 94°C; 30 s at 58°C; 1 min at 72°C) using the following primers: mLPLf 5′-GCCCTAAGGACCCC TGAAGACACA-3′ (nucleotides 323 – 346) and mLPLr 5′-CAAACCAGTAATTCT ATTGA CC-3′ (nucleotides 724 – 745) (MWG Biotech, Ebersberg, Germany). Primer sequence were extracted from GenBank (reference number XM_005191). For human LPL (hLPL), 25-μl PCR reactions contained 0.4 mM dNTPs, appropriate oligonucleotide primers at 10 μM, 1× PCR buffer (Finnzymes Oy, Vienna, Austria), 1 mM MgCl2, 4% DMSO and 0.5 unit of DyNAzyme II DNA polymerase (Finnzymes Oy, Vienna, Austria). Reaction mix was heated at 94°C for 2 min, and subsequently amplification was carried out for 20 cycles (30 s at 94°C, 60 s at 68°C−0.5°C/cycle, 60 s at 72°C) and 10 cycles (30 s at 94°C, 60 s at 58°C, 60 s at 72°C) using the following primers: hLPLf 5′-TGC CCA CTT CTA GCT GCC CT-3′ (nucleotides 29 – 48) and hLPLr 5′-CAC ACG GCC AGA GTC AGC AC-3′ (nucleotides 196 – 215) (Life Technologies Inc, Vienna, Austria).
Free fatty acid composition analysis
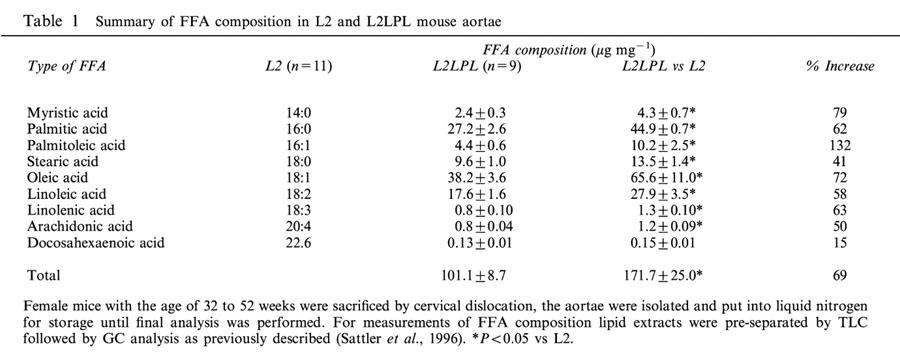
Free fatty acid (FFA) composition analysis was carried out in lipid extracts of aortic segments of the L2 and the L2LPL mice using TLC followed by GC analysis of fatty acid methyl ester as previously described (Sattler et al., 1996). Concentrations of the individual free fatty acids were calculated by peak-area comparison with the internal standard (heptadecanoic acid) and were normalized to μg FFA per mg tissue (w w).
Lipid analysis
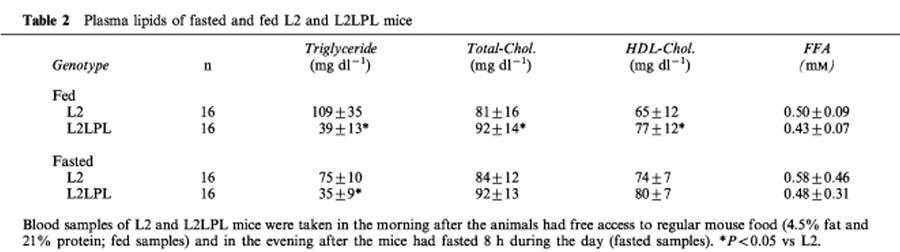
Control and transgenic animals were fed regular mouse chow (4.5% fat and 21% protein). Blood was taken in the morning after the animals had free access to food (fed samples) and in the evening after they had fasted 8 h during the day (fasted samples). Plasma levels of triglyceride, total cholesterol, HDL-cholesterol and FFA were determined with commercially available kits.
Immunoblotting
Standard immunoblotting procedures were used. Tissues were put in lysis buffer containing 2-mercaptoethanol and kept for 10 min at 95°C. Following electrophoretic transfer nitrocellulose sheets were blocked by 5% nonfat dried milk in phosphate-buffered saline containing 0.05% Tween-20 and incubated overnight at 4°C with the antibody for either endothelial nitric oxide synthase (eNOS) or inducible nitric oxide synthase (iNOS) followed by a peroxidase-coupled goat anti-rabbit IgG and immunoreactive bands were detected by enhanced chemiluminescence (ECL, Amersham Life Science Ltd., Vienna, Austria).
Organ bath studies
Organ bath experiments were performed as previously described (Fleischhacker et al., 1999; 2000). Two 2-mm segments of the thoracic aorta were taken and mounted separately in a two chamber tissue bath containing 5 ml Krebs solution (KHS; in mM): NaCl 118.4, KCl 4.7, KH2PO4 1.2, MgCl2 1.2, NaHCO3 11.9, CaCl2 2.5 and D-glucose 10.1, pH=7.4. This physiological salt solution was bubbled continuously with a mixture of 95% O2 and 5% CO2 at 37°C. For recordings of the mechanical activity, the aortic segments were mounted on a isometric transducer connected to a two channel interface (Myograph 410A) and a two pen recorder. Prior to each experiment an initial tension of 2 g was applied for calibration followed by an equilibration time of 90 to 120 min. Blood vessel contraction was initiated by KCl performing a cumulative-response curve in the range of 10 to 40 mM. After the highest KCl concentration was washed out the vessels rested for a period of 30 to 40 min, followed by a cumulative concentration-response curve for PE in the range of 10−9 to 10−4 M. For testing endothelium-dependent relaxation the vessels were precontracted twice with 1 μM PE in the presence of 10 μM indomethacin. The stable tension observed in the second contraction (approximately after 20 min equilibration time) was taken as the maximum contraction. To achieve endothelium-dependent relaxation, ACh and ATP were added cumulatively in a range of 10−9 to 3×10−7 and 10−9 to 2×10−8 M, respectively. To verify endothelial dependency of the observed relaxation, endothelium denuded vessels were used. To remove the endothelium, air was blown through the intact vessel followed with an insertion of a metal probe into the lumen. The absence of endothelium was confirmed by the lack of relaxation in response to 1 μM A23187.
Cell isolation
For endothelial cell isolation (Graier et al., 1998b), the thoracic and abdominal aorta (approximately 1.5 to 2 cm) was filled for 2 h with DMEM (pH=7.4) containing collagenase (200 u ml−1; type II) plus dilutions of essential and non-essential amino acids (0.02 v v−1; Gibco BRL, Life Technologies, Vienna, Austria), vitamins (0.01 v v−1; Gibco BRL, Life Technologies, Vienna, Austria), donor horse serum (5%) and bovine serum albumin (2 mg ml−1) as described previously (Fleischhacker et al., 1999). The isolated cells were stored in DMEM containing dilutions of essential and non-essential amino acids (0.02 v v−1; Gibco BRL, Life Technologies, Vienna, Austria), vitamins (0.01 v v−1; Gibco BRL, Life Technologies, Vienna, Austria), donor horse serum (5%) and bovine serum albumin (2 mg ml−1). Cells were used for the experiments within 4 h. To ensure that endothelial cells are tested but not smooth muscle cells, single endothelial cells were selected under the microscope by the cell shape. Moreover, the lack of smooth muscle/fibroblast L-type Ca2+ channels was tested by incubation with 20 mM KCl containing solution prior to each experiment.
Ca2+ measurements
Intracellular Ca2+ concentrations were examined in single endothelial cells freshly isolated from mouse aortae using the microfluorometric setup described previously (Graier et al., 1995). After cell isolation as described above, the cells were loaded with fura-2 by incubating the cells in DMEM containing 2 μM fura-2/am. After 45 min at room temperature, cells were centrifuged, washed twice and resuspended in DMEM. Prior to the experiments, cells were equilibrated for 20 min in the dark at room temperature. For experiments, cells were transferred into an experiment chamber and the DMEM was replaced by KHS by constant superfusion (1 ml min−1). Maximal fluorescence intensities of fura-2 to control differences in dye accumulation/distribution were measured after each experiment by the addition of 3 μmol l−1 ionomycin in the presence of 2.5 mM extracellular Ca2+. In addition, minimal fluorescence and autofluorescence were monitored in the presence of 3 μM ionomycin in nominal Ca2+-free solution containing either 1 mM EGTA or Mn2+. Dye distribution was monitored using digital confocal microscopy as described previously (Fleischhacker et al., 1999).
Data acquisition
Fura-2: Single cell Ca2+ was recorded using a microfluorometer (Sturek et al., 1991) which excited with 360 and 380 nm alternatively. Emission light was detected at 510 nm using photon counting technique. Fluorescence intensity for each pair of excitation/emission wavelength was converted to analogue by an optical processor and registered by a PC running AxoBASIC®1.0 (Axon Instruments, Foster City, CA, U.S.A.)
Determination of superoxide anion release
The release of O2− was determined photometrically by measuring the SOD-sensitive reduction of ferricytochrome c as previously described (Graier et al., 1996). After the wet weight of the aorta with or without endothelium was determined, the aortae were transferred into 1.5 ml phosphate buffered solution (PBS; in mM): NaCl 137, KCl 2.7, Na2HPO4 8, KH2PO4 1.5 and EGTA 0.1; pH adjusted at 7.4 plus 10 μM ferricytochrome c (horse heart type III). After 2 min, 1 ml PBS was aspirated and placed into a photometer, the reduction of ferricytochrome c was followed photometrically at 550 nm (Et1) and the solution was readded to the aorta. This procedure was repeated every 10 min for 1 h (Et2, … Et6). After the fourth reading (i.e. Et4) 500 u ml−1 superoxide dismutase (SOD) were added to ensure that the observed reduction of ferricytochrome c was due to O2−, and this reduction was monitored for an additional 20 min. The differences in absorption between Et1 and Et2, Et3 and Et4 were calculated and normalized to 10 min. From the mean increase in the extinction/10 min the mean extinction increase/min in the presence of SOD was subtracted to calculate the SOD-sensitive reduction of ferricytochrome c. The concentration of O2− was calculated using the molar extinction coefficient (e=21.000) of the reduced ferricytochrome c (Steinbrecher, 1988).
Drugs and chemicals
Cell culture chemicals were obtained from Life Technologies, Vienna, Austria and foetal calf serum (premium quality) and donor horse serum were from PAA Laboratories, Linz, Austria. Petri dishes and cell culture plastic wear were from Corning, Vienna, Austria. Fura-2/am was purchased from Molecular Probes, Leiden. GF109203X was from Tocris, Bristol, U.K. Buffer salts were from Merck, Vienna, Austria. All other materials were from Sigma, Vienna, Austria.
Statistics
The given ‘n' values express the number of mice, whose aortae and freshly isolated endothelial cells were tested in quadruplicate (organ bath) and quintuplicate to nonaduplicate (Ca2+ experiments). EC50 values were calculated of at least five single concentration response curves from different mice and were expressed as the mean with 95% confidential intervals in parenthesis. All other data represent the means±s.e.mean. Analysis of variance (ANOVA) was used for data evaluation including post hoc Tukey test. Differences were considered to be statistical significant at P<0.05.
Results
Expression of human lipoprotein lipase and mouse lipoprotein lipase in mouse aortae
RT – PCR with total RNA from mouse aortae of L2 and L2LPL animals were performed using primers specific for human LPL and mouse LPL mRNA (Figure 1). Only in the aortae of L2LPL animals human LPL was detectable (Figure 1A). Expression of mouse LPL (Figure 1B) did not differ between L2 and L2LPL animals.
Figure 1.
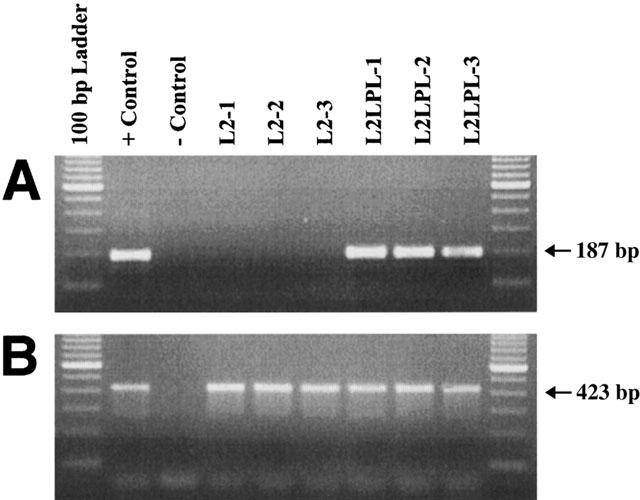
RT – PCR showing expression of human LPL (hLPL; A) and mouse LPL (mLPL: B) in aortas of control (L2) and transgenic mice (L2-LPL2). RT – PCR with total RNA from mouse aortas was performed using primers specific for hLPL and mLPL mRNA as described in Methods. Total RNA from the cardiac muscle of L2-LPL mice was used as a positive control for both hLPL and mLPL. PCR products were separated on a 2% agarose gel. The positions of 423 bp mLPL- and 187 bp hLPL-amplification products are indicated on the right.
FFA composition
Northern blot analysis of mRNA isolated from aortae obtained from control and transgenic animals revealed the expression of human LPL (3.6 kb) (Levak-Frank et al., 1999). As a consequence of the expression of the human LPL in the aorta the overall FFA content increased by 69% compared with the non-transgenic littermates (Table 1). A detailed analysis of single FFA indicated the largest increase in myristic (14 : 0), palmitoleic (16 : 1) and oleic (18 : 1) acids.
Table 1.
Summary of FFA composition in L2 and L2LPL mouse aortae
Lipid analysis
In L2LPL mice, the plasma triglyceride levels were reduced by 64 and 53% in fed and fasted animals, respectively (Table 2). In contrast, plasma levels of total cholesterol, HDL-cholesterol and FFA were slightly decreased in the L2LPL animals compared with their non-transgenic littermates (Table 2).
Table 2.
Plasma lipids of fasted and fed L2 and L2LPL mice
Vessel contractility
Blood vessel reactivity of mouse aortae was tested by K+-induced membrane depolarization and the inositol 1,4,5-trisphosphate (IP3) generating α1-receptor agonist phenylephrine. In L2LPL mice (n=8) the stable, non-oscillatory contraction in response to various KCl concentration was increased by 33% compared to that observed in the non-transgenic littermates (L2, n=8, P<0.05; Figure 2). In contrast, the absolute aorta contraction in response to phenylephrine (PE) was not altered significantly in the L2LPL animals (Figure 3A; L2: EC50=9.3 (3.2 – 26.8)×10−8 M, n=9; L2LPL: EC50=15.3 (4.53 – 51.6)×10−8 M, n=9). However, the kinetics of tonus development of the PE-mediated oscillatory contraction differed between aortae from L2LPL and L2 mice indicated by an increase in the frequency by 104% (Figure 3B; P<0.05) and a decrease in oscillation amplitude of the changes in tension during oscillation by 23% (data not shown; P<0.05). The oscillatory tonus evoked by PE was not affected by endothelium removal or L-nitroarginine.
Figure 2.
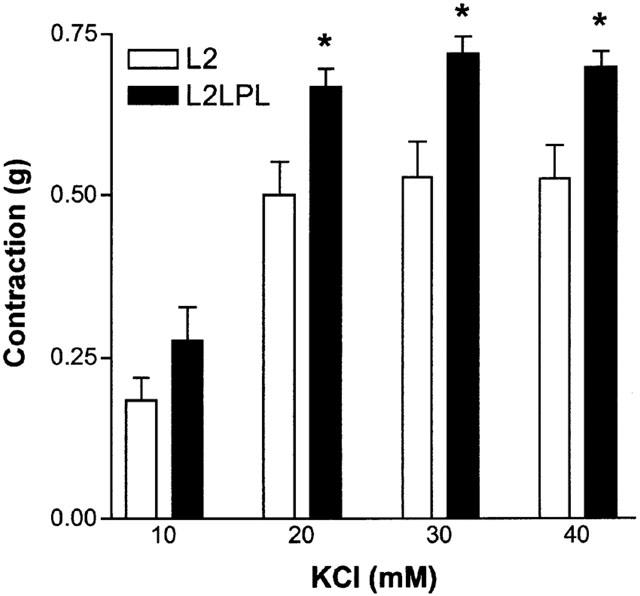
Effect of a tissue-specific expression of human LPL in the mice circulatory system on KCl-evoked contractile responses of mouse aortae. The L2 (n=8) and the L2LPL mice (n=8) were sacrificed by cervical dislocation and their aortae removed and immediately preserved in DMEM. The contractile responses to KCl were measured isometrically using the myograph technique for investigating resistant vessels. After mounting all vessels were allowed to equilibrate for 90 min in the organ bath containing KHS that was constantly bubbled with 95% O2 and 5% CO2 at 37°C. All contractile responses were calculated in ‘g'. Each column represents the mean±s.e.mean. *P<0.05 vs the response obtained in aortae of the L2 animals.
Figure 3.
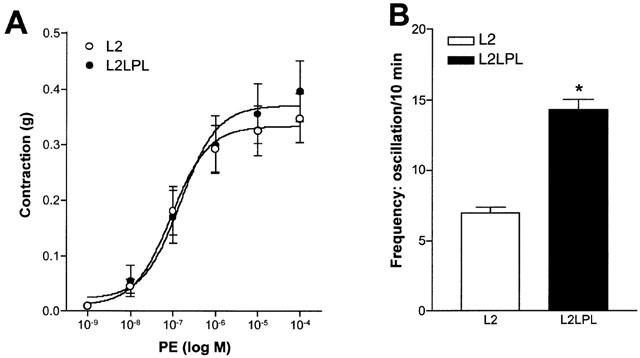
Effect of the α1 receptor agonist phenylephrine (PE) on blood vessel tone of aortae that were freshly isolated from L2 and L2LPL mice. (A) Cumulative concentration response curve for PE was commenced. If oscillatory tone occurred, the mean tension was taken. Curve fitting and the statistical evaluation was preformed using Prizm GraphPad. All contractile responses were calculated in ‘g'. Each point represents the mean±s.e.mean (n=9 in each group). (B) Analysis of the frequency in tone oscillation in response to 1 μM PE in aortae from L2 (n=9) and L2LPL (n=9) mice. Each point represents the mean±s.e.mean. *P<0.05 vs oscillations in tone obtained in aortae from the L2 animals.
Endothelium-dependent relaxation
Maximal relaxation in response to ACh was reduced by 47% in female L2LPL mice in comparison to the L2 animals. The EC50 of ACh was increased from 1.07 (0.2 – 6.0) in the L2 (n=8) to 14.2 (4.1 – 49.1) nM in the L2LPL (n=8; P<0.05; Figure 4A). In male mice, maximal ACh-induced relaxation was reduced in the L2LPL animals by 48% compared with male L2 animals (Figure 4B). ACh-evoked relaxation in male and female mice were very similar and no significant differences between the female and male L2LPL could be observed.
Figure 4.
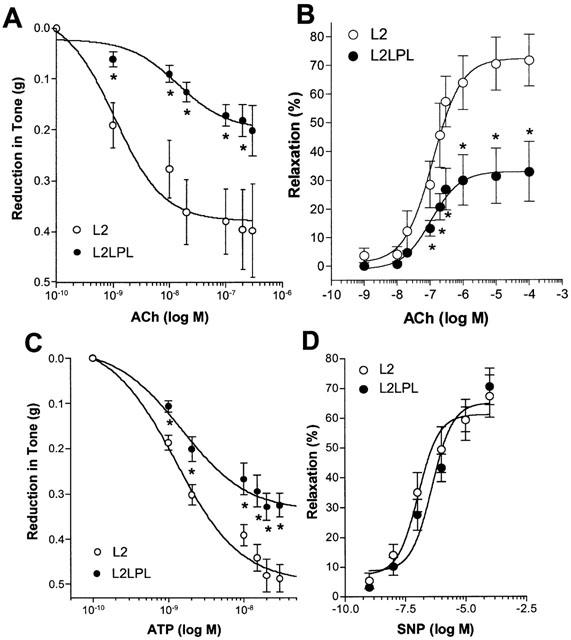
Endothelium-dependent relaxation in response to acetylcholine (ACh) in female (A) and male mice (B). In addition, endothelium-dependent relaxation to ATP (C) and endothelium-independent relaxation to SNP (D) were tested in aortae from female L2 and L2LPL mice. In the presence of 10 μM indomethacin the tissue was precontracted with 1 μM PE to achieve a stable contraction. A cumulative concentration response curve was conducted using ACh (female: n=8 each group; male: L2, n=6 and L2LPL, n=7), ATP (n=14 each group) and SNP (L2, n=9; L2LPL, n=7). Blood vessel relaxation is indicated as the reduction in tone in ‘g' achieved by the concentration of the agonist indicated (A,C) and as % relaxation (B,D). Each point represents the mean±s.e.mean. *P<0.05 vs relaxation obtained in aortae of the non-transgenic littermates (L2).
In agreement with these findings on ACh-induced relaxation, the relaxation to ATP was reduced by 32% (Figure 4C; P<0.05). In contrast to our results with ACh, the EC50 values for ATP to initiate relaxation did not differ in both mouse types (L2: 1.2 (0.7 – 2.2) nM (n=14); L2LPL: 1.5 (0.9 – 2.4) nM (n=14). Notable, neither ACh nor ATP initiated relaxation in endothelium-denuded aortic segments or in the presence of the eNOS inhibitor L-nitroarginine (30 μM data not shown), thus indicating that the autacoid-evoked relaxation is due to endothelial •NO formation.
Endothelium-independent relaxation
In contrast to the reduced endothelium-dependent relaxation in response to ACh and ATP, endothelium-independent relaxation evoked by SNP did not differ between the L2 and the L2LPL2 (Figure 4D).
Autacoid-stimulated Ca2+ signalling
Endothelial Ca2+ signalling in response to 10 μM ATP was measured in single endothelial cells freshly isolated from mouse aortae. As shown in Figure 5, endothelial Ca2+ signalling was increased by 41% in cells isolated from aortae of L2LPL mice (n=16) compared with that from the L2 mice (n=8). In contrast, basal Ca2+ levels did not differ between endothelial cells from the L2 and the L2LPL mice.
Figure 5.
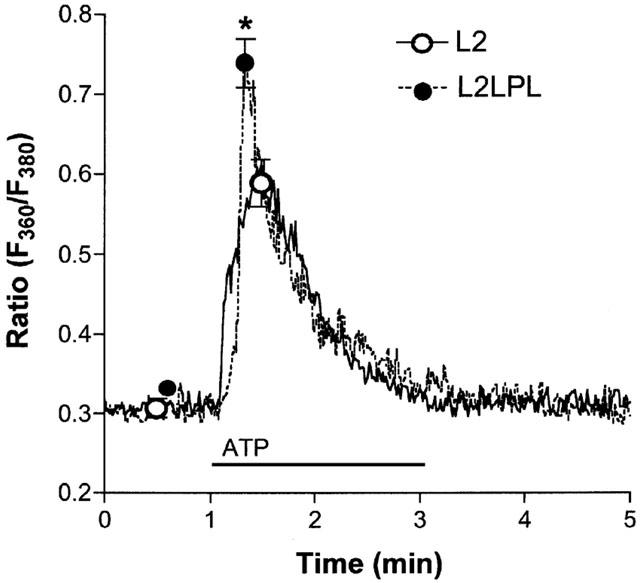
ATP-initiated Ca2+ signalling in single endothelial cells freshly isolated from aortae of L2 and L2LPL mice. Cells were isolated by enzymatic digestion, loaded with fura-2 and resuspended in KHS containing 2.5 mM CaCl2. In a microflorometer intracellular Ca2+ concentration in single endothelial cells was monitored as the ratio of 360 and 380 nm excitation at 510 nm emission (Ratio F360/F380). As indicated, 10 μM ATP was added to the superfusion. Tracings show representative experiments and points indicate the mean±s.e.mean (L2: n=8; L2LPL: n=16). *P<0.05 vs the effect of ATP in cells from L2 mice.
Superoxide anion release
Tissue-specific expression of human LPL in the circulatory system of the mouse resulted in increased release of superoxide anions (•O2−) in the aortic segments. As shown in Figure 6A, •O2− release from the intact aortae of L2LPL animals (n=24) was increased by 21% (P<0.05) compared with that from L2 mice (n=20). In contrast, in endothelium-denuded aortae no further difference in •O2− release was observed (L2: n=24; L2LPL: n=20; Figure 6B). If the differences of the •O2− release of intact and denuded aortae is calculated to represent the amount of •O2− released by the endothelium, in L2LPL aortae an elevation of •O2− release by 79% (P<0.05) was found compared with the non-transgenic littermates (L2; Figure 6C). In the presence of the NAD(P)H oxidase inhibitor diphenylene iodonium (DPI, 10 μM) aortic •O2− release was reduced compared with respective solvent control (DMSO) in the L2 and L2LPL mice by 34 and 48%, respectively (Figure 7A). No further difference in aortic •O2− release appeared between the L2 and the L2LPL mice in the presence of DPI. Preincubation of mouse aortae with the protein kinase C (PKC) inhibitor GF109203X (1 μM) for 30 min had no effect on •O2− release from aortae of L2 animals, while inhibition of PKC normalized •O2− release in L2LPL animals (Figure 7A). In order to test changes in the expression of iNOS in the L2LPL transgenic animals, Western blot analysis for iNOS were performed in aortae isolated from L2 and L2LPL animals. As shown in Figure 7B, no induction of iNOS could be observed in the L2LPL animals. In agreement with these findings, the eNOS and iNOS inhibitor L-nitroarginine (30 μM) had no effect on •O2− release in L2 and L2LPL mice (data not shown).
Figure 6.
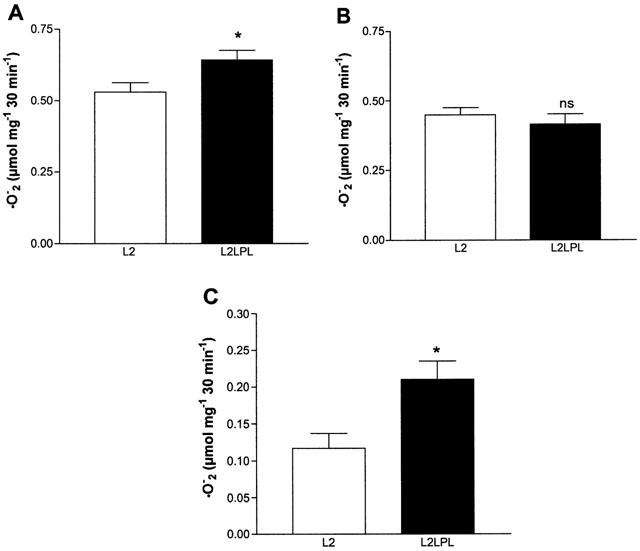
Release of superoxide anions (•O2−) from aortae freshly isolated from L2 and L2LPL mice. Release of the •O2− was measured as the SOD-sensitive reduction of ferricytochrome c that was photometrically monitored at 550 nm as described under Methods. The release of •O2− from intact mouse aortae (A) and endothelium denuded vessels (B) was measured over 30 min and is indicated as the μmol •O2− produced within 10 min per mg wet weight (w w−1). Each column represent the mean±s.e.mean (L2: n=24; L2LPL: n=20). (C) Release of •O2− from the endothelium in aortae from L2 and L2LPL mice calculated as the difference in •O2− release in intact and endothelium-denuded vessels. *P<0.05 vs results obtained in aortae of L2 animals.
Figure 7.
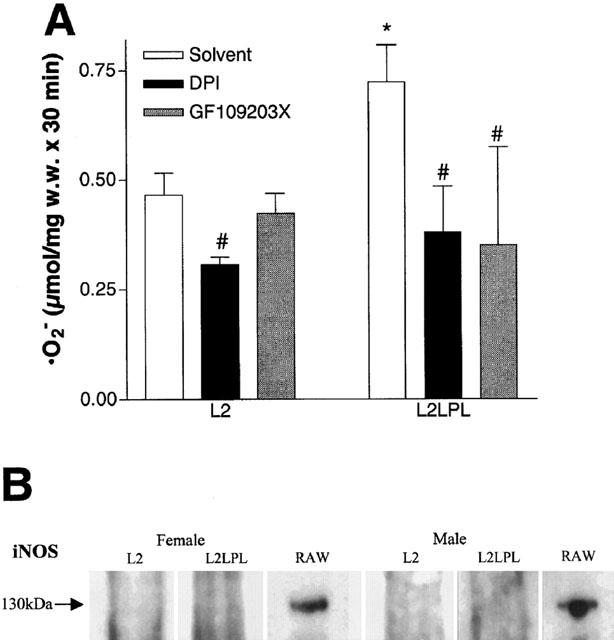
(A) Release of superoxide anions (•O2−) from aortae freshly isolated from L2 and L2LPL mice in the absence (solvent: 1% DMSO) or presence of 10 μM diphenylen iodonium (DPI) or after preincubation for 30 min with 1 μM GF109203X. Release of the •O2− was measured as the SOD-sensitive reduction of ferricytochrome c that was photometrically monitored at 550 nm. Columns represent the means±s.e.mean (L2: n=8; L2LPL: n=8). *P<0.05 vs results obtained in aortae of L2 animals and #P>0.05 vs the respective solvent control. (B) Western blot analysis on the expression of iNOS in aortae of female and male L2 and L2LPL mice. As a positive control, the cell lysate of the mouse macrophage cell line RAW 264.7 stimulated for 12 h IFNγ (10 ng ml−1) and LPS (1 μg ml−1) was used (BD Transduction Laboratories, Life Science Research, Vienna, Austria).
In the presence of 300 u ml−1 SOD, endothelial-dependent relaxation in response to 100 nM ACh was increased in the L2 and the L2LPL animals as well (Figure 8). Nevertheless, the effect of SOD to increase vasodilation was much more pronounced in aortae of the L2LPL animals (+164%; n=5) compared with the effect found in aortae of the non-transgenic littermates (+27%; n=5). In the presence of SOD no further difference in ACh-induced relaxation in aortae of L2 and L2LPL animals was found.
Figure 8.
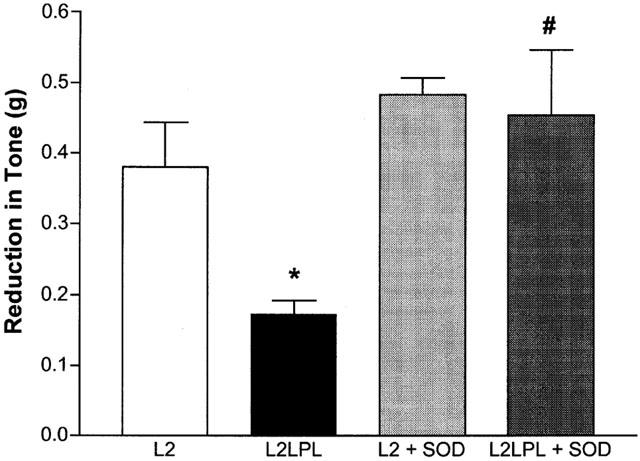
Effect of SOD on ACh-evoked endothelium-dependent relaxation in aortae isolated from L2 and L2LPL mice. Blood vessels were precontracted in the presence of 10 μM indomethacin with 5 μM PE. After reaching a stable tone, relaxation was initiated in L2 (n=5) and L2LPL (n=5) by 100 nM ACh in the absence and in the presence (+SOD) of 300 u ml−1 SOD. Each column represents the mean±s.e.mean. *P<0.05 vs relaxation in aortae of the L2 mice in the absence of SOD; #P<0.05 vs relaxation in aortae of L2LPL animals in the absence of SOD.
Discussion
The risk of hyperlipoproteinaemia for vascular dysfunction and atherosclerosis is evident from numerous experimental and clinical studies. While the role of macrophage-derived LPL in atherogenesis has been clearly highlighted to be responsible for macrophage lipoprotein uptake that can lead to the formation of foam cells, a role of vascular smooth muscle-derived LPL in atherogenesis and the development of vascular dysfunction has not been studied so far. During the present study we have used a transgenic mouse model expressing human LPL in a tissue specific manner (Levak-Frank et al., 1999). This unique model enables the selective expression of human LPL in cardiac myocytes and the vascular system but not in adipose tissue and skeletal muscles by expressing human LPL under the control of the mouse LPL promoter by fusing an 8 kb DNA fragment 5′-upstream of the transcriptional start site of the mouse LPL gene with a human LPL minigene (Levak-Frank et al., 1995; 1997). The high and moderate expression of human LPL in the cardiac muscle and vascular smooth muscles was associated with decreased fasting triglycerides and slightly increased HDL-cholesterol levels. Moreover, the fatty acid composition of the cardiac myocyte did not differ from that of the non-transgenic littermates (L2), indicating that in cardiac myocytes the absorbed FFAs are immediately used for energy consumption (Levak-Frank et al., 1999). In the present study we show that in the aortic tissue of the human LPL-transgenic animals (L2LPL) expression of human LPL occurs that does not affect expression of the mouse LPL. This overall increase in LPL protein content might account for the increased overall FFA content in the vascular wall of the human LPL-transgenic animals (L2LPL) compared with that found in the L2 mice. These results emphasize that smooth muscle cells, like the cardiac myocytes absorb FFAs that are generated by the hydrolysis of triglycerides by LPL bound at the endothelial cells. However, in contrast to the cardiac myocytes, the FFAs are, at least in part, stored within the vascular wall.
The effect of FFAs on endothelial cells has been studied frequently. Particular dietary supplementation with docosahexaenoic acid (DHA; 22 : 6) was found to provide atheroprotective effects such as a reduction in adhesion molecule expression (De-Caterina et al., 2000), increase in prostacyclin formation (De-Caterina et al., 2000), attenuation of prostaglandin H synthase (Lagrade et al., 1980) and augmentation of endothelium-dependent relaxation in pig coronary arteries (Shimokawa et al., 1988). However, the increase in aortic FFAs in human LPL transgenic mice (L2LPL) was least pronounced for DHA (15% increase compared with the L2 animals), while (on a quantitative basis) the largest increases were found for 16 : 0, 18 : 1 and 18 : 2. These data indicate that despite the unchanged levels of lipoproteins (Levak-Frank et al., 1999), expression of human LPL resulted in considerable accumulation of FFAs in the aortic tissue. In contrast to the beneficial effect of polyunsaturated FFAs, saturated and mono unsaturated FFAs were found to initiate vascular dysfunction and to contribute to atherogenesis (Egan et al., 1999).
In line with these findings, the data presented herein indicate clear changes in vascular function as suggested to appear in the initial steps of atherogenesis: an enhanced contractile responsiveness of the vascular smooth muscle cells (Cox et al., 1996; Dam et al., 1997; Kim et al., 1994; Lamping, 1997; Pfister et al., 1996), that is associated with a reduced endothelium-dependent relaxation (Flavahan, 1992; Freiman et al., 1986; Garland et al., 1995; Jayakody et al., 1987; Ross, 1986; Shimokawa & Vanhoutte, 1989; Zeiher et al., 1991).
Notably, we have found an increased contractile response to an activation of L-type Ca2+ channels by KCl-mediated membrane depolarization. These findings are in line with reports of an enhanced contractility of vascular smooth muscle cells found in hypercholesterolaemic humans (Lamping, 1997; Fleischhacker et al., 2000) and animals (Cox & Cohen, 1996; Dam et al., 1997; Kim et al., 1994; Pfister & Campbell, 1996). In addition, significant changes in the α1-agonist- (PE-) mediated oscillatory tonus generation that was independent from the endothelium, indicate a general enhancement of contractile properties in the blood vessels of L2LPL animals, while maximal contraction remained unchanged. These data are in contrast to our own findings in uterine arteries of hypercholesterolaemic patients (Fleischhacker et al., 2000) and that found in hypercholesterolaemic rabbits (Kim et al., 1994). These differences might be related to the oscillatory tonus development in mouse aortae in response to the α1-agonist, while stable tonus is observed in the other reports.
Besides the increase in smooth muscle contractility, endothelium-dependent relaxation was blunted in L2LPL animals. We have found that the relaxation in response to ACh and ATP was reduced, suggesting that a down regulation of certain receptors seems unlikely. These findings are in line with several reports in animals tissue as well as increased FFAs (de kreutzenberg et al., 2000) and hypercholesterolaemia (Lüscher et al., 1992; 1993; Lüscher & Noll, 1994; Zeiher et al., 1991; 1993). Since in the presence of the inhibitor of endothelial nitric oxide synthase (eNOS) L-nitroarginine no further relaxation was found, these data indicate that autocoid-induced relaxation in the mouse aorta is mediated by nitric oxide (•NO). Our findings that the ACh-induced relaxation was normalized in the presence of SOD suggest that the reduced endothelium-dependent relaxation in the L2LPL mice might be due to •O2−-mediated scavenging of •NO rather than reduced •NO production. Furthermore, since the endothelium-independent relaxation to the •NO donor SNP was not altered in the L2LPL mice, a non-specific generalized dysfunction of the smooth muscle to •NO seems unlikely.
The production of •NO in response to both compounds tested (ACh, ATP) critically depends on an elevation in endothelial free Ca2+ concentration (Busse et al., 1991; 1993). Our data that endothelial Ca2+ signalling in response to ATP was even increased in single endothelial cells freshly isolated from the L2LPL mice compared with that isolated from the L2 animals clearly excludes a reduction of endothelial Ca2+ signalling to be responsible for the reduced endothelium-dependent relaxation in the transgenic animals. Hence the increased Ca2+ signalling is in agreement with other reports indicating that some FFAs enhance Ca2+ uptake in human arterial endothelial cells (Kummerow et al., 1999).
Recently, such an increase in autacoid-induced Ca2+ signalling was shown to occur after treatment with •O2− (Graier et al., 1998a) or an enhanced endothelial •O2− production during hyperglycaemia (Graier et al., 1996). Since we report here that in L2LPL mice an increased •O2− formation takes place, one might suggest that the reported increased endothelial Ca2+ signalling is, at least in part, due to the formation of this radical.
Our findings that the removal of the endothelium normalized blood vessel •O2− release suggest the vascular endothelium to be responsible for the production of this free oxygen species. These findings are in line with the report of Ohara et al. (1993) who described an increased endothelial •O2− formation in rabbits during hypercholesterolaemia. In contrast, in human arteries the smooth muscle cells were found to be responsible for the overshooting •O2− production found in hypercholesterolaemia (Miller et al., 1998; Fleischhacker et al., 2000). Whether or not these differences are related to species variation needs to be further elucidated, but, in any case, the increased •O2− production is thought to be responsible for the reduced bioactivity of •NO (Kimura et al., 1995; Miller et al., 1998; Mügge et al., 1994). In line with these previous reports, endothelium-dependent relaxation of aortae from the L2LPL mice was restored in the presence of SOD that protects NO from •O2−.
Our findings that the NAD(P)H oxidase inhibitor DPI, but not the eNOS/iNOS inhibitor L-nitroarginine prevented the enhanced •O2− release in the aortae of L2LPL animals suggests an involvement of the NAD(P)H oxidase but not eNOS/iNOS in the increased •O2− release in the L2LPL transgenic animals. In line with these findings, expression of iNOS could not be detected in the L2LPL animals, thus indicating that, at least in our model, FFA-loading of the vascular wall did not yield to the induction of iNOS as a possible additional source of •O2−. These data are consistent with many reports on different models of hypercholesterolaemia (Böger et al., 1995; Inoue et al., 1998; Miller et al., 1998; Mügge et al., 1994; Ohara et al., 1993), which indicate that the enhanced vascular •O2− release is mainly due to NAD(P)H oxidase in endothelial and smooth muscle cells (Azzumi et al., 1999; Mohazzab & Wolin, 1994; Mohazzab et al., 1994; Pagano et al., 1995). Furthermore, the inhibitory effect of the PKC inhibitor GF109203X on the enhanced •O2− release in the L2LPL animals may indicate that in the L2LPL animals NAD(P)H oxidase is activated in a PKC-dependent manner. This finding is in line with a recent report in cultured vascular cells that demonstrated that FFA stimulated formation of •O2− through PKC-dependent activation of NAD(P)H oxidase (Inoguchi et al., 2000). Additionally/alternatively, the elevated concentration of Ca2+ and archidonic acid found in the L2LPL animals may mutually trigger NAD(P)H oxidase activation (Henderson et al., 1995).
This study points to a crucial role of LPL-mediated FFAs loading in the development of vascular dysfunction that finally leads to atherosclerosis. Remarkably, despite unchanged lipoprotein levels, an increased FFA content in the vascular wall due to an increased overall LPL activity is already suitable to initiate changes in vascular functions that represent the initial step in atherogenesis. Thus, besides high cholesterol/LDL levels, vascular LPL activity might represent a primary risk factor for atherosclerosis that is independent from the plasma levels of LDL.
Acknowledgments
We thank Ing. Helga Reicher for the excellent analysis of the FFAs and Mag. Michaela Hiden for her great help with the animals. This work was supported by the Austrian Funds (SFB 714, SFB713, SFB702, and P-14586-PHA), the Austrian National bank (P7542 and P7902), the Kamillo-Eisner Stiftung (Hergiswil, Switzerland) and the Franz Lanyar Foundation. V.E. Esenabhalu was supported by the Austrian Academic Exchange Program (OeAD) and M. Frieden was supported by the Swiss National Funds.
Abbreviations
- ACh
acetylcholine
- DPI
diphenylene iodonium
- DMEM
Dulbecco's minimal essential medium
- eNOS
endothelial nitric oxide synthase
- GF109203X
2-[-1-(3-dimethylaminopropyl)indol-3-yl]-3-(indol-3-yl)maleimide
- FFA
free fatty acid
- iNOS
inducible nitric oxide synthase
- KHS
Krebs solution
- LPL
lipoprotein lipase
- •NO
nitric oxide
- PBS
phosphate buffered solution
- PE
phenylephrine
- SNP
sodium nitroprusside
- •O2−
superoxide anion
- SOD
superoxide dismutase
References
- AZZUMI H., INOUE N., TAKESHITA S., RIKITAKE Y., KAWASHIMA S., HAYASHI Y., ITOH H., YOKOYAMA M. Expression of NADH/NADPH oxidase P22phox in human coronary arteries. Circulation. 1999;100:1494–1498. doi: 10.1161/01.cir.100.14.1494. [DOI] [PubMed] [Google Scholar]
- BABAEV V.R., FAZIO S., GLEAVES L.A., CARTER K.J., SEMENKOVICH C.F., LINTON M.F. Macrophage lipoprotein lipase promotes foam cell formation and atherosclerosis in vivo. J. Clin. Invest. 1999;103:1697–1705. doi: 10.1172/JCI6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARTH S., KLEINHAPPL B., GUTSCHI A., JELOVCAN S., MARTH E. In vitro cytokine mRNA expression in normal human peripheral blood mononuclear cells. Inflamm. Res. 2000;49:266–274. doi: 10.1007/PL00000206. [DOI] [PubMed] [Google Scholar]
- BÖGER R.H., BODE-BÖGER S.M., MÜGGE A., KIENKE S., BRANDES R., DWENGER A., FRÖLICH J.C. Supplementation of hypercholesterolaemic rabbits with L-arginine reduces the vascular release of superoxide anions and restores NO production. Atherosclerosis. 1995;117:273–284. doi: 10.1016/0021-9150(95)05582-h. [DOI] [PubMed] [Google Scholar]
- BUSSE R., LÜCKHOFF A., MÜLSCH A. Cellular mechanisms controlling EDRF/NO formation in endothelial cells. Basic Res. Cardiol. 1991;86 Suppl. 2:7–16. doi: 10.1007/978-3-642-72461-9_2. [DOI] [PubMed] [Google Scholar]
- BUSSE R., MÜLSCH A., FLEMMING I., HECKER M. Mechanisms of nitric oxide release from the vascular endothelium. Circulation. 1993;87 Suppl. V:V18–V25. [Google Scholar]
- CARLUCCIO M.A., MASSARO M., BONFRATE C., SICULELLA L., MAFFIA M., NIOLARDI G., DISTANTE A., STORELLI C., DE CATERINA R. Oleic acid inhibits endothelial activation – a direct vascular antiatherogenic mechanism of a nutritional component in the Mediterranean diet. Arterioscler. Thromb. Vasc. Res. 1999;19:220–228. doi: 10.1161/01.atv.19.2.220. [DOI] [PubMed] [Google Scholar]
- COX D.A., COHEN M.L. Selective enhancement of 5-hydroxytryptamine-induced contraction of porcine artery by oxidized low-density lipoprotein. J. Pharmacol. Exp. Ther. 1996;276:1095–1103. [PubMed] [Google Scholar]
- DAM J.P., VLEEMING W., RIEZEBOS J., POST M.J., PORSIUS A.J., WEMER J. Effects of hypercholesterolemia on the contractions to angiotensin II in the isolated aorta and iliac artery of the rabbit: role of arachidonic acid metabolites. J. Cardiovasc. Pharmacol. 1997;30:118–123. doi: 10.1097/00005344-199707000-00017. [DOI] [PubMed] [Google Scholar]
- DE CATERINA R., CYBULSKY M.I., CLINTON S.K., GIMBRONE M.A.J., LIBBY P. The ω-3 fatty acid docosahexaaenoate reduces cytokine-induced expression of pro-atherogenic and pro-inflammatory proteins in human endothelial cells. Arterioscler. Thromb. 1994;14:1829–1836. doi: 10.1161/01.atv.14.11.1829. [DOI] [PubMed] [Google Scholar]
- DE CATERINA R., CYBULSKY M.I., CLINTON S.K., GIMBRONE M.A.J., LIBBY P. Omega-3 fatty acids and endothelial leucocyte adhesion molecules. Prostaglandins Leukot. Essent. Fatty Acids. 1995;52:192–195. doi: 10.1016/0952-3278(95)90021-7. [DOI] [PubMed] [Google Scholar]
- DE KREUTZENBERG S.V., CREPALDI C., MARCHETTO S., CALO L., TIENGO A., DEL PRATO S., AVOGARO A. Plasma free fatty acids and endothelium-dependent vasodilation: effect of chain-length and cyclooxygenase inhibition. J. Clin. Endocrinol. Metab. 2000;85:793–798. doi: 10.1210/jcem.85.2.6352. [DOI] [PubMed] [Google Scholar]
- DE-CATERINA R., LIAO J.K., LIBBY P. Fatty acid modulation of endothelial activation. Am. J. Clin. Nutr. 2000;71:213S–223S. doi: 10.1093/ajcn/71.1.213S. [DOI] [PubMed] [Google Scholar]
- EGAN B.M., LU G., GREENE E.L. Vascular effects of non-esterified fatty acids: implications for the cardiovascular risk factor cluster. Prostaglandins Leukot. Essent. Fatty Acids. 1999;60:411–420. doi: 10.1016/s0952-3278(99)80022-2. [DOI] [PubMed] [Google Scholar]
- ENDRESEN M.J., TOSTI E., HEIMLI H., LORENTZEN B., HENRIKSEN T. Effects of free fatty acids found increased in women who develop pre-eclampsia on the ability of endothelial cells to produce prostacyclin, cGMP and inhibit platelet aggregation. Scand. J. Clin. Lab. Invest. 1994;54:549–557. doi: 10.3109/00365519409088567. [DOI] [PubMed] [Google Scholar]
- FLAVAHAN N.A. Atherosclerosis or lipoprotein-induced endothelial dysfunction. Circulation. 1992;85:1927–1940. doi: 10.1161/01.cir.85.5.1927. [DOI] [PubMed] [Google Scholar]
- FLEISCHHACKER E., ESENABHALU V.E., HOLZMANN S., SKRABAL F., KOIDL B., KOSTNER G.M., GRAIER W.F. In human hypercholesterolemia increased reactivity of smooth muscle cells is due to altered subcellular Ca2+ distribution. Atherosclerosis. 2000;149:33–42. doi: 10.1016/s0021-9150(99)00290-7. [DOI] [PubMed] [Google Scholar]
- FLEISCHHACKER E., HOLZMANN S., SKRABAL F., KOIDL B., KOSTNER G.M., GRAIER W.F. Diabetes mellitus is associated with hyperreactivity of smooth muscle cells due to altered subcellular Ca2+ distribution in the human uterine artery. Diabetes. 1999;48:1323–1330. doi: 10.2337/diabetes.48.6.1323. [DOI] [PubMed] [Google Scholar]
- FREIMAN P.C., MITCHELL G.G., HEISTAD D.D., ARMSTRONG M.L., HARRISON D.G. Atherosclerosis impairs endothelium-dependent vascular relaxation to acetylcholine and thrombine in primates. Circ. Res. 1986;58:783–789. doi: 10.1161/01.res.58.6.783. [DOI] [PubMed] [Google Scholar]
- GARLAND C.J., PLANE F., KEMP B.K., COCKS T.M. Endothelium-dependent hyperpolarization: a role in the control of vascular tone. Trends Pharmacol. Sci. 1995;16:23–30. doi: 10.1016/s0165-6147(00)88969-5. [DOI] [PubMed] [Google Scholar]
- GOLDBERG I.J. Lipoprotein lipase and lipolysis: central roles in lipoprotein metabolism and atherogenesis. J. Lipid Res. 1996;37:693–707. [PubMed] [Google Scholar]
- GRAIER W.F., HOEBEL B.G., PALTAUF-DOBURZYNSKA J., KOSTNER G.M. Effects of superoxide anions on endothelial Ca2+ signaling pathways. Arterioscler. Thromb. Vasc. Biol. 1998a;18:1470–1479. doi: 10.1161/01.atv.18.9.1470. [DOI] [PubMed] [Google Scholar]
- GRAIER W.F., PALTAUF-DOBURZYNSKA J., HILL B., FLEISCHHACKER E., HOEBEL B.G., KOSTNER G.M., STUREK M. Submaximal stimulation of porcine endothelial cells causes focal Ca2+ elevation beneath the cell membrane. J. Physiol. Lond. 1998b;506:109–125. doi: 10.1111/j.1469-7793.1998.109bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRAIER W.F., SIMECEK S., KUKOVETZ W.R., KOSTNER G.M. High-D-glucose-induced changes in endothelial Ca2+/EDRF signaling is due to generation of superoxide anions. Diabetes. 1996;45:1386–1395. doi: 10.2337/diab.45.10.1386. [DOI] [PubMed] [Google Scholar]
- GRAIER W.F., SIMECEK S., STUREK M. Cytochrome P450 mono-oxygenase-regulated signaling of endothelial Ca2+ entry. J. Physiol. London. 1995;482:259–274. doi: 10.1113/jphysiol.1995.sp020515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HENDERSON L.M., BANTING G., CHAPPELL J.B. The arachidonate-activable, NADPH oxidase-associated H+ channel. J. Biol. Chem. 1995;270:5909–5916. [PubMed] [Google Scholar]
- INOGUCHI T., LI P., UMEDA F., YU H.Y., KAKIMOTO M., IMAMURA M., AOKI T., ETOH T., HASHIMOTO T., NARUSE M., SANO H., UTSUMI H., NAWATA H. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49:1939–1945. doi: 10.2337/diabetes.49.11.1939. [DOI] [PubMed] [Google Scholar]
- INOUE N., OHARA Y., FUKAI T., HARRISON D.G., NISHIDA K. Probucol improves endothelial-dependent relaxation and decreases vascular superoxide production in cholesterol-fed rabbits. Am. J. Med. Sci. 1998;315:342–347. doi: 10.1097/00000441-199804000-00005. [DOI] [PubMed] [Google Scholar]
- JAYAKODY L., SENARATNE M., THOMSON A., KAPPAGODA T. Endothelium-dependent relaxation in experimental atherosclerosis in the rabbit. Circ. Res. 1987;60:251–264. doi: 10.1161/01.res.60.2.251. [DOI] [PubMed] [Google Scholar]
- KIM J.H., KLYACHKIN M.L., SVENDSEN E., DAVIES M.G., HAGEN P.O., CARSON C.C. Experimental hypercholesterolemia in rabbits induces cavernosal atherosclerosis with endothelial and smooth muscle cell dysfunction. J. Urol. 1994;151:198–205. doi: 10.1016/s0022-5347(17)34916-9. [DOI] [PubMed] [Google Scholar]
- KIMURA H., MINAKAMI H., KIMURA S., SAKURAI T., NAKAMURA T., KURASHIGE S., NAKANO M., SHOJI A. Release of superoxide radicals by mouse macrophages stimulated by oxidative modification of glycated low density lipoproteins. Atherosclerosis. 1995;118:1–8. doi: 10.1016/0021-9150(95)05587-m. [DOI] [PubMed] [Google Scholar]
- KUMMEROW F.A., ZHOU Q., MAHFOUZ M.M. Effect of trans fatty acids on calcium influx into human endothelial cells. Am. J. Clin. Nutr. 1999;70:832–838. doi: 10.1093/ajcn/70.5.832. [DOI] [PubMed] [Google Scholar]
- LAGRADE M., BURTIN M., BERICIAUD P., BLANC M., VELARDO B., DECHAVANNE M. Increase of platelet thromboxane A2 formation and of its plasmic half-life in diabetes mellitus. Thromb. Res. 1980;19:823–830. doi: 10.1016/0049-3848(80)90010-9. [DOI] [PubMed] [Google Scholar]
- LAMPING K.G. Hypercontractility of vascular muscle in atherosclerosis. Circulation. 1997;96:4131–4132. [PubMed] [Google Scholar]
- LEVAK-FRANK S., HOFMANN W., WEINSTOCK P.H., RADNER H., SATTLER W., BRESLOW J.L., ZECHNER R. Induced mutant mouse lines that express lipoprotein lipase in cardiac muscle, but not in skeletal muscle and adipose tissue, have normal plasma triglyceride and high-density lipoprotein-cholesterol levels. Proc. Natl. Acad. Sci. U.S.A. 1999;96:3165–3170. doi: 10.1073/pnas.96.6.3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEVAK-FRANK S., RADNER H., WALSH A., STOLLBERGER R., KNIPPING G., HOEFLER G., SATTLER W., WEINSTOCK P.H., BRESLOW J.L., ZECHNER R. Muscle-specific overexpression of lipoprotein lipase causes a severe myopathy characterized by proliferation of mitochondria and peroxisomes in transgenic mice. J. Clin. Invest. 1995;96:976–986. doi: 10.1172/JCI118145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEVAK-FRANK S., WEINSTOCK P.H., HAYEK T., VERDERY R., HOFMANN W., RAMAKRISHAN R., SATTLER W., BRESLOW J.L., ZECHNER R. Induced mutant mice expressing lipoprotein lipase exclusively in muscle have subnormal triglycerides yet reduced high density lipoprotein cholesterol levels in plasma. J. Biol. Chem. 1997;272:17182–17190. doi: 10.1074/jbc.272.27.17182. [DOI] [PubMed] [Google Scholar]
- LÜSCHER T.F., DOHI Y., TSCHUDI M. Endothelium-dependent regulation of resistance arteries: alterations with aging and hypertension. J. Cardiovasc. Pharmacol. 1992;19 Suppl. 5:S34–S42. [PubMed] [Google Scholar]
- LÜSCHER T.F., NOLL G. Endothelium dysfunction in the coronary circulation. J. Cardiovasc. Pharmacol. 1994;24 Suppl. 3:S16–S26. [PubMed] [Google Scholar]
- LÜSCHER T.F., TANNER F.C., TSCHUD M.R., NOLL G. Endothelial dysfunction in coronary artery disease. Annu. Rev. Med. 1993;44:395–418. doi: 10.1146/annurev.me.44.020193.002143. [DOI] [PubMed] [Google Scholar]
- MATTSSON L., JOHANSSON H., OTTOSSON M., BONDJIERS G., WIKLUND O. Expression of lipoprotein lipase mRNA and secretion in macrophages isolated from human atherosclerotic aorta. J. Clin. Invest. 1993;92:1759–1765. doi: 10.1172/JCI116764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEAD J.R., CRYER A., RAMJI D.P. Lipoprotein lipase, a key role in atherosclerosis. FEBS Lett. 1999;462:1–6. doi: 10.1016/s0014-5793(99)01495-7. [DOI] [PubMed] [Google Scholar]
- MILLER F.J., GUTTERMAN D.D., RIOS C.D., HEISTAD D.D., DAVIDSON B.L. Superoxide production in vascular smooth muscle contributes to oxidative stress and impairs relaxation in atherosclerosis. Circ. Res. 1998;82:1298–1305. doi: 10.1161/01.res.82.12.1298. [DOI] [PubMed] [Google Scholar]
- MOHAZZAB K.M., KAMINSKI P.M., WOLIN M.S. NADH oxidoreductase is a major source of superoxide anion in bovine coronary artery endothelium. Am. J. Physiol. 1994;266:H2568–H2572. doi: 10.1152/ajpheart.1994.266.6.H2568. [DOI] [PubMed] [Google Scholar]
- MOHAZZAB K.M., WOLIN M.S. Sites of superoxide anion production detected by lucigenin in calf pulmomary artery smooth muscle. Am. J. Physiol. 1994;267:L815–L822. doi: 10.1152/ajplung.1994.267.6.L815. [DOI] [PubMed] [Google Scholar]
- MÜGGE A., BRANDES R.P., BÖGER R.H., DWEGER A., BODE-BÖGER S., KIENKE S., FRÖLICH J.C., LICHTLEN P.R. Vascular release of superoxide radicals is enhanced in hypercholesterolemic rabbits. J. Cardiovasc. Pharmacol. 1994;24:994–998. doi: 10.1097/00005344-199424060-00019. [DOI] [PubMed] [Google Scholar]
- NIU X.L., LIU L.Y., HU M.L., CHEN X. Some similarities in vascular effects of oleic acid and oxidized low-density lipoproteins on rabbit aorta. J. Mol. Cell. Cardiol. 1995;27:531–539. doi: 10.1016/s0022-2828(08)80048-x. [DOI] [PubMed] [Google Scholar]
- OHARA Y., PETERSON T.E., HARRISON D.G. Hypercholesterolemia increases endothelial superoxide anion production. J. Clin. Invest. 1993;91:2546–2551. doi: 10.1172/JCI116491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OLIVECRONA G., OLIVECRONA T. Triglyceride lipases and atherosclerosis. Curr. Opin. Lipidol. 1995;6:291–305. doi: 10.1097/00041433-199510000-00009. [DOI] [PubMed] [Google Scholar]
- PAGANO P.J., ITO Y., TORNHEIM K., GALLOP P., COHEN R.A. An NADPH oxidase superoxide generating system in the rabbit aorta. Am. J. Physiol. 1995;268:H2274–H2280. doi: 10.1152/ajpheart.1995.268.6.H2274. [DOI] [PubMed] [Google Scholar]
- PFISTER S.L., CAMPBELL W.B. Contribution of arachidonic acid metabolites to reduced norepinephrine-induced contractions in hypercholesterolemic rabbit aortas. J. Cardiovasc. Pharmacol. 1996;28:784–791. doi: 10.1097/00005344-199612000-00008. [DOI] [PubMed] [Google Scholar]
- ROSS R. The pathogenesis of atherosclerosis – An uptake. New Engl. J. Med. 1986;314:488–500. doi: 10.1056/NEJM198602203140806. [DOI] [PubMed] [Google Scholar]
- SATTLER W., LEVAK-FRANK S., RADNER H., KOSTNER G.M., ZECHNER R. Muscle-specific overexpression of lipoprotein lipase in transgenic mice results in increased a-tocopherol levels in skeletal muscle. Biochem. J. 1996;318:15–19. doi: 10.1042/bj3180015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIMOKAWA H., AARHUS L.L., VANHOUTTE P.M. Dietary omega 3 polyunsaturated fatty acids augment endothelium-dependent relaxation to bradykinin in coronary microvessels of the pig. Br. J. Pharmacol. 1988;95:1191–1203. doi: 10.1111/j.1476-5381.1988.tb11755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIMOKAWA H., VANHOUTTE P. Impaired endothelium-dependent relaxation to aggregating platelets and related vasoactive substances in porcine coronary arteries in hypercholesterolemia and atherosclerosis. Circ. Res. 1989;64:900–914. doi: 10.1161/01.res.64.5.900. [DOI] [PubMed] [Google Scholar]
- STEINBRECHER U.P. Role of superoxide in endothelial-cell modification of low-density lipoproteins. Biochim. Biophys. Acta. 1988;959:20–30. doi: 10.1016/0005-2760(88)90145-2. [DOI] [PubMed] [Google Scholar]
- STUREK M., CALDWELL W.M., HUMPHREY D.A., WAGNER-MANN C.Methods for simultaneous voltage-clamp, microfluorimetry, and video of cells. I. Electronic and optical instrumentation Ion Channels of Vascular Smooth Muscle Cells and Endothelial Cells 1991Elsevier: New York; 239–267.eds. Sperelakis, N. & Kuriyama, H [Google Scholar]
- VOSSEN R.C.R.M., VAN-DAM-MIERAS M.C.E., LEMMENS P.J.M.R., HORNSTRA G., ZWAAL R.F.A. Membrane fatty acid composition and endothelial cell functional properties. Biochim. Biophys. Acta. Lipids. Lipid. Metab. 1991;1083:243–251. doi: 10.1016/0005-2760(91)90078-v. [DOI] [PubMed] [Google Scholar]
- YLÄ-HERTTUALA S., LIPTON B.A., ROSENFELD M.E., GOLDBERG I.J., STEINBERG D., WITZTUM J.L. Macrophages and smooth muscle cells express lipoprotein lipase in human and rabbit atherosclerotic leasons. Proc. Natl. Acad. Sci. U.S.A. 1991;88:10143–10147. doi: 10.1073/pnas.88.22.10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YLÄ-HERTTUALA S., PALINSKI W., ROSENFELD M.E., PARTHASARATHY S., CAREW T.E., BUTLER S., WITZTUM J.L., STEINBERG D. Evidence for the presence of oxidatively modified low density lipoprotein in atherosclerotic lesions of rabbit and man. J. Clin. Invest. 1989;84:1086–1095. doi: 10.1172/JCI114271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZECHNER R. The tissue-specific expression of lipoprotein lipase: implications for energy and lipoprotein metabolism. Cur. Opin. Lipidol. 1997;8:77–88. doi: 10.1097/00041433-199704000-00005. [DOI] [PubMed] [Google Scholar]
- ZEIHER A., DREXLER H., SAURBIER B., JUST H. Endothelium-mediated coronary blood flow modulation in humans. J. Clin. Invest. 1993;92:652–662. doi: 10.1172/JCI116634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZEIHER A., DREXLER H., WOLLSCHLÄGER H., JUST H. Progressive endothelial dysfunction with different stages of early coronary atherosclerosis. Circulation. 1991;83:391–401. doi: 10.1161/01.cir.83.2.391. [DOI] [PubMed] [Google Scholar]
- ZILVERSMIT D.B. Atherogenesis: a postprandial phenomenon. Circulation. 1979;60:473–485. doi: 10.1161/01.cir.60.3.473. [DOI] [PubMed] [Google Scholar]