

Abstract
The efficacy of the free radical trapping agent NXY-059 in reducing the infarct volume following both transient and permanent focal ischaemia has been examined in rats.
In the transient ischaemia model, rats were subjected to a 2 h occlusion of the middle cerebral artery (MCA). Intravenous infusion of NXY-059 (1, 10 and 30 mg kg−1 h) for 21.75 h starting 2.25 h after the occlusion, produced a dose-dependent decrease in both neurological impairment and the histologically measured infarct volume (a mean 59% decrease at 10 mg kg−1 h).
In the permanent ischaemia model, animals were injected (s.c.) with a loading dose of NXY-059 of 32.5, 53.8 or 75.4 mg kg−1 and osmotic minipumps were implanted which had been primed to deliver respectively 30, 50 or 70 mg kg−1 h. When treatment was initiated 5 min after MCA occlusion there was a dose dependent protection of both cortical and sub-cortical tissue (cortex: 63% at the mid-range dose). Protection was related linearly to plasma concentration (plasma unbound NXY-059 concentration at 1 h: 37±16 μmol l−1 at the mid-range dose).
When the mid range dose was administered between 5 min – 4 h after MCA occlusion, a marked and statistically significant protection was seen at all time points (44% protection in cortex at 4 h).
These data demonstrate the substantial neuroprotective efficacy of NXY-059 at plasma concentrations that can be achieved clinically and indicate that NXY-059 also has a therapeutic window of opportunity that is clinically relevant.
Keywords: Stroke, NXY-059, transient cerebral ischaemia, permanent cerebral ischaemia, radical trapping agent, therapeutic window, nitrone
Introduction
There is substantial evidence that one of the major mechanisms involved in neuronal cell death following an ischaemic insult is free radical mediated neurotoxicity (Schulz et al., 1995a; Facchinetti et al., 1998). This suggests that compounds that trap free radicals should be a valid therapeutic approach to the treatment of acute ischaemic stroke and there is good experimental evidence to support this contention. The free radical trapping nitrone compound α-phenyl-N-tert-butylnitrone (PBN) attenuates the neuronal damage produced in rodents by a period of cerebral ischaemia. This was first demonstrated in the gerbil model of global ischaemia (Oliver et al., 1990; Phillis & Clough-Helfman, 1990). Subsequent studies demonstrated the efficacy of this compound in focal models of ischaemia involving occlusion of the middle cerebral artery (MCA). PBN markedly reduced infarct volume in the rat model of transient focal ischaemia (Kuroda et al., 1996; Zhao et al., 1994) and Cao & Phillis (1994) also demonstrated efficacy following permanent MCA occlusion.
The structurally related nitrone NXY-059 (disodium 4-[(tert-butylimino) methyl] benzene-1, 3-disulphonate N-oxide) is more water soluble than PBN and has recently been examined in a rat transient MCA occlusion model (Kuroda et al., 1999). This study examined the effect of NXY-059 on infarct size in rats, measured by use of 2,3,5-triphenyltetrazolium chloride (TTC) staining and the effect of treatment on the functional consequences of the ischaemic insult by use of a neurological assessment score (Bederson et al., 1986). Kuroda et al. (1999) showed that NXY-059 had greater efficacy than PBN in the transient MCA occlusion model using both infarct size and neurological deficits as outcome measures. This study also demonstrated that NXY-059 had a large window of therapeutic opportunity, being effective even when given 5 – 8 h after the onset of ischaemia, which is 3 – 6 h after the start of recirculation (Kuroda et al., 1999).
The Kuroda et al. (1999) study did not measure the plasma drug concentration required for neuroprotection. We have, therefore, now undertaken a study to examine the dose-response characteristics of NXY-059 in a transient MCA occlusion model and also to examine the plasma concentration of NXY-059 required to produce effective neuroprotection. This part of the study was performed by administering the drug by continuous intravenous infusion as had been performed by Kuroda et al. (1999).
In addition we have now examined the effect of NXY-059 on cerebral infarct volume following permanent MCA occlusion in the rat. In this study the method of drug administration was simplified by giving the drug by subcutaneously implanted Alzet minipumps, thereby removing the problem of indwelling intravenous giving sets and keeping the lines patent. Both the dose dependence of its neuroprotective effect and the therapeutic window of opportunity were examined.
Methods
Chemicals and animals
NXY-059 (disodium 4-[(tert-butylimino) methyl] benzene-1,3-disulphonate N-oxide) was obtained from AstraZeneca R&D Södertälje, Södertälje, Sweden.
Adult male Wistar Kyoto rats (Harlan, Indianapolis, U.S.A) weighing 290 – 335 g (approximate age 3 – 4 months) were used. They were housed in groups of three in plastic cages under the following environmental conditions: temperature 20 – 22°C, humidity 40 – 60%, ventilation 15 – 20 changes per hour and artificial lighting 12 h light/dark cycle (lights on: 0600 h) and given free access to food (vitamin E reduced) and tap water. The vitamin E reduced diet was used as we have previously found that animals fed this diet have a much more consistent ischaemia-induced lesion in the brain than those fed with the normal diet (Sydserff, unpublished observations). The study was performed in compliance with ARP 179 issued by the AstraZeneca U.S.A. Institutional Animal Care and Use Committee.
Animal preparation and middle cerebral artery occlusion
Animals were anaesthetized using a mixture of halothane, oxygen and nitrous oxide. Induction of anaesthesia was achieved by placing the rats into a sealed plastic chamber and introducing halothane (6%), O2 (1.0 l min−1) and N2O (1.5 1 min−1). Anaesthetized animals were placed supine on a heated operating mat and anaesthesia maintained with halothane (2%), O2 (1.0 l min−1) and N2O (1.5 l min−1). A rectal thermometer was inserted and the temperature was monitored.
Transient MCA occlusion was performed using a published method (Sydserff et al., 1995), with minor modification. The primary carotid arteries (common carotid [CCA], external carotid [ECA] and internal carotid [ICA]) were exposed. The CCA was dissected free from the vagus nerve and controlled with a loosely held silk suture. The carotid bifurcation was exposed allowing access to the carotid angle (origin of ICA, ECA and occipital artery). An atraumatic aneurysm clip was placed across the proximal portion of the ECA to maintain haemostasis and the remaining branches of the ECA (superior thyroid, ascending pharyngeal and distal segments of the ECA) divided using diathermy leaving a free arterial stump of around 3 – 4 mm in length. A second aneurysm clip was placed on the most distal portion of the arterial stump and the proximal clip removed.
The pterygopalatine branch of the ICA was exposed and ligated with 6/0 silk. Aneurysm clips were then placed across the lumens of the CCA and ICA (2 – 3 mm each side of the bifurcation) and a 26 G needle held in self-retaining forceps were used to make an arteriotomy in the stump of the ECA allowing a length of poly-L-lysine coated 3/0 blue monofilament ‘DERMILON' suture with a flared rounded tip to be introduced into the ECA and then into the proximal ICA. A silk suture was tied around the ECA stump to maintain haemostasis and the microvascular clips removed from the CCA and ICA. The endovascular suture was then passed up the lumen of the ICA, into the intracranial circulation where it lodged after travelling about 20 – 22 mm depending on rat size. This is consistent with the passage of the tip of the suture into the narrow portion of the proximal anterior cerebral artery occluding the middle cerebral artery (MCA) at its origin. Two hours after the onset of MCA occlusion, the cervical wound was reopened and a clip placed across the CCA. The monofilament was withdrawn from the ICA / ECA and the arteriotomy closed. Following removal of the CCA clip the cervical wound was closed with autoclips and the animal allowed to recover from anaesthesia.
In the case of permanent MCA occlusion the procedures detailed above were followed but the filament was left in place and the arteriotomy in the ECA was closed with diathermy, and if necessary the vessel was wrapped in haemostatic gauze (Surgical Johnson & Johnson Medical). The wound was closed with autoclips and the animal allowed to recover from anaesthesia. All operated rats were administered saline (4 ml kg−1 s.c.) 15 min following wound closure to assist in rehydration during recovery.
During both techniques, a rectal thermometer was inserted immediately following anaesthesia. Body temperature was monitored and adjusted with heating lamps and a heated operating mat if the temperature fluctuated beyond pre-determined limits of 37±1°C. Body temperature was monitored throughout the intra-operative period and the values in one representative experiment are shown in Table 1 to illustrate the high degree of temperature control achieved. Following this the animals were allowed to recover in a warm (27 – 30°C), quiet environment and allowed free access to mashed food, whole pellet food and water.
Table 1.
The rectal temperature (°C) of vehicle and NXY-059 (50 mg−1 kg−1 h) treated rats (drug administered 5 min post-occlusion) in the 120 min following permanent MCA occlusion

Mortality following either the transient or permanent MCA occlusion procedure was very low being <4% of operated animals.
Administration of NXY-059 following transient MCA occlusion
Ninety minutes following MCA occlusion, the animals were anaesthetized with halothane and an incision made in the abdomen running from the left groin to the knee. The left femoral vein was cannulated with PE50 renthane tubing and the cannula passed subcutaneously around to the nape of the neck where it emerged and was secured to a tether button. The cannula was connected via a secure tethering system to a 60 ml syringe coupled to a syringe pump. Three separate experiments were performed each comparing the effect of infusion with NXY-059 to that of controls (vehicle infusion). All experiments followed an identical format. The drug was dissolved in water and the volume infused was identical in both control and NXY-059-treated rats. Increased dosing was achieved by increasing the concentration of the drug, not the volume infused. Twenty-four hours following the start of reperfusion the neurological impairment was assessed using the scoring method detailed below and histological assessment was made following sacrifice of the animals at 48 h after reperfusion.
Administration of NXY-059 following permanent MCA occlusion
In the study on the neuroprotective response of rats to increasing doses of NXY-059 following a permanent MCA occlusion, NXY-059 was dissolved in saline at 460 mg ml−1, buffered to a neutral pH with sodium bicarbonate and loaded into Alzet model 2001D minipumps (220 μl volume at 7.6 μl h−1). Each pump delivered approximately 3.5 μl h−1 (or approximately 10.5 μl kg−1 h for a standard 330 g rat). Three, five and seven pumps were implanted respectively for doses of 30, 50 and 70 mg kg−1 h. Buffered saline (vehicle) or NXY-059 was administered subcutaneously for 23 h 55 min via implanted osmotic minipumps. A bolus dose of 32.5, 53.8 or 75.4 mg kg−1 h was given immediately prior to pump implantation.
In the study on the therapeutic window of opportunity, animals were administered the mid-range dose used in the dose response experiment (50 mg kg−1 h), with both the loading dose and pump implantation being performed at various times (5 – 240 min) after the start of the MCA occlusion.
Histological measurement of neuronal damage
Twenty-four hours after the commencement of permanent ischaemia or 48 h after transient ischaemia the rats were re-anaesthetized with halothane. When the animal had been terminally anaesthetized the brain was rapidly removed and coronally cut into 2 mm thick blocks using the Harvard adult rat brain matrix. These blocks were placed into dishes containing a 1.5% solution of triphenyl tetrazolium chloride (TTC) at 37°C for 10 – 15 min until normal tissue stained a plum red colour. When the stain had developed, the tissue blocks were removed into 10% formalin for analysis. The area of unstained tissue was demarcated onto a matching stereotaxic map taken from the rat brain atlas of Paxinos & Watson (1986) and the areas of damage digitized and matched against position from interaural line to give a volumetric assessment of damage. TTC stains both neuronal and glial cells with a deep red pigment. In areas where neuronal loss occurs TTC does not stain and tissue remains white. If there is massive microglial infiltration in the lesion site then TTC staining can provide an erroneous description of the lesion size. However it is our experience, confirmed histologically, that at 24 h microglial proliferation is not a significant factor.
Assessment of neurological deficit
Neurological impairment in the stroked animals was examined by a modification of the method published by Bederson et al. (1986). Briefly, forelimb flexion, spontaneous rotation and absence of response to contralateral whisker stimulation were scored on a 0 – 2 scale (0=normal behaviour, 2=severely impaired). In addition, torsion of the body towards the contralateral side was assessed on a 0 – 2 scale. Thus the maximum impairment score was 8.
Analysis of unchanged NXY-059 in plasma
NXY-059 was assayed by coupled column liquid chromatography and detected by u.v. detection at 299 nm. A blood sample (0.2 – 0.3 ml) was taken from the tail vein immediately before cessation of the intravenous infusion of vehicle or saline, and centrifuged to separate the plasma. Plasma samples were stored in screw cap glass vials at −20°C prior to analysis. The plasma samples were treated with methanol followed by centrifugation, evaporation of the supernatant and reconstitution in buffer before injection into the liquid chromatographic system. The limit of quantitation was 0.5 μmol l−1, accuracy 93 – 103% and intra-assay coefficient of variation 3.8%.
Free (unbound) drug concentration in the plasma was estimated from the total drug concentration based on a figure of 70% protein binding obtained in other studies.
Previous studies on the protein binding characteristics of the drug in rat plasma have demonstrated that NXY-059 is 70% protein bound (J. Lundstrom, Department of Drug Metabolism and Pharmacokinetics, AstraZeneca R&D Södertälje, Sweden, personal communication). This value was, therefore, used for calculation in the present investigation.
Statistical analysis
Data was analysed for statistical significance using analysis of variance and group differences were further analysed using the Newman-Keuls test. All data are shown grouped as mean±standard error of mean (mean±s.e.mean) with P values and test used as appropriate.
Results
Effect of NXY-059 in transient focal ischaemia
The transient occlusion of the MCA for 2 h resulted in a neurological deficit 24 h after the start of the occlusion period (Figure 1a). This deficit was attenuated by administration of all three doses of NXY-059 (3, 10 and 30 mg kg−1 h for 21.75 h) examined (Figure 1a). The ischaemic episode also produced a large volume of damage in the brain, encompassing both the cortex and striatum. In the study using the lowest dose of NXY-059 (3 mg kg−1 h) the damage in the vehicle treated group was less than that seen in the control groups associated with the studies using higher doses of NXY-059 (Figure 1b). Administration of NXY-059 (3 mg kg−1 h) failed to produce a significant decrease in infarct volume (Figure 1b) but infusion doses of NXY-059 of 10 mg kg−1 h and 30 mg kg−1 h both produced a substantial decrease in the infarct volume (Figure 1b).
Figure 1.

The neurological deficit (a) and infarct volume (b) of groups of rats (n=9 – 10) administered various infusion doses of NXY-059 (3, 10 and 30 mg kg−1 h for 21.75 h) starting 2.25 h post-occlusion, together with the respective control groups. *Shows a significant difference from vehicle treatment at P<0.05 (ANOVA with Newman-Keuls post-hoc analysis).
The plasma concentration of NXY-059 in blood taken from the tail vein at the end of the 24 h infusion period in rats dosed with 10 mg kg−1 h was 28.5±3.0 μmol l−1 (n=8). Assuming a linear relationship between plasma concentration and dose as seen in another part of the study then the relationship between plasma concentration and degree of neuroprotection at all three doses used in this experiment can be calculated and is shown in Figure 2. The concentration of the estimated plasma ‘free' (unbound) drug is also shown (Figure 2, insert).
Figure 2.
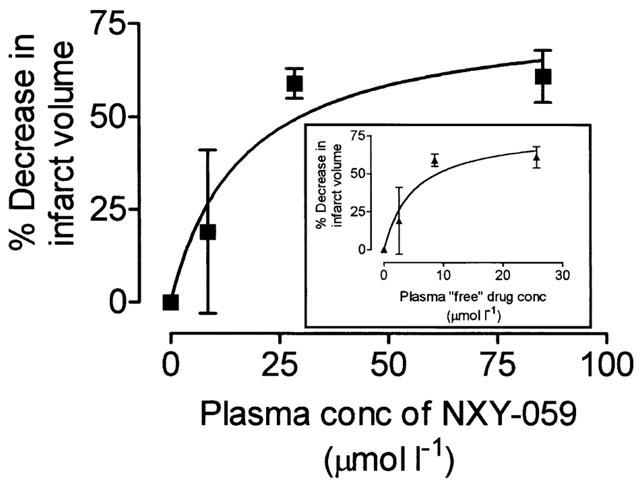
The relationship between the per cent decrease in infarct volume and the estimated plasma concentration of NXY-059, calculated from actual values obtained using the mid-range dose (10 mg kg−1 h). This assumed a linear relationship between dose and plasma concentration (as was confirmed in the study reported in Figure 4.) The insert shows the same data but reports plasma concentrations in terms of the protein unbound (‘free') concentration.
Effect of NXY-059 in permanent focal ischaemia: dose dependence study
Twenty-four hours after permanent occlusion of the left middle cerebral artery ischaemic damage was evident in the neocortex, ranging from the piriform cortex into the primary motor and sensory areas. The caudate putamen was extensively involved and damage extended into the ventral pallidum and lateral preoptic areas. In several instances the lateral hypothalamus was within the infarct boundary however damage to this region did not result in a change to rectal temperature. Rats treated with the loading dose of NXY-059 followed by a s.c. dose of 30, 50 and 70 mg kg−1 h had significantly reduced total ischaemic damage (Figure 3). The neuroprotection was linearly dose-dependent (Figure 3d). Protective effects with NXY-059 were observed in both cortex and sub-cortex, except in the 30 mg kg−1 h group where protection was statistically significant only in the sub-cortical region which resulted in a significant decrease in the total damage volume assessment (Figure 3a – c).
Figure 3.

The effect of increasing doses of NXY-059 on the volume of ischaemic damage in (a) the cortex and (b) the sub-cortex following permanent MCA occlusion in rats. (c) Shows the effect of the drug on the total volume of damage and (d) the per cent protection against damage (using the total volume data) versus the dose of NXY-059 administered. The number of animals in each group=8. *Shows a significant difference from vehicle treatment at P<0.05 (ANOVA with Newman-Keuls post-hoc analysis).
Animals treated with the infusion dose of NXY-059 of 30, 50 and 70 mg kg−1 h NXY-059 had plasma concentrations of NXY-059 at 24 h of respectively: 196±35, 387±52 and 450±68 μmol l−1. The plasma concentration of NXY-059 thus appears to be linearly related to dose (Figure 4).
Figure 4.
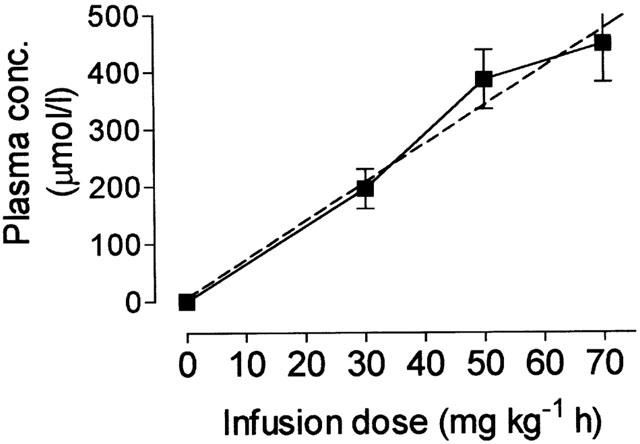
The relationship between the total drug plasma concentration of NXY-059 at the end of the 24 h infusion period and the infusion dose. The dashed line shows the regression line (r2=0.98; n=8 at each point).
Plasma concentration of NXY-059 at various times after pump implantation in permanent MCA occlusion experiments
The plasma concentration at 24 h was close to that predicted from kinetic calculations. However, these calculations were based on earlier intravenous data, whereas the pumps deliver drug subcutaneously. Furthermore, no data were available as to how rapidly the peak concentration was achieved. A study was therefore undertaken where plasma was taken from groups of four animals (two control and two permanent MCA occluded) at various times after pump implantation. Since no significant difference was seen in the plasma concentration at the different times between the control and MCA occluded rats, data have been pooled for calculation of mean±s.e.mean values.
The total concentrations of NXY-059 following a loading dose of 53.8 mg kg−1 followed by a subcutaneous dose of 50 mg kg−1 h did not rise above 200 μmol l−1 in the first 4 h (Figure 5a). The concentration at 24 h was 321 μmol 1−1.
Figure 5.

(a) The total drug plasma concentration of NXY-059 in groups of four rats (two vehicle-treated and two permanent MCA occlusion treated) at various times following administration of a s.c. bolus injection of NXY-059 (53.8 mg kg−1) and implantation of osmotic minipumps delivering 50 mg kg−1 h). The per cent reduction of infarct volume of NXY-059 treatment following three doses of NXY-059 in rats with a permanent MCA occlusion and the calculated plasma free concentration shown at (b) 1 h and (c) 24 h of the infusion period. This was estimated from the measured total concentration for the mid-range dose at 1 h and 24 h (shown in Figure 5a) and the other values calculated from that value based on the linear relationship between dose and plasma concentration (Figure 2). The dashed line shows the regression line for both graphs (r2=0.97).
Data from the first 4 h of this study were then applied to doses of 30 and 70 mg kg−1 h, and the corresponding plasma drug concentration values estimated, since there was a linear relationship between dose and plasma concentration (Figure 4). Values for the estimated total and free (unbound) drug concentration at 1 h and the measured total and estimated ‘free' drug concentrations 24 h after the start of infusion versus the degree of neuroprotection observed previously (Figure 3) are shown in Figure 5b,c.
Window of therapeutic opportunity following permanent MCA occlusion
In the next study animals were administered the mid-range dose (50 mg kg−1) used in the first experiment, with both the loading dose and pump implantation being performed at various times (5 – 240 min) after the start of the MCA occlusion. Twenty-four hours after commencing vehicle treatment similar damage was observed to that seen in the first study (Figure 6). Treatment with NXY-059 (50 mg kg−1 h) produced a significant reduction in ischaemic damage when the pump was implanted at 5, 30, 60, 120 and 240 min delay after MCA occlusion, with neuroprotection apparent in both the cortex and the sub-cortex (Figure 6). In a further experiment a group of animals were treated with saline or NXY-059 at the same dose but starting 360 min post-occlusion. While the mean volume of total damage in the NXY-059 treated group was 22% smaller than the control group, this decrease was not statistically significant (P=0.23).
Figure 6.

The effect of delayed administration of NXY-059 (a bolus injection of 53.8 mg kg−1, followed immediately by pump implantation delivering 50 mg kg−1 h) on the total volume of ischaemic damage and the volume of ischaemic damage in the cortex and sub-cortex of rats with a permanent MCA occlusion. The time is that at which the drug administration began following the MCA occlusion. The time of damage analysis was the same in all cases. The number of animals at every time point is eight, except 4 h (n=7). *Shows a significant difference from vehicle treatment at P<0.05 (ANOVA with Newman-Keuls post-hoc analysis).
Discussion
At the onset it is worth stating that we are aware that a limitation of our study is that measurement of physiological perameters (other than rectal temperature) was not undertaken. Temperature is a vital measure because of the body of evidence indicating that even modest hypothermia can be neuroprotective (Busto et al., 1987; Corbett et al., 1990; Nurse & Corbett, 1996). The body temperature was therefore checked and adjusted if necessary both during and immediately following the operative period.
Previous studies on the effect of NXY-059 during transient ischaemia in rats (Kuroda et al., 1999) and permanent ischaemia in both rats (Zhao et al., 2001) and marmosets (Marshall et al., 2001) have failed to detect any effect of NXY-059 on body temperature. Similarly none of these three groups (Kuroda et al., 1999; Marshall et al., 2001; Zhao et al., 2001) observed any effect of the drug on pCO2, pO2, blood glucose, blood pressure, blood pH or heart rate rendering it unlikely that changes in these perameters were occurring and producing the neuroprotective efficacy of NXY-059 seen in the current study. However evidence that brain temperature can also influence protection (Dowden et al., 1999) does indicate that this perameter should be measured in any future study on mechanistic aspects of NXY-059.
These results show NXY-059 to be a very effective compound in attenuating damage caused by permanent occlusion of the MCA. It is generally acknowledged that occlusion of the MCA is a model for producing ischaemic damage that has relevance to stroke in humans, because an infarct in the region of the MCA is the most common cause of stroke in man (Mohr et al., 1986). While many infarcts do reperfuse over time, recanalization may be slow to occur (Ringelstein et al., 1992). Therefore, it is vital that potential therapeutic compounds are able to ameliorate the consequences of permanent occlusion in animal models. The evidence for efficacy of NXY-059 in a permanent MCA occlusion model in rats are complimentary to a recent study by Zhao et al. (2001) who demonstrated efficacy in a spontaneously hypertensive rat permanent MCA occlusion model. The compound has also been shown to be powerfully neuroprotective in a primate permanent MCA occlusion model (Marshall et al., 2001). In this marmoset model the drug produced not only a greater than 50% decrease in infarct volume, but also markedly attenuated the ischaemia-induced motor deficits and spatial hemineglect.
The current study also confirms and extends the report of Kuroda et al. (1999) who demonstrated that NXY-059 was an effective neuroprotective agent against cerebral damage following transient focal ischaemia in the rat. While Kuroda et al. (1999) showed histological evidence of efficacy of NXY-059 at infused doses of 0.3, 3.0 and 30 mg kg−1 h, our study was only able to demonstrate efficacy at doses of 10 and 30 mg kg−1 h. The failure to demonstrate significant protection at 3 mg kg−1 h may have been due to comparison with a control group which showed a low degree of damage compared to that seen in the other control groups (see Figure 1b). Neuroprotection was however demonstrable at this low dose when efficacy was measured by a neurological deficit score (Figure 1a) which suggests that there is not a strict correlation between neurological improvement and histological measures. A further reason for the Kuroda et al. (1999) study observing efficacy at lower dose than the current investigation may be the fact that this group gave a loading dose of drug at the start of infusion. At the higher doses the histological protection observed in the present study was similar to that reported by Kuroda et al. (1999). In their study, neuroprotection was 57% following 3 mg kg−1 h and 76% following 30 mg kg−1 h of NXY-059 while in our current study it was 59% following 10 mg kg−1 h and 61% following 30 mg kg−1 h.
It is clear that the doses of compound required for effective neuroprotection in permanent focal ischaemia are higher than those required for attenuation of damage in transient ischaemia. In the transient model, a dose of 30 mg kg−1 h provided substantial neuroprotection. However, this dose produced a modest effect in the permanent occlusion model. The protection by NXY-059 following permanent MCA occlusion was linearly dose-dependent and the highest dose given produced a reduction in the volume of damage of around 80%.
The permanent MCA occlusion experiments were planned using a dosing schedule designed to produce a rapid steady state concentration that would be sustained over 24 h. Measurement of the plasma levels at 24 h suggested that the projected value was achieved at that time. However, this result did not indicate how soon this value was reached after the start of the infusion. A further study was therefore undertaken with measurement of plasma concentration in groups of animals at several time points after the start of the infusion. These results demonstrated that plasma drug concentrations in the first few hours following administration were considerably lower than the 24 h value. It is reasonable to conclude that the first few hours following administration and MCA occlusion are the time when the plasma drug levels are most relevant to the therapeutic effect. These data have been re-plotted in terms of the plasma ‘free' drug concentration at 1 and 24 h and the degree of neuroprotection (Figure 5). This figure shows that substantial neuroprotection was achieved in rats at initial plasma free drug concentrations that were safely tolerated in stroke patients (45 μmol l−1) in a Phase II clinical study (Lees et al., 2001). The current animal data however suggest that further increasing the clinical plasma concentration might be therapeutically valuable.
Since the transient and permanent occlusion studies were performed in the same laboratory and under similar conditions our results obtained allow comparison of the volume of damage following permanent occlusion with that seen after a period of 2 h occlusion followed by reperfusion (Figure 7). This comparison suggests that reperfusion, if it occurs after 2 h of hypoxic-ischaemia, ameliorates the damage by a figure of around 27%. This is in close agreement with the study of Brinker et al. (1999) who observed that administration of recombinant tissue plasminogen activator (rt-PA) at 1.5 h reduced damage produced by MCA clot embolism by 35%. It can be also seen that administration of NXY-059 (50 mg kg−1 h) at 4 h produced a greater mean percentage neuroprotection value (Figure 7). These data are encouraging for the further clinical evaluation of NXY-059 in acute ischaemic stroke, since t-PA has demonstrable clinical efficacy (The NINDS t-PA Stroke Study Group, 1995; 1997; 1999).
Figure 7.

The volume of ischaemic damage (±s.e.mean) in a group of rats subjected to a permanent occlusion of the MCA for 24 h (n=16), a group subjected to an MCA occlusion for 2 h followed by reperfusion for 22 h (n=28) and a group subjected to a permanent MCA occlusion but administered NXY-059 (50 mg kg−1 h for 24 h) starting 4 h post-occlusion (n=8). Data demonstrate that both reperfusion and administration of NXY-059 have similar efficacy in decreasing the size of infarct damage and that both treatments significantly (* P<0.001) decrease the volume of damage.
Another major observation made in the current investigation was the time window of therapeutic opportunity that is available with NXY-059 administration. Kuroda et al. (1999), using a transient MCA occlusion model, showed that NXY-059 produced a substantial decrease in infarct volume when given 5 h after the occlusion (3 h after the start of reperfusion). We have now found significant benefit when the compound is given 4 h after the start of a permanent MCA occlusion (and a non-significant benefit when given 6 h later). This means that it is effective in rodents in a time window that is relevant to clinical practice, a feature that contrasts strongly with NMDA antagonists which have a window in rodents of around 1 h in permanent models of ischaemia (Foster et al., 1988; Bakker & Foster, 1991; Massieu et al., 1993). We recently confirmed this short time window using the stroke model utilized in the current experiments in a study on the non-competitive NMDA antagonist AR-R15896 (Sydserff & Cross, unpublished observations). This difference in the window of opportunity between glutamate antagonists and nitrone trapping agents was also observed in the malonate model of striatal neurodegeneration by Schulz et al. (1995b), who reported that dizocilpine only protected when given up to 1 h post malonate administration, while 2-sulphophenyl-N-tert-butylnitrone (S-PBN) was efficacious 6 h post administration.
One question arising from these findings of robust neuroprotective effects in transient and permanent models of ischaemia is the site of action of NXY 059. Kuroda et al. (1999) provided evidence suggesting that, in contrast to PBN, NXY-059 had poor brain penetration after transient (2 h) focal ischaemia. While it might, initially, appear surprising that a drug with poor brain penetration can be neuroprotective there is other evidence to support the notion that nitrones with poor brain penetration are nevertheless neuroprotective. In the malonate injection model, Schulz et al. (1995b) used S-PBN, another nitrone that has been suggested to have poor brain penetration, based on its physico-chemical properties (see Yang et al., 2000), and found it to be an effective neuroprotective agent. Yang et al. (2000) also examined S-PBN in their focal embolic cerebral ischaemia model in rats and reported it to have similar neuroprotective efficacy to the brain penetrating nitrone PBN.
There is compelling evidence for an interaction between endothelial cells and inflammatory cells such as polymorphonuclear leucocytes and macrophages and that these are major sources of free radicals during reperfusion following focal ischaemia (Betz, 1996; Hallenbeck, 1996). Kuroda et al. (1999) speculated that this mechanism may, through the up-regulation of adhesion molecules on the endothelial surface, be involved in reperfusion injury. Furthermore, they suggested that NXY-059 might reduce reactive oxygen species produced during reperfusion damage by interfering with the interaction between endothelial cells and inflammatory cells or by directly reacting with reactive oxygen species produced by circulating inflammatory cells or the endothelium.
There is little evidence at present for a prominent role of endothelial/inflammatory cell interactions in producing neuronal death following permanent ischaemia (Clark et al., 1991; Zhang et al., 1995; Carcia et al., 1995; Kato et al., 1996; Relton & Rothwell, 1996). This purported site of action may therefore be less relevant for the neuroprotective effect of NXY-059 during permanent ischaemia. The extent to which NXY-059 penetrates the brain in models of permanent ischaemia is not known at present.
If there is also limited penetration of NXY-059 in permanent ischaemia an alternative explanation for the neuroprotective effects of NXY-059 is that high concentrations in circulating blood might protect the blood brain barrier from damage. Recently Leinonen et al. (2000) reported that low plasma antioxidant activity was associated with high lesion volume and neurological impairment in a group of 22 cerebral ischaemic stroke patients, which suggests that the antioxidant activity of plasma might be an important factor in neuroprotection. There is also evidence that early disruption of the blood brain barrier to large molecules is mediated by free oxygen radicals (Tasdemiroglu et al., 1994) and protecting the blood brain barrier from ischaemia-induced damage by use of a Type IV phophodiesterase inhibitor has been shown to significantly improve the neurological score of rats subjected to transient MCA occlusion (Belayev et al., 1998). Therefore, the evidence of poor brain penetration of NXY-059 following an ischaemic insult might be reflective of part of its therapeutic mechanism of action. Further mechanistic studies are clearly necessary, together with studies on the integrity of the blood brain barrier during an ischaemic episode.
There is now substantial evidence for the neuroprotective action of NXY-059 in both transient and permanent ischaemia models of stroke. It decreases the volume of ischaemic damage following transient MCA occlusion in rats (Kuroda et al., 1999; this paper) and permanent MCA occlusion in rats (Zhao et al., 2001; this paper) and marmosets (Marshall et al., 2001). It also attenuates neurological deficits in rats following a haemorrhagic stroke (Peeling et al., 2001). The therapeutic window appears longer than for most other agents in both transient MCA and permanent MCA occlusion in rats (Kuroda et al., 1999; this paper). All these data demonstrate efficacy of the drug when administered at doses that produce plasma NXY-059 concentrations which are known to be well tolerated by stroke patients (Lees et al., 2001). NXY-059 therefore appears to be worthy of clinical development as a treatment for acute ischaemic stroke.
Acknowledgments
We would like to thank Kerstin Lanbeck Vallén, BSc, for undertaking the bioanalysis in this study. AstraZeneca is developing NXY-059 under a Licence agreement with Centaur Pharmaceuticals, Inc. (Santa Clara, CA, U.S.A).
Abbreviations
- AR-R15896
S-(+)-α-phenyl-2-pyridine ethanamide dihydrochloride
- CCA
common carotid artery
- ECA
external carotid artery
- ICA
internal carotid artery
- MCA
middle cerebral artery
- NXY-059
disodium 4-[(tert-butylimino) methyl] benzene - 1, 3 - disulphonate N-oxide
- PBN
α - phenyl - N - tert-butylnitrone
- S - PBN
2-sulphophenyl-N-tert-butylnitrone
- TTX
2,3,5-triphenyltetrazolium chloride
References
- BAKKER M.H., FOSTER A.C. An investigation of the mechanisms of delayed neurodegeneration caused by direct injection of quinolinate into the rat striatum in vivo. Neuroscience. 1991;42:387–395. doi: 10.1016/0306-4522(91)90383-y. [DOI] [PubMed] [Google Scholar]
- BEDERSON J., PITTS L., TSUJI M., NISHIMURA M., DAVIS R., BARTKOWSKI H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurological examination. Stroke. 1986;17:472–476. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- BELAYEV L., BUSTO R., IKEDA M., RUBIN L.L., KAJIWARA A., MORGAN L., GINSBERG M.D. Protection against blood-brain barrier disruption in focal cerebral ischemia by the type IV phosphodiesterase inhibitor BBB022: a quantitative study. Brain Res. 1998;787:277–285. doi: 10.1016/s0006-8993(97)01499-6. [DOI] [PubMed] [Google Scholar]
- BETZ A.L. Alterations in cerebral endothelial cell function in ischemia. Adv. Neurol. 1996;71:301–313. [PubMed] [Google Scholar]
- BRINKER G., FRANKE C., HOEHN M., UHLENKUKEN U., HOSSMANN K.A. Thrombolysis of cerebral clot embolism in rat: effect of treatment delay. Neuroreport. 1999;10:3269–3272. doi: 10.1097/00001756-199911080-00004. [DOI] [PubMed] [Google Scholar]
- BUSTO R., DIETRICH W.D., GLOBUS M.Y., VALDOS A., STEINBERG P., GINSBERG W.D. Small differences in intraischemic brain temperature critically determine the extent of ischemic brain injury. J. Cereb. Blood Flow Metab. 1987;7:729–738. doi: 10.1038/jcbfm.1987.127. [DOI] [PubMed] [Google Scholar]
- CARCIA J.H., LUI K.F., RELTON J.K. Interleukin-1 receptor antagonist decreases the number of necrotic neurons in rats with middle cerebral artery occlusion. Am. J. Pathol. 1995;146:1477–1486. [PMC free article] [PubMed] [Google Scholar]
- CAO X., PHILLIS J. α-Phenyl-tert-butyl-nitrone reduces cortical infarct and edema in rats subjected to focal ischemia. Brain Res. 1994;644:267–272. doi: 10.1016/0006-8993(94)91689-6. [DOI] [PubMed] [Google Scholar]
- CLARK W.M., MADDEN K.P., ROTHLEIN R., ZIVIN J.A. Reduction of central nervous system ischemic injury by monoclonal antibody to intracellular adhesion molecule. J. Neurosurg. 1991;75:623–627. doi: 10.3171/jns.1991.75.4.0623. [DOI] [PubMed] [Google Scholar]
- CORBETT D., EVANS S., THOMAS L., WANG D., JONAS R. MK 801 reduces cerebral ischemic injury by inducing hypothermia. Brain Res. 1990;514:300–304. doi: 10.1016/0006-8993(90)91424-f. [DOI] [PubMed] [Google Scholar]
- DOWDEN J., REID C., DOOLEY P, CORBETT D. Diazepam-induced neuroprotection: dissociating the effects of hypothermia following global ischemia. Brain Res. 1999;829:1–6. doi: 10.1016/s0006-8993(99)01229-9. [DOI] [PubMed] [Google Scholar]
- FACCHINETTI F., DAWSON V.L., DAWSON T.M. Free radicals as mediators of neuronal injury. Cell. Mol. Neurobiol. 1998;18:667–682. doi: 10.1023/A:1020685903186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FOSTER A.C., GILL R., WOODRUFF G.N. Neuroprotective effects of MK-801 in vivo: Selectivity and evidence for delayed degeneration mediated by NMDA receptor activation. J. Neurosci. 1988;8:4745–4754. doi: 10.1523/JNEUROSCI.08-12-04745.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HALLENBECK J.M. Inflammatory reactions at the blood-endothelial interface in acute stroke. Adv. Neurol. 1996;71:281–300. [PubMed] [Google Scholar]
- KATO H., KOGURE K., LUI X.H., ARAKI T., ITOYAMA Y. Progressive expression of immunomolecules on activated microglia and invading leukocytes following focal ischemia in the rat. Brain Res. 1996;734:203–212. [PubMed] [Google Scholar]
- KURODA S., KATSURA K., HILLERED L., BATES T.E., SIESJÖ B.K. Delayed treatment with α-phenyl-N-tert-butyl nitrone (PBN) attenuates secondary mitochondrial dysfunction after transient focal cerebral ischemia in the rat. Neurobiol. Dis. 1996;3:149–157. doi: 10.1006/nbdi.1996.0015. [DOI] [PubMed] [Google Scholar]
- KURODA S., TSUCHIDATE R., SMITH M-L., MAPLES K.R., SIESJÖ B.K. Neuroprotective effects of a novel nitrone, NXY-059, after transient focal ischemia in the rat. J. Cereb. Blood Flow Metab. 1999;19:778–787. doi: 10.1097/00004647-199907000-00008. [DOI] [PubMed] [Google Scholar]
- LEES K.R., SHARMA A.K., BARER D., FORD G.A., KOSTULAS V., CHENG Y.F., ODERGREN T. Tolerability and pharmacokinetics of the nitrone NXY-059 in patients with acute stroke. Stroke. 2001;32:675–680. doi: 10.1161/01.str.32.3.675. [DOI] [PubMed] [Google Scholar]
- LEINONEN J.S., AHONEN J.P., LONNROT K., JEHKONEN M., DASTIDAR P., MOLNAR G., ALHO H. Low plasma antioxidant activity is associated with high lesion volume and neurological impairment in stroke. Stroke. 2000;31:33–39. doi: 10.1161/01.str.31.1.33. [DOI] [PubMed] [Google Scholar]
- MARSHALL J.W.B., DUFFIN K.J., GREEN A.R., RIDLEY R.M. NXY-059, a free radical trapping agent, substantially lessens the functional disability resulting from cerebral ischemia in a primate species. Stroke. 2001;32:190–198. doi: 10.1161/01.str.32.1.190. [DOI] [PubMed] [Google Scholar]
- MASSIEU L., THEDINGA K.H., MCVEY M., FAGG G.E. A comparative analysis of the neuroprotective properties of competitive and uncompetitive N-methyl-D-aspartate receptor antagonists in vivo: Implications for the process of excitotoxic degeneration and its therapy. Neuroscience. 1993;55:883–892. doi: 10.1016/0306-4522(93)90305-y. [DOI] [PubMed] [Google Scholar]
- MOHR J.P., GAUTIER J.C., HIER D., STEIN R.W.Middle cerebral artery Stroke: Pathophysiology, Diagnosis and Management 1986Vol.1New York: Churchill Livingstone; 377–450.eds Barnett HJM, Stein BM, Mohr JP Yatsu FM [Google Scholar]
- NURSE S., CORBETT D. Neuroprotection after several days of mild drug-induced hypothermia. J. Cereb. Blood Flow Metab. 1996;16:474–480. doi: 10.1097/00004647-199605000-00014. [DOI] [PubMed] [Google Scholar]
- OLIVER C., STARKE-REED P., STADTMAN E., LUI G., CARNEY J.M., FLOYD R.A. Oxidative damage to brain proteins, loss of glutamine synthetase activity, and production of free radicals during ischemia/reperfusion-induced injury to gerbil brain. Proc. Natl. Acad. Sci. U.S.A. 1990;87:5144–5147. doi: 10.1073/pnas.87.13.5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAXINOS G., WATSON C. The rat brain in stereotaxic co-ordinates 1986London: Academic Press; 2nd edn [Google Scholar]
- PEELING J., DEL BIGIO M.R., CORBETT D., GREEN A.R., JACKSON D.M. Efficacy of disodium 4-[(tert-butylimino)methyl]benzene-1,3-disulfonate N-oxide NXY-059), a free radical trapping agent, in a rat model of hemorrhagic stroke. Neuropharmacology. 2001;40:433–439. doi: 10.1016/s0028-3908(00)00170-2. [DOI] [PubMed] [Google Scholar]
- PHILLIS J., CLOUGH-HELFMAN C. Protection from cerebral ischemia injury in gerbils with the spin trap agent N-tert-butyl-α-phenylnitrone (PBN) Neurosci. Letts. 1990;116:315–319. doi: 10.1016/0304-3940(90)90093-o. [DOI] [PubMed] [Google Scholar]
- RELTON J.K., ROTHWELL N.J. Interleukin-1 receptor antagonist inhibits ischemic and excitotoxic neuronal damage in the rat. Brain Res. Bull. 1996;29:243–246. doi: 10.1016/0361-9230(92)90033-t. [DOI] [PubMed] [Google Scholar]
- RINGELSTEIN E.B., BINIEK R., WEILLER C., AMMELING B., NOLTE P.N., THRON A. Type and extent of hemispheric brain infarctions and clinical outcome in early and delayed middle cerebral artery recanalization. Neurology. 1992;42:289–298. doi: 10.1212/wnl.42.2.289. [DOI] [PubMed] [Google Scholar]
- SCHULZ J.B., HENSHAW D.R., SIWEK D., JENKINS B.G., FERRANTE R.H., CIPOLONI P.B., KOWALL N.W., ROSEN B.R., BEAL M.F. Involvement of free radicals in excitotoxicity in vivo. J. Neurochem. 1995a;64:2239–2247. doi: 10.1046/j.1471-4159.1995.64052239.x. [DOI] [PubMed] [Google Scholar]
- SCHULZ J.B., MATHEWS R.T., JENKINS B.G., BRAR P., BEAL M.F. Improved therapeutic window for treatment of histotoxic hypoxia with a free radical spin trap. J. Cereb. Blood Flow Metab. 1995b;15:948–952. doi: 10.1038/jcbfm.1995.120. [DOI] [PubMed] [Google Scholar]
- SYDSERFF S.G., CROSS A.J., WEST K.J., GREEN A.R. The effect of chlormethiazole on neuronal damage in a model of transient focal ischaemia. Br. J. Pharmacol. 1995;114:1631–1635. doi: 10.1111/j.1476-5381.1995.tb14950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TASDEMIROGLU E., CHRISTENBERRY P.D., ARDELL J.L., CHRONISTER R.B., TAYLOR A.E. Effects of antioxidants on the blood-brain barrier and postischemic hyperemia. Acta Neurochir. 1994;131:302–309. doi: 10.1007/BF01808631. [DOI] [PubMed] [Google Scholar]
- THE NINDS T-PA STROKE STUDY GROUP Tissue plasminogen activator for acute ischemic stroke. New Engl. J. Med. 1995;333:1581–1587. doi: 10.1056/NEJM199512143332401. [DOI] [PubMed] [Google Scholar]
- THE NINDS T-PA STROKE STUDY GROUP Generalized efficacy of t-PA for acute stroke. Subgroup analysis of the NINDS t-PA Stroke Trial. Stroke. 1997;28:2119–2125. doi: 10.1161/01.str.28.11.2119. [DOI] [PubMed] [Google Scholar]
- THE NINDS T-PA STROKE STUDY GROUP Effects of tissue plasminogen activator for acute ischemic stroke at one year. New Engl. J. Med. 1999;340:1781–1787. doi: 10.1056/NEJM199906103402302. [DOI] [PubMed] [Google Scholar]
- YANG Y., LI Q., SHUAIB A. Neuroprotection by 2-h postischemia administration of two free radical scavengers, alpha-phenyl-n-tert-butyl-nitrone (PBN) and N-tert-butyl-(2-sulfophenyl)-nitrone (S-PBN), in rats subjected to focal embolic cerebral ischemia. Exp. Neurol. 2000;163:39–45. doi: 10.1006/exnr.2000.7364. [DOI] [PubMed] [Google Scholar]
- ZHANG R.L., CHOPP M., JIANG N. Anti-intercellular adhesion molecule-1 antibody reduces ischemic cell damage after transient but not permanent middle cerebral artery occlusion in the Wistar rat. Stroke. 1995;26:1438–1443. doi: 10.1161/01.str.26.8.1438. [DOI] [PubMed] [Google Scholar]
- ZHAO Q., PAHLMARK K., SMITH M.L., SIESJÖ BK. Delayed treatment with the spin trap α-phenyl-tert-butyl-nitrone (PBN) reduces infarct size following transient middle cerebral artery occlusion in rats. Acta Physiol. Scand. 1994;152:349–350. doi: 10.1111/j.1748-1716.1994.tb09816.x. [DOI] [PubMed] [Google Scholar]
- ZHAO A., CHENG M., MAPLES K.R., MA J.Y., BUCHAN A.M. NXY-059, a novel free radical trapping compound, reduces cortical infarction after permanent focal cerebral ischemia in the rat. Brain Res. 2001;909:46–50. doi: 10.1016/s0006-8993(01)02618-x. [DOI] [PubMed] [Google Scholar]