

Abstract
We investigated why angiotensin (Ang) I and II induce vasoconstriction with similar potencies, although Ang I-II conversion is limited.
Construction of concentration-response curves to Ang I and II in porcine femoral arteries, in the absence or presence of the AT1 or AT2 receptor antagonists irbesartan and PD123319, revealed that the ≈2 fold difference in potency between Ang I and II was not due to stimulation of different AT receptor populations by exogenous and locally generated Ang II.
Measurement of Ang I and II and their metabolites at the time of vasoconstriction confirmed that, at equimolar application of Ang I and II, bath fluid Ang II during Ang I was ≈18 times lower than during Ang II and that Ang II was by far the most important metabolite of Ang I. Tissue Ang II was 2.9±1.5% and 12.2±2.4% of the corresponding Ang I and II bath fluid levels, and was not affected by irbesartan or PD123319, suggesting that it was located extracellularly.
Since ≈15% of tissue weight consists of interstitial fluid, it can be calculated that interstitial Ang II levels during Ang II resemble bath fluid Ang II levels, whereas during Ang I they are 8.8 – 27 fold higher. Consequently at equimolar application of Ang I and II, the interstitial Ang II levels differ only 2 – 4 fold.
Interstitial, rather than circulating Ang II determines vasoconstriction. Arterial Ang I, resulting in high interstitial Ang II levels via its local conversion by ACE, may be of greater physiological importance than arterial Ang II.
Keywords: Angiotensin, interstitial fluid, femoral artery, angiotensin receptor antagonists
Introduction
Angiotensin (Ang) I and II both induce vasoconstriction via stimulation of AT1 receptors, the former following its conversion to Ang II by ACE and/or chymase (Urata et al., 1990; Wei et al., 1999). In healthy volunteers, during intrabrachial Ang I infusion, one third of arterially delivered Ang I is converted to Ang II in the forearm vascular bed (Saris et al., 2000), whereas in isolated human coronary arteries mounted in organ baths, the Ang II levels reached in the organ bath at the time of contraction are <1% of the levels of Ang I (Maassenvandenbrink et al., 1999). Yet, despite the relatively low Ang II levels during Ang I administration, the potencies of Ang I and II in vivo as well as in vitro are similar (Borland et al., 1998; Maassenvandenbrink et al., 1999; Saris et al., 2000; Voors et al., 1998). Likewise, in isolated perfused rat hindquarters, renin infusion causes equal increases in perfusion pressure as Ang II infusion, although the Ang II levels during renin perfusion are five times lower than during Ang II perfusion (Hilgers et al., 1998).
Several explanations may solve these discrepancies. Firstly, in view of the vasodilator effects of AT2 receptors (Muller et al., 1998; Zwart et al., 1998), it is possible that locally generated Ang II reaches different AT receptors than exogenously applied Ang II. For instance, exogenous Ang II may stimulate both endothelial (vasodilatory) AT2 receptors and AT1 receptors on smooth muscle cells, whereas locally generated Ang II might stimulate the latter predominantly. Secondly, Ang I and II are degraded to vasoconstrictor (Ang-(2 – 8) and Ang-(3 – 8), also known as Ang III and IV) and vasodilator (Ang-(1 – 7)) metabolites (Li et al., 1995; Ferrario et al., 1997; Tom et al., 2001), and this may affect their vasoconstrictor potency. Thirdly, the Ang II levels in the circulation and organ bath may not be representative for the tissue (interstitial fluid) levels in the immediate vicinity of the AT receptors that actually cause vasoconstriction. This theory has also been put forward to explain the beneficial effects of ACE inhibitors on blood pressure and cardiac remodelling in the absence of clear reductions in circulating Ang II (Nussberger et al., 1986; van Kats et al., 2000). Fourthly, results obtained with Ang I and II in organ baths, as well as during systemic infusion in vivo, may be of little physiological relevance, since normally vascular Ang II (i.e., the Ang II that is responsible for vasoconstriction) is largely derived from local synthesis by renin and converting enzymes from angiotensinogen (Campbell, 1987; Danser et al., 1992b; Admiraal et al., 1993). In this respect, it is of interest to note that the majority of vascular Ang I-II conversion, at least in vitro, is mediated by chymase, a converting enzyme that is located in the adventitia (Urata et al., 1993; Borland et al., 1998; Maassenvandenbrink et al., 1999).
To investigate these issues, we constructed Ang I and II concentration-response curves (CRCs) in porcine femoral and carotid arteries mounted in organ baths. Ang I and Ang II-mediated effects were also studied following preconstriction, to facilitate the detection of vasodilator effects, and in the presence of AT1 and AT2 receptor antagonists. Additional studies with Ang I were performed in the presence of the ACE inhibitor quinaprilat, to investigate the importance of ACE versus chymase. Subsequently, we compared the tissue and bath fluid levels of Ang I and II and their metabolites at the time of maximal vasoconstriction. The amount of vascular Ang II presented in interstitial fluid was estimated after correction of the tissue levels for Ang II bound to AT receptors (van Kats et al., 1997). Finally, we studied the vasoconstrictor effects of Ang I and II applied to the adventitial side of perfused carotid arteries, thereby mimicking the vascular origin of Ang II mediating vasoconstriction.
Methods
Chemicals
Angiotensin I and II, prostaglandin F2α (PGF2α), 9,11-dideoxy-11α,9α-epoxymethano-prostaglandin F2α (U46619) and substance P (acetate salt) were purchased from Sigma (Zwijndrecht, The Netherlands). Irbesartan was a kind gift of Bristol-Myers Squibb (Princeton, NJ, U.S.A.) PD123319 and quinaprilat were a kind gift of Parke Davis (Hoofddorp, The Netherlands). Irbesartan was dissolved in ethanol and quinaprilat in dimethylsulphoxide. All other chemicals were dissolved in distilled water.
Tissue collection
Porcine femoral and carotid arteries were obtained from 33 2 – 3 month-old pigs (Yorkshire x Landrace, weight 10 – 15 kg). The pigs had been used in in-vivo experiments studying the effects of α-adrenoceptor and serotonin receptor (ant)agonists under pentobarbital (600 mg, i.v.) anaesthesia (Willems et al., 2001). The Ethics Committee of the Erasmus University Rotterdam dealing with the use of animals for scientific experiments approved the protocol for this investigation. After sacrificing the animal with an overdose of pentobarbital, the right femoral artery and both carotid arteries were removed and stored overnight in a cold, oxygenated Krebs bicarbonate solution of the following composition (mM): NaCl 118, KCl 4.7, CaCl2 2.5, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25 and glucose 8.3; pH 7.4.
Organ bath studies with femoral arteries
To study Ang I- and II-induced vasoconstriction and/or vasodilatation, femoral arteries were cut into segments of approximately 3 – 4 mm length, suspended on stainless steel hooks in 15-ml organ baths containing Krebs bicarbonate solution, aerated with 95% O2/5% CO2, and maintained at 37°C. All vessel rings were allowed to equilibrate for at least 30 min and the organ bath fluid was refreshed every 15 min during this period. Changes in tissue contractile force were recorded with a Harvard isometric transducer (South Natick, MA, U.S.A.) The vessel rings, stretched to a stable force of about 50 mN, were exposed to 30 mM K+ twice. Subsequently, the tissue was exposed to 100 mM K+to determine the maximal contractile response to K+. The rings were then allowed to equilibrate in fresh organ bath fluid for 30 min. Next, the vessel rings were pre-incubated for 30 min with or without the AT1 antagonist irbesartan (1 μM), the AT2 antagonist PD123319 (1 μM) or the ACE inhibitor quinaprilat (10 μM). Thereafter, Ang I or Ang II (0.3 nM – 10 μM) CRCs were constructed. To facilitate detection of vasodilatation due to AT2 receptor stimulation, Ang I or II CRCs were also constructed following preconstriction with 1 μM PGF2α or 10 nM U46619. Preconstrictions amounted to approximately 40 – 50% of the maximal contraction induced by 100 mM K+. In the presence of irbesartan, the amount of PGF2α or U46619 required to obtain such preconstrictions was approximately 5 – 6 times higher, because, at a concentration of 1 μM, irbesartan acts as an antagonist of the thromboxane A2/prostaglandin endoperoxide receptor (Li et al., 2000). After the addition of Ang I or II, it took 10 – 15 min to reach a stable contraction. Subsequent angiotensin concentrations were applied as soon as a stable contraction had been reached. Construction of the CRC was discontinued when desensitization occurred, i.e. when subsequent angiotensin doses elicited no response or decreased contraction. At the end of the experiment the functional integrity of the endothelium was verified by observing relaxation to 1 nM substance P or 100 nM bradykinin after pre-constriction with PGF2α or U46619. To measure the release of newly formed Ang II into the organ bath fluid during Ang I CRCs, bath fluid samples (150 μl) for Ang II measurements were taken as soon as a stable contraction had been reached. All samples were collected in chilled tubes containing 15 μl inhibitor solution (125 mM disodium EDTA and 25 mM 1,10-ortho-phenanthroline) and 15 μl 0.1% bovine serum albumin (BSA) in distilled water. The samples were stored at −80°C until analysis.
Metabolism studies with femoral arteries
To study vascular Ang I-II conversion and Ang I and II metabolism in further detail, femoral arteries were cut into segments of approximately 3 – 4 mm length (weight 10 – 45 mg; mean 22 mg), put into test tubes containing 1.0 ml Krebs bicarbonate solution (‘incubation fluid'), aerated with 95% O2/5% CO2, and maintained at 37°C. After a 10-min equilibration period, vessel rings were pre-incubated for 30 min with or without irbesartan (1 μM), PD123319 (1 μM) or quinaprilat (10 μM). Next, Ang I (10 or 100 pmol) or Ang II (10 pmol), randomly combined with 125I-Ang I (100,000 c.p.m.) or 125I-Ang II (100,000 c.p.m.), were added to the incubation fluid. The use of radiolabelled angiotensins facilitates the detection of the various angiotenin metabolites, by gamma-counting after their high-performance liquid chromatographic (HPLC) separation (see below). After either 15 or 60 min the vessel segments were removed, washed in fresh Krebs solution and dried on tissue paper. The remaining incubation fluid (≈1 ml) as well as the dried segment were rapidly frozen in liquid nitrogen and stored at −80°C until analysis.
Perfusion and organ bath studies with carotid arteries
To study whether adventitial application of Ang I and II elicits a response that is different from the combined luminal and adventitial application obtained after addition of Ang I or II to the organ bath in the above set-up, carotid arteries (diameter 2 – 3 mm) were divided in 3 – 4 mm rings as well as 1 – 1.5 cm sections. The rings were mounted in organ baths as described above. The vessels sections, which did not contain side branches, were mounted horizontally in a double-jacketed 4-ml bath containing a carbogenated Krebs bicarbonate solution and maintained at 37°C, as described by Hulsmann et al. (1992). Krebs solution was perfused through the vessels using a roller pump (Ismatec IPS, Zürich, Switzerland). This approach allows the adventitial application (i.e., into the bath) of drugs. Fluids were refreshed every 15 min. Changes in pressure were recorded with a Viggo-spectramed disposable pressure transducer (Bilthoven, The Netherlands). The vessel sections and rings, at a stable force of 44 mmHg and 20 mN, respectively, were exposed to 30 mM K+ twice. Subsequently, the tissues were exposed to 100 mM K+ to determine the maximal contractile response to K+. The vessels were then allowed to equilibrate in fresh organ bath fluid for 30 min. Thereafter, Ang I or II (1 nM – 1 μM) CRCs were constructed as described above. At the end of the experiment the functional integrity of the endothelium of the vessel rings was verified by observing relaxation to 100 nM bradykinin after pre-constriction with 10 nM U46619.
Measurements of angiotensins
In the metabolism studies, Ang I and II and radiolabelled Ang I and II in vascular tissue and incubation fluid were measured as described previously (Danser et al., 1992a; van Kats et al., 1997), using SepPak extraction and HPLC separation. In short, frozen vessel segments were homogenized in 4 ml ice-cold 0.1 M HCl/80% ethanol. The homogenate was centrifuged at 20,000×g for 10 min at 4°C. Ethanol in the supernatant was evaporated under constant air flow. The remainder of the supernatant was diluted in 8 ml 1% ortho-phosphoric acid and concentrated on SepPak cartridges. SepPak extracts were dissolved in 100 μl HPLC elution buffer and injected into the HPLC column. Incubation fluid was directly applied to the column without prior SepPak extraction. The concentrations of Ang I and II and of radiolabelled Ang I and II in the HPLC eluate fractions were measured by radioimmunoassays and gamma counting, respectively. Measurements were not corrected for losses occurring during extraction. These losses were maximally 20 – 30% (van Kats et al., 1997). The lowest Ang I levels that could be measured were 50 fmol g−1 tissue and 10 fmol ml−1 incubation fluid. The lowest Ang II levels that could be measured were 25 fmol−1 tissue and 5 fmol ml−1 incubation fluid. The lowest 125I-Ang I and 125I-Ang II levels that could be measured were 500 c.p.m. g−1 tissue and 100 c.p.m. ml−1 incubation fluid.
In the organ bath studies, Ang I and II in organ bath fluid were measured by radioimmunoassay without prior SepPak extraction or HPLC separation (Maassenvandenbrink et al., 1999). The lowest Ang I and II levels that could be measured in these experiments were 20 and 10 fmol ml−1 bath fluid, respectively.
Data presentation and statistical analysis
Data are given as mean±s.e.mean. Data of the functional studies are expressed as a percentage of the maximal contraction to 100 mM K+ (88±5 mN in the organ bath studies with femoral arteries (n=15); 220±20 mmHg in the perfusion studies with carotid arteries (n=7) and 111±8 mN in the organ bath studies with the carotid arteries (n=7)). Data of metabolism experiments are expressed as percentage of the angiotensin concentration in the bath fluid at the start (t=0) of the experiment, assuming that 1 g of tissue equals 1 ml of fluid (de lannoy et al., 1998). To allow statistical evaluation, angiotensin levels that were below the detection limit were taken to be equal to this limit.
CRCs were analysed using the logistic function described by de lean et al. (1978) to obtain pEC50 (−10logEC50) values. Statistical analysis was by ANOVA, followed by post hoc evaluation according to Dunnett. P values <0.05 were considered significant. Statistics were performed using the software package SigmaStat.
Results
Organ bath studies with femoral arteries
In non-preconstricted femoral arteries (Figure 1), Ang I and II displayed similar maximal effects (Emax 31±2% and 37±7%, respectively, n=5). Ang I was 2 fold less potent than Ang II (pEC50 8.21±0.13 and 8.59±0.09, respectively; P<0.05). In femoral arteries that had been preconstricted with PGF2α or U46619 to 34±4 mN (or ≈45% of the maximal contraction induced by 100 mM K+; n=13), Ang I and II increased contraction further to 63±9 and 62±6% (Figure 1). The potencies of Ang I and II in the preconstricted vessels (pEC50 8.08±0.11 and 8.51±0.05, respectively) were not different from those in the non-preconstricted vessels.
Figure 1.
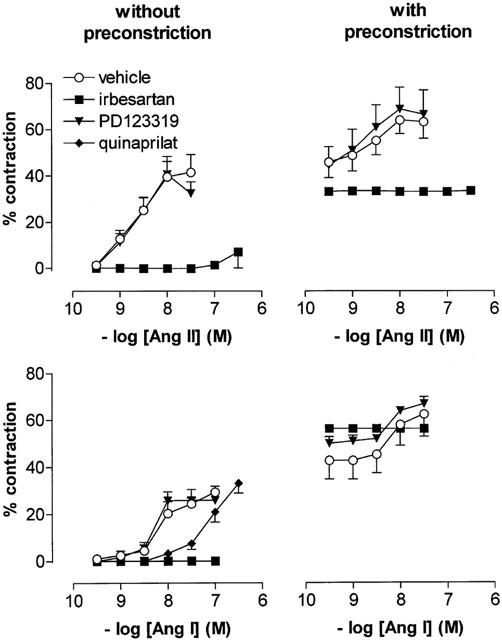
Contractions of femoral arteries, at baseline and after preconstriction with 1 μM PGF2α or 10 nM U46619, to Ang II (upper panels; n=8) or Ang I (lower panels; n=5) in the absence or presence of 1 μM irbesartan, 1 μM PD123319 or 10 μM quinaprilat. Data are expressed as a percentage (mean±s.e.mean) of the response to 100 mM K+.
The AT1 receptor antagonist irbesartan abolished all contractile responses to both Ang I and II, whereas the AT2 receptor antagonist PD123319 was ineffective under all circumstances. In non-preconstricted vessels, the ACE inhibitor quinaprilat shifted the Ang I CRC approximately 10 fold to the right (pEC50 7.12±0.10, n=5; P<0.001 vs control).
Despite the small difference in potency between Ang I and II, the Ang II levels measured in the organ bath fluid during the Ang I CRC, at the time a stable contraction had been reached, were <3% of the levels of Ang I. Figure 2 (left panel) relates the organ bath fluid levels of Ang II measured during the Ang I CRC (‘endogenous Ang II') to the contractile response. For comparison the CRC obtained with exogenous Ang II is also shown in this Figure. The pEC50 value of endogenous Ang II (9.84±0.14) illustrates that, for a given contractile response, the organ bath fluid levels of endogenous Ang II are ≈18 fold lower than for exogenous Ang II.
Figure 2.
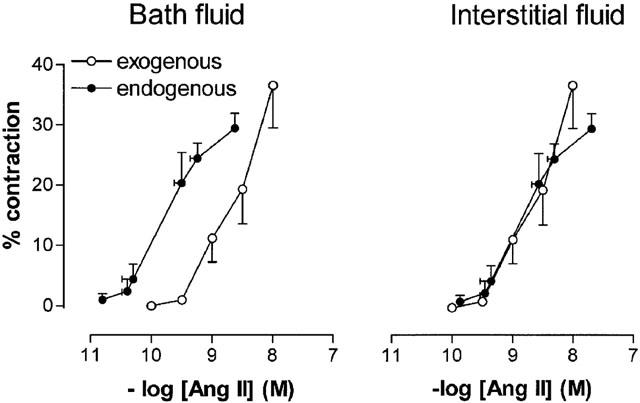
Contractile responses of femoral arteries versus organ bath fluid Ang II levels (left panel) or interstitial fluid Ang II levels (right panel), during Ang I (‘endogenous') or Ang II (‘exogenous') application. Contractions are expressed as a percentage (mean±s.e.mean; n=5) of the response to 100 mM K+. Interstitial fluid levels of endogenous and exogenous Ang II were calculated by multiplying the Ang II levels in the organ bath with the interstitial fluid/incubation fluid Ang II concentration ratios obtained 15 min following Ang I (8.5±4.4) or Ang II (1.1±0.2) administration in the metabolism studies.
Metabolism studies with femoral arteries
Incubation fluid
Femoral artery segments slowly metabolized 125I-Ang I and Ang I (Figure 3). After 1 h of incubation at 37°C, 56±6% (n=4) and 64±9% (n=6) of the incubation fluid levels at t=0 was still present as intact 125I-Ang I and Ang I, respectively. Metabolism was partly due to Ang I-II conversion, as evidenced by the rise in the 125I-Ang II and Ang II levels over time (Figure 3). After 1 h, the incubation fluid levels of 125I-Ang II and Ang II were 4.5±2.6 and 5.4±2.0% of the respective 125I-Ang I and Ang I levels at t=0. HPLC separation of the incubation fluid samples revealed that 125I-Ang II was by far the most important metabolite of 125I-Ang I (Figure 4). The metabolism of 125I-Ang II and Ang II occurred at a similarly slow rate as the metabolism of 125I-Ang I and Ang I, resulting in incubation fluid 125I-Ang II and Ang II levels after 1 h that were >60% of the levels at t=0 (data not shown). Metabolites that could be detected in the incubation fluid during incubation with 125I-Ang II were 125I-Ang-(4 – 8) and 125I-tyrosine. Irbesartan and PD123319 did not affect the metabolism of 125I-Ang I, 125I-Ang II, Ang I or II (Figure 3). Quinaprilat virtually completely prevented the appearance of 125I-Ang II and Ang II in the incubation fluid during incubation of the vessel segments with 125I-Ang I and Ang I, but did not significantly affect the decrease in 125I-Ang I or Ang I over time (Figure 3), thereby indicating that Ang I-II conversion is not the only metabolic pathway which results in destruction of Ang I.
Figure 3.
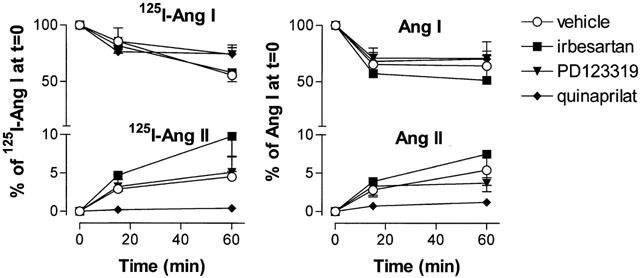
Metabolism of 125I-Ang I and generation of 125I-Ang II (left panel) and metabolism of Ang I and generation of Ang II (right panel) by femoral artery rings incubated in 1 ml incubation fluid containing 125I-Ang I (100,000 c.p.m.) or Ang I (10 or 100 pmol) in the absence or presence of 1 μM irbesartan, 1 μM PD123319 or 10 μM quinaprilat. Incubation fluid levels are expressed as a percentage (mean±s.e.mean, n=4 – 6) of the 125I-Ang I and Ang I levels at t=0.
Figure 4.
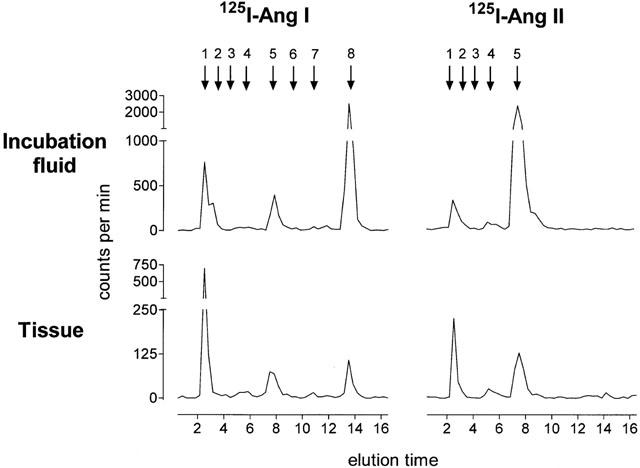
HPLC elution profile of 125I-labelled antiotensins in femoral artery tissue (22 mg) and incubation fluid (100 μl) obtained after incubation of femoral artery rings at 37°C for 60 min in 1 ml incubation medium containing 100,000 c.p.m. 125I-Ang I or 125I-Ang II. The retention times of 125I-labelled standards are indicated by arrows at the top (1, 125I-tyrosine; 2, 125I-Ang-(1 – 7); 3, 125I-Ang III; 4, 125I-Ang-(4-8); 5, 125I-Ang II; 6, 125I-Ang IV, 7, 125I-Ang-(2 – 10); 8, 125I-Ang I).
Vessel segment
Under all conditions that were investigated, the angiotensin levels measured in the vessel segment at t=15 min were not different from those at t=60 min. Therefore, the data in Figure 5 represent the average of the measurements at these two time points (‘steady-state' levels). The 125I-Ang I and Ang I levels in the vessel segments (expressed per g tissue) were 3 – 6 times lower than the concomitant levels in the incubation fluid (expressed per ml fluid). In contrast, the tissue 125I-Ang II and Ang II levels, measured after the addition of 125I-Ang I and Ang I to the incubation fluid, we as high as (or slightly higher than) the incubation fluid 125I-Ang II and Ang II levels. Quinaprilat reduced these levels by >75% (data not shown).
Figure 5.
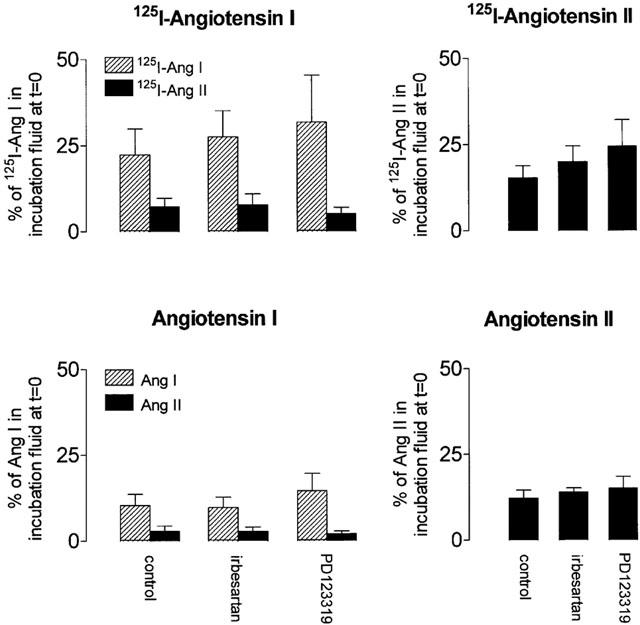
Steady-state tissue 125I-Ang I, 125I-Ang II, Ang I and II levels in femoral artery rings incubated with 125I-Ang I (100,000 c.p.m.) or Ang I (10 or 100 pmol) (left panels) or with 125I-Ang II (100,000 c.p.m.) or Ang II (10 pmol) (right panels) in the absence or presence of 1 μM irbesartan or 1 μM PD123319. Data are expressed as a percentage of the radiolabelled and endogenous Ang I or II levels in incubation fluid at t=0 (mean±s.e.mean, n=4 – 6).
The tissue 125I-Ang II and Ang II levels, measured after the addition of 125I-Ang II and Ang II to the incubation fluid were 5 – 6 times lower than the concomitant levels in the incubation fluid. Other metabolites that could be detected in the vessel segments during 125I-Ang I or 125I-Ang II application were 125I-Ang-(2 – 10), 125I-Ang-(4 – 8) and 125I-tyrosine (Figure 4).
Neither irbesartan nor PD123319 affected the tissue 125I-Ang II and Ang II levels under any of the conditions studied, thereby indicating that the majority of tissue 125I-Ang II and Ang II was not bound to cell surface – or internalized AT receptors. Assuming therefore that all tissue 125I-Ang II and Ang II is located extracellularly (i.e., present in vascular interstitial fluid, which accounts for ≈15% of tissue weight (Linde, 1975; Plank et al., 1976; Reed & Wiig, 1984; Katz et al., 1997), it can be calculated that during 125I-Ang I and Ang I application, the interstitial 125I-Ang II and Ang II levels are 49±17 and 19±10%, respectively, of the initial 125I-Ang I and Ang I levels in the incubation fluid. Moreover, during 125I-Ang II and Ang II application, the interstitial 125I-Ang II and Ang II levels are 103±23 and 81±16%, respectively, of the initial 125I-Ang II and Ang II levels in the incubation fluid. In other words, 15 min after the application of 125I-Ang I and Ang I, the interstitial fluid 125I-Ang II and Ang II levels are, respectively, 27±13 and 8.8±4.4 times higher than the 125I-Ang II and Ang II levels in the incubation fluid (the latter levels are shown in Figure 3), whereas 15 min after the application of 125I-Ang II and Ang II, the interstitial fluid 125I-Ang II and Ang II levels equal the levels in the incubation fluid.
Perfusion and organ bath studies with carotid arteries
Ang I and II displayed similar maximal effects, both in the organ bath studies and the perfusion experiments (Figure 6). Under both conditions, Ang I tended to be less potent than Ang II (pEC50 7.53±0.11 vs 7.76±0.08 in the organ bath studies and 7.73±0.07 vs 8.00±0.11 in the perfusion experiments), but the difference did not reach statistical significance. Ang II, but not Ang I, was more potent after adventitial application (perfusion experiment) than after application to the organ bath (P<0.05).
Figure 6.
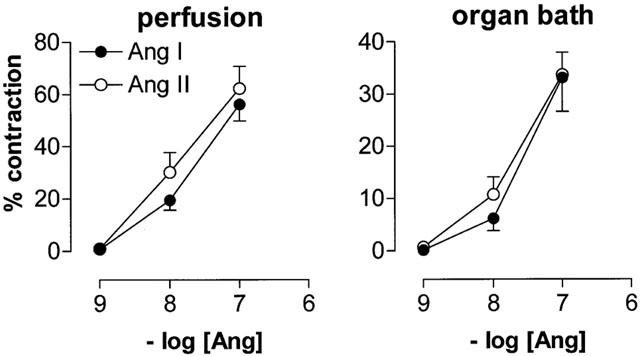
Contractions of carotid arteries to Ang I or II added adventitially to perfused vessel sections (left panel) or into the bath fluid of vessel rings mounted in organ baths (right panel). Data are expressed as a percentage (mean±s.e.mean; n=7) of the response to 100 mM K+.
Discussion
In the present in-vitro study, in agreement with previous studies measuring Ang II following Ang I administration (Danser et al., 1995; Maassenvandenbrink et al., 1999; Saris et al., 2000), the levels of Ang II in the organ bath during the construction of the Ang I CRC in femoral artery rings were <3% of the Ang I levels at the time of maximal vasoconstriction. Yet, in femoral, as well as in carotid arteries, the potency of Ang I was only 2 fold lower than that of Ang II.
The results obtained with the AT1 receptor antagonist irbesartan and the AT2 receptor antagonist PD123319 do not support the possibility that exogenous Ang II and locally generated Ang II stimulate different AT receptors. Neither at baseline, nor following preconstriction, we were able to demonstrate Ang I- or II-induced vasodilatation via AT2 receptors in femoral arteries. Thus, either such receptors do not exist in porcine femoral arteries, or AT2 receptors in these vessels mediate other, non-blood pressure-related effects in this vessel (e.g., effects on vascular growth and remodelling (Stoll et al., 1995; Yamada et al., 1996; van Kesteren et al., 1997). Measurement of radiolabelled angiotensin metabolites during incubation of femoral artery rings with 125I-Ang I and 125I-Ang II, revealed that the metabolism of Ang I and II by these rings is relatively slow (half life >1 h), and that Ang II is by far the most important metabolite of Ang I. The enzyme responsible for Ang I-II conversion is ACE, since the ACE inhibitor quinaprilat shifted the Ang I CRC 10 fold to the right and prevented the appearance of Ang II in the organ bath. The latter contrasts with previous findings in human vessels (Borland et al., 1998; Maassenvandenbrink et al., 1999), where chymase was by far the most important converting enzyme. Neither the vasoconstrictor metabolites Ang III and IV (Li et al., 1995), nor the vasodilator metabolite Ang-(1 – 7) (Ferrario et al., 1997; Tom et al., 2001) were present in detectable amounts. Thus, the similar potencies of Ang I and II are not related to the generation of different vasoconstrictor and/or vasodilator metabolites from Ang I and II, nor to differences in the rapidity of their metabolism. Moreover, the rightward shift of Ang I CRC caused by quinaprilat, and the similar potencies of Ang I and II despite the >100 fold lower affinity of Ang I for the AT1 receptor (Whitebread et al., 1989; Dudley et al., 1990), exclude the possibility that Ang I mediated its vasoconstrictor effects directly, independent of its conversion to Ang II.
During adventitial application to perfused carotid arteries, Ang I and II were equipotent, thereby showing that their similar potency in organ bath studies (Borland et al., 1998; Voors et al., 1998; Maassenvandenbrink et al., 1999) or following arterial infusion in vivo (Saris et al., 2000) is not a consequence of the experimental set-up. The perfusion studies in the present investigation were performed with carotid arteries, since the many sidebranches of femoral arteries did not allow such studies with these vessels. The slightly higher potency of Ang II following adventitial application as compared to combined adventitial+luminal application (i.e., into the organ bath) is in agreement with earlier studies showing a higher metabolic clearance rate of angiotensins after luminal application than after adventitial application (Danser et al., 1995). Apparently, the majority of the vascular angiotensin metabolizing enzymes is located on endothelial cells.
Based on the above findings, we reasoned that Ang I administration results in high tissue Ang II levels with limited Ang II release into the surrounding bath fluid, and that these high tissue Ang II levels might explain why the potency of Ang I is similar to that of Ang II. We therefore expected the vascular Ang II levels during Ang I administration to be much higher than the organ bath fluid Ang II levels. As shown in Figures 3 and 5, this was not the case. Fifteen minutes after Ang I administration (i.e., at the time maximal vasoconstriction had occurred), the vascular Ang II levels were approximately as high as the bath fluid levels. The tissue concentrations at 15 min represented steady-state levels, since similar concentrations were measured after 60 min. Tissue Ang II, however, is localized in one or more different compartments, and a low tissue level therefore does not argue against the possibility that Ang II is present at high concentrations in a specific tissue compartment. For instance, previous in-vivo (Siragy et al., 1995; Dell'italia et al., 1997) and in-vitro (de lannoy et al., 1997; 1998) studies have shown that interstitial fluid Ang II may be higher than circulating Ang II. In addition, tissue Ang II is, at least partly, localized intracellularly, because of its internalization via AT1 receptors (van Kats et al., 1997; 2001). Such internalization does not appear to occur after binding to AT2 receptors (Matsubara, 1998; van Kats et al., 1997), nor via non-AT1, non-AT2 receptor-mediated mechanisms (van Kats et al., 2001). Finally, Ang II may be bound to AT receptors on the cell surface. Receptor binding protects Ang II against rapid metabolism by degrading enzymes (Schuijt et al., 1999). Since in pigs, like in humans, there is currently no evidence for the existence of AT receptors other than the AT1 and AT2 receptor, we distinguished cellular (membrane bound or internalized) and extracellular Ang II by measuring tissue Ang II in the presence of irbesartan or PD123319. No significant differences were found in comparison with the control situation. This suggests that only a small fraction of tissue Ang II in femoral artery rings is bound to cell surface – and/or internalized AT receptors, and, thus, that the majority of vascular Ang II is localized in interstitial fluid. Assuming therefore that all vascular Ang II is present in the interstitial space, and taking into account that this compartment represents ≈15% of tissue weight (Linde & Chisolm, 1975; Plank et al., 1976; Reed & Wiig, 1984; Katz et al., 1997), it follows that interstitial Ang II during Ang I administration is 8.8 – 27 times higher than bath fluid Ang II.
The latter was not the case during Ang II administration. Tissue Ang II following Ang II administration was also restricted to the interstitial fluid compartment (i.e., not affected by irbesartan or PD123319), and the interstitial Ang II fluid levels during Ang II administration were not different from the bath fluid Ang II levels. The latter contrasts with earlier findings in isolated perfused rat Langendorff hearts, where interstitial Ang II during Ang II infusion was <35% of arterial Ang II (de lannoy et al., 1998), and with in-vivo studies in pigs (Schuijt et al., 1999) and dogs (Dell'italia et al., 1997), where interstitial fluid Ang II during Ang II infusion was <5% of arterial Ang II. These differences are most likely related to the amount of metabolizing enzymes present under the various experimental conditions. In addition, chronic exposure to Ang II is known to affect the level of angiotensin-metabolizing enzymes, including ACE (Schunkert et al., 1993).
Why are the interstitial Ang II levels during Ang I administration higher than the bath fluid Ang II levels? In porcine arteries, Ang I-II conversion occurs both after luminal and after adventitial Ang I administration (Danser et al., 1995), suggesting that ACE is present both in the endothelium and the adventitia. In addition, ACE has been demonstrated on rat and human vascular smooth muscle cells (Coulet et al., 2001). Most likely therefore, Ang I-II conversion occurs by ACE within the interstitial space. This does not exclude Ang I-II conversion on either the endothelial or adventitial surface. In fact, in view of the much larger volume of the organ bath (15 ml) as compared to the interstitial fluid volume of a 22 mg vessel segment (≈3 μl), it can be calculated that the majority (>95%) of Ang II is present in the organ bath, and thus is probably generated on the vascular surface. In the artery segment, the interstitial space is continuously supplied, via diffusion (de lannoy et al., 1997), with Ang I from the bath fluid, and this Ang I will be converted by ACE into Ang II in close proximity of the AT1 receptors. The small volume of the interstitial space allows a rapid rise in the interstitial Ang II levels, resulting in a steady state within 15 min. The rate-limiting factor in this process is most likely the diffusion of Ang I into the interstitial space (de lannoy et al., 2001). Although the bath fluid Ang II levels continued to rise over time, previous studies (Danser et al., 1995; Maassenvandenbrink et al., 1999) have shown that it is unlikely that these levels will become as high as the interstitial Ang II levels. This is due to the small volume of the interstitial space, thereby allowing interstitial Ang II to contribute only marginally to the organ bath levels of Ang II. Indeed, in previous perfusion studies with porcine carotid arteries, we found no Ang II in the perfusion fluid upon adventitial Ang I administration, nor was Ang II detectable in bath fluid upon luminal Ang I administration (Danser et al., 1995). Moreover, we were also unable to demonstrate significant release of Ang II from tissue sites into the circulation (Danser et al., 1992b; Admiraal et al., 1993).
Taken together, the similar potencies of Ang I and II in the present and previous studies (Danser et al., 1995; Maassenvandenbrink et al., 1999; Saris et al., 2000), can be fully explained on the basis of the interstitial Ang II levels that are reached in the vessel wall during Ang I and II application. During Ang I application, at the time of vasoconstriction, these levels are almost one order of magnitude higher than the levels in the organ bath, whereas during Ang II application the interstitial and bath fluid Ang II levels are virtually equal. The right panel of Figure 2 illustrates the consequences of this concept. Our results not only explain why circulating Ang II does not always represent tissue (interstitial) Ang II (Nussberger et al., 1986; Campbell, 1987; van Kats et al., 2000), but they also raise the possibility that circulating Ang I, through its local conversion into Ang II, is of greater physiological importance than circulating Ang II. When considering the latter, it should be kept in mind that the circulating Ang I levels are higher than those of Ang II (Danser et al., 1992b; Admiraal et al., 1993).
Abbreviations
- ACE
angiontensin-converting enzyme
- Ang
angiotensin
- AT1
angiotensin II type 1
- AT2
angiotensin II type 2
- CRC
concentration response curve
- PGF2α
prostaglandin F2α
- U46619
9,11-dideoxy-11α,9α-epoxymethano-prostaglandin F2α
References
- ADMIRAAL P.J.J., DANSER A.H.J., JONG M.S., PIETERMAN H., DERKX F.H.M., SCHALEKAMP M.A.D.H. Regional angiotensin II production in essential hypertension and renal artery stenosis. Hypertension. 1993;21:173–184. doi: 10.1161/01.hyp.21.2.173. [DOI] [PubMed] [Google Scholar]
- BORLAND J.A.A., CHESTER A.H., MORRISON K.A., YACOUB M.H. Alternative pathways of angiotensin II production in the human saphenous vein. Br. J. Pharmacol. 1998;125:423–428. doi: 10.1038/sj.bjp.0702018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAMPBELL D.J. Circulating and tissue angiotensin systems. J. Clin. Invest. 1987;79:1–6. doi: 10.1172/JCI112768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COULET F., GONZALEZ W., BOIXEL C., MEILHAC O., PUEYO M.E., MICHEL J.B. Endothelium-independent conversion of angiotensin I by vascular smooth muscle cells. Cell Tissue Res. 2001;303:227–234. doi: 10.1007/s004410000309. [DOI] [PubMed] [Google Scholar]
- DANSER A.H.J., CHOWDURY S., DE LANNOY L.M., VAN DER GIESSEN W.J., SAXENA P.R., SCHALEKAMP M.A.D.H. Conversion and degradation of [125I] labelled angiotensin I in isolated perfused porcine coronary and carotid arteries. Cardiovasc. Res. 1995;29:789–795. [PubMed] [Google Scholar]
- DANSER A.H.J., KONING M.M.G., ADMIRAAL P.J.J., DERKX F.H.M., VERDOUW P.D., SCHALEKAMP M.A.D.H. Metabolism of angiotensin I by different tissues in the intact animal. Am. J. Physiol. 1992a;263:H418–H428. doi: 10.1152/ajpheart.1992.263.2.H418. [DOI] [PubMed] [Google Scholar]
- DANSER A.H.J., KONING M.M.G., ADMIRAAL P.J.J., SASSEN L.M.A., DERKX F.H.M., VERDOUW P.D., SCHALEKAMP M.A.D.H. Production of angiotensins I and II at tissue sites in intact pigs. Am. J. Physiol. 1992b;263:H429–H437. doi: 10.1152/ajpheart.1992.263.2.H429. [DOI] [PubMed] [Google Scholar]
- DE LANNOY L.M., DANSER A.H.J., BOUHUIZEN A.M.B., SAXENA P.R., SCHALEKAMP M.A.D.H. Localization and production of angiotensin II in the isolated perfused rat heart. Hypertension. 1998;31:1111–1117. doi: 10.1161/01.hyp.31.5.1111. [DOI] [PubMed] [Google Scholar]
- DE LANNOY L.M., DANSER A.H.J., VAN KATS J.P., SCHOEMAKER R.G., SAXENA P.R., SCHALEKAMP M.A.D.H. Renin-angiotensin system components in the interstitial fluid of the isolated perfused rat heart. Local production of angiotensin I. Hypertension. 1997;29:1240–1251. doi: 10.1161/01.hyp.29.6.1240. [DOI] [PubMed] [Google Scholar]
- DE LANNOY L.M., SCHUIJT M.P., SAXENA P.R., SCHALEKAMP M.A.D.H., DANSER A.H.J. Angiotensin-converting enzyme is the main contributor to angiotensin I-II conversion in the interstitium of isolated perfused rat heart. J. Hypertens. 2001;19:959–965. doi: 10.1097/00004872-200105000-00017. [DOI] [PubMed] [Google Scholar]
- DE LEAN A., MUNSON P.J., RODBARD D. Simultaneous analysis of families of sigmoidal curves: application to bioassay, radioligand assay, and physiological dose-response curves. Am. J. Physiol. 1978;235:E97–E102. doi: 10.1152/ajpendo.1978.235.2.E97. [DOI] [PubMed] [Google Scholar]
- DELL'ITALIA L.J., MENG Q.C., BALCELLS E., WEI C.C., PALMER R., HAGEMAN G.R., DURAND J., HANKES G.H., OPARIL S. Compartmentalization of angiotensin II generation in the dog heart. Evidence for independent mechanisms in intravascular and interstitial spaces. J. Clin. Invest. 1997;100:253–258. doi: 10.1172/JCI119529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUDLEY D.T., PANEK R.L., MAJOR T.C., LU G.H., BRUNS R.F., KLINKEFUS B.A., HODGES J.C., WEISHAAR R.E. Subclasses of angiotensin II binding sites and their functional significance. Mol. Pharmacol. 1990;38:370–377. [PubMed] [Google Scholar]
- FERRARIO C.M., CHAPPELL M.C., TALLANT E.A., BROSNIHAN K.B., DIZ D.I. Counterregulatory actions of angiotensin-(1-7) Hypertension. 1997;30:535–541. doi: 10.1161/01.hyp.30.3.535. [DOI] [PubMed] [Google Scholar]
- HILGERS K.F., BINGENER E., STUMPF C., MULLER D.N., SCHMIEDER R.E., VEELKEN R. Angiotenases restrict locally generated angiotensin II to the blood vessel wall. Hypertension. 1998;31:368–372. doi: 10.1161/01.hyp.31.1.368. [DOI] [PubMed] [Google Scholar]
- HULSMANN A.R., RAATGEEP H.R., BONTA I.L., STIJNEN T., KERREBIJN K.F., DE JONGSTE J.C. The perfused human bronchiolar tube characteristics of a new model. J. Pharmacol. Toxicol. Methods. 1992;28:29–34. doi: 10.1016/1056-8719(92)90062-6. [DOI] [PubMed] [Google Scholar]
- KATZ S.A., OPSAHL J.A., LUNZER M.M., FORBIS L.M., HIRSCH A.T. Effect of bilateral nephrectomy on active renin, angiotensinogen, and renin glycoforms in plasma and myocardium. Hypertension. 1997;30:259–266. doi: 10.1161/01.hyp.30.2.259. [DOI] [PubMed] [Google Scholar]
- LI P., FUKUHARA M., DIZ D.I., FERRARIO C.M., BROSNIHAN K.B. Novel angtotensin II AT1 antagonist irbesartan prevents thromboxane A2-induced vasoconstriction in canine coronary arteries and human platelet aggregation. J. Pharmacol. Exp. Ther. 2000;292:238–246. [PubMed] [Google Scholar]
- LI Q., ZHANG L., PFAFFENDORF M., VAN ZWIETEN P.A. Comparative effects of angiotensin II and its degradation products angiotensin III and angiotensin IV in rat aorta. Br. J. Pharmacol. 1995;116:2963–2970. doi: 10.1111/j.1476-5381.1995.tb15951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LINDE B., CHISOLM G. The interstitial space of adipose tissue as determined by single injection and equilibration techniques. Acta Physiol. Scand. 1975;95:383–390. doi: 10.1111/j.1748-1716.1975.tb10065.x. [DOI] [PubMed] [Google Scholar]
- MAASSENVANDENBRINK A., DE VRIES R., SAXENA P.R., SCHALEKAMP M.A.D.H., DANSER A.H.J. Vasoconstriction by in-situ formed angiotensin II: role of ACE and chymase. Cardiovasc. Res. 1999;44:407–415. doi: 10.1016/s0008-6363(99)00249-7. [DOI] [PubMed] [Google Scholar]
- MATSUBARA H. Pathophysiological role of angiotensin II type 2 receptor in cardiovascular and renal diseases. Circ. Res. 1998;83:1182–1191. doi: 10.1161/01.res.83.12.1182. [DOI] [PubMed] [Google Scholar]
- MULLER C., ENDLICH K., HELWIG J. AT2 antagonist-sensitive potentiation of angiotensin II-induced constriction by NO blockade and its dependence on endothelium and P450 eicosanoids in rat renal vasculature. Br. J. Pharmacol. 1998;124:946–952. doi: 10.1038/sj.bjp.0701906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NUSSBERGER J., BRUNNER D.B., WAEBER B., BRUNNER H.R. Specific measurement of angiotensin metabolites and in vitro generated angiotensin II in plasma. Hypertension. 1986;8:476–482. doi: 10.1161/01.hyp.8.6.476. [DOI] [PubMed] [Google Scholar]
- PLANK B., RABERGER G., BRUGGER G., ADLER-KASTNER L. The determination of the myocardial extracellular space in the cat in vivo: a comparative methodological study. Basic Res. Cardiol. 1976;71:173–178. doi: 10.1007/BF01927869. [DOI] [PubMed] [Google Scholar]
- REED R.K., WIIG H. Compliance of the interstitial space in rats. III. Contribution of skin and skeletal muscle interstitial fluid volume to changes in total extracellular fluid volume. Acta Physiol. Scand. 1984;121:57–63. doi: 10.1111/j.1748-1716.1984.tb10457.x. [DOI] [PubMed] [Google Scholar]
- SARIS J.J., VAN DIJK M.A., KROON I., SCHALEKAMP M.A.D.H., DANSER A.H.J. Functional importance of angiotensin-converting enzyme-dependent in situ angiotensin II generation in the human forearm. Hypertension. 2000;35:764–768. doi: 10.1161/01.hyp.35.3.764. [DOI] [PubMed] [Google Scholar]
- SCHUIJT M.P., VAN KATS J.P., DE ZEEUW S., DUNCKER D.J., VERDOUW P.D., SCHALEKAMP M.A.D.H., DANSER A.H.J. Cardiac interstitial fluid levels of angiotensin I and II in the pig. J. Hypertens. 1999;17:1885–1891. doi: 10.1097/00004872-199917121-00017. [DOI] [PubMed] [Google Scholar]
- SCHUNKERT H., INGELFINGER J.R., HIRSCH A.T., PINTO Y., REMME W.J., JACOB H., DZAU V.J. Feedback regulation of angiotensin converting enzyme activity and mRNA levels by angiotensin II. Circ. Res. 1993;72:312–318. doi: 10.1161/01.res.72.2.312. [DOI] [PubMed] [Google Scholar]
- SIRAGY H.M., HOWELL N.L., RAGSDALE N.V., CAREY R.M. Renal interstitial fluid angiotensin. Modulation by anesthesia, epinephrine, sodium depletion, and renin inhibition. Hypertension. 1995;25:1021–1024. doi: 10.1161/01.hyp.25.5.1021. [DOI] [PubMed] [Google Scholar]
- STOLL M., STECKELINGS U.M., PAUL M., BOTTARI S.P., METZGER R., UNGER T. The angiotensin AT2-receptor mediates inhibition of cell proliferation in coronary endothelial cells. J. Clin. Invest. 1995;95:651–657. doi: 10.1172/JCI117710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOM B., DE VRIES R., SAXENA P.R., DANSER A.H.J. Bradykinin potentiation by angiotensin-(1-7) and angiotensin-converting enzyme (ACE) inhibitors correlates with ACE C- and N-domain blockade. Hypertension. 2001;38:95–99. doi: 10.1161/01.hyp.38.1.95. [DOI] [PubMed] [Google Scholar]
- URATA H., BOEHM K.D., PHILIP A., KINOSHITA A., GABROVSEK J., BUMPUS F.M., HUSAIN A. Cellular localization and regional distribution of an angiotensin II-forming chymase in the heart. J. Clin. Invest. 1993;91:1269–1281. doi: 10.1172/JCI116325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- URATA H., HEALY B., STEWART R.W., BUMPUS F.M., HUSAIN A. Angiotensin II-forming pathways in normal and failing human hearts. Circ. Res. 1990;66:883–890. doi: 10.1161/01.res.66.4.883. [DOI] [PubMed] [Google Scholar]
- VAN KATS J.P., DUNCKER D.J., HAITSMA D.B., SCHUIJT M.P., NIEBUUR R., STUBENITSKY R., BOOMSMA F., SCHALEKAMP M.A.D.H., VERDOUW P.D., DANSER A.H.J. Angiotensin-converting enzyme inhibition and angiotensin II type 1 receptor blockade prevent cardiac remodeling in pigs after myocardial infarction: role of tissue angiotensin II. Circulation. 2000;102:1556–1563. doi: 10.1161/01.cir.102.13.1556. [DOI] [PubMed] [Google Scholar]
- VAN KATS J.P., VAN MEEGEN J.R., VERDOUW P.D., DUNCKER D.J., SCHALEKAMP M.A.D.H., DANSER A.H.J. Subcellular localization of angiotensin II in kidney and adrenal. J. Hypertens. 2001;19:583–589. doi: 10.1097/00004872-200103001-00010. [DOI] [PubMed] [Google Scholar]
- VAN KESTEREN C.A.M., VAN HEUGTEN H.A.A., LAMERS J.M.J., SAXENA P.R., SCHALEKAMP M.A.D.H., DANSER A.H.J. Angiotensin II-mediated growth and antigrowth effects in cultured neonatal rat cardiac myocytes and fibroblasts. J. Mol. Cell. Cardiol. 1997;29:2147–2157. doi: 10.1006/jmcc.1997.0448. [DOI] [PubMed] [Google Scholar]
- VOORS A.A., PINTO Y.M., BUIKEMA H., URATA H., OOSTERGA M., ROOKS G., GRANDJEAN J.G., GANTEN D., VAN GILST W.H. Dual pathway for angiotensin II formation in human internal mammary arteries. Br. J. Pharmacol. 1998;125:1028–1032. doi: 10.1038/sj.bjp.0702150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEI C.C., MENG Q.C., PALMER R., HAGEMAN G.R., DURAND J., BRADLEY W.E., FARRELL D.M., HANKES G.H., OPARIL S., DELL'ITALIA L.J. Evidence for angiotensin-converting enzyme- and chymase-mediated angiotensin II formation in the interstitial fluid space of the dog heart In vivo. Circulation. 1999;99:2583–2589. doi: 10.1161/01.cir.99.19.2583. [DOI] [PubMed] [Google Scholar]
- WHITEBREAD S., MELE M., KAMBER B., DE GASPARO M. Preliminary biochemical characterization of two angiotensin II receptor subtypes. Biochem. Biophys. Res. Commun. 1989;163:284–291. doi: 10.1016/0006-291x(89)92133-5. [DOI] [PubMed] [Google Scholar]
- WILLEMS E.W., HEILIGERS J.P.C., DE VRIES P., TOM B., KAPOOR K., VILLALON C.M., SAXENA P.R. A61603-induced vasoconstriction in porcine carotid vasculature: involvement of a non-adrenergic mechanism. Eur. J. Pharmacol. 2001;417:195–201. doi: 10.1016/s0014-2999(01)00898-6. [DOI] [PubMed] [Google Scholar]
- YAMADA T., HORIUCHI M., DZAU V.J. Angiotensin II type 2 receptor mediates programmed cell death. Proc. Natl. Acad. Sci. U.S.A. 1996;93:156–160. doi: 10.1073/pnas.93.1.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZWART A.S., DAVIS E.A., WIDDOP R.E. Modulation of AT1 receptor-mediated contraction of rat uterine artery by AT2 receptors. Br. J. Pharmacol. 1998;125:1429–1436. doi: 10.1038/sj.bjp.0702210. [DOI] [PMC free article] [PubMed] [Google Scholar]