

Abstract
This study was performed to determine the effect and action mechanisms of sodium butyrate (NaB) on the growth of breast cancer cells.
Butyrate inhibited the growth of all breast cancer cell lines analysed. It induced cell cycle arrest in G1 and apoptosis in MCF-7, MCF-7ras, T47-D, and BT-20 cells, as well as arrest in G2/M in MDA-MB-231 cells.
Transient transfection of MCF-7 and T47-D cells with wild-type and antisense p53 did not modify butyrate-induced apoptosis. Pifithrin-α, which inhibits the transcriptional activity of P53, did not modify cell growth or apoptosis of MCF-7 and T47-D cells treated with butyrate. These results indicate that P53 was not involved in butyrate-induced growth inhibition of breast cancer cells.
Treatment of MCF-7 cells with anti-Fas agonist antibody induced cell death, indicating that Fas was functional in these cells. Moreover, butyrate potentiated Fas-induced apoptosis, as massive apoptosis was observed rapidly when MCF-7 cells were treated with butyrate and anti-Fas agonist antibody. In addition, butyrate-induced apoptosis in MCF-7 cells was considerably reduced by anti-Fas antagonist antibody. Western blot analysis showed that butyrate increased Fas and Fas ligand levels (Fas L), indicating that butyrate-induced apoptosis may be mediated by Fas signalling.
These results demonstrate that butyrate inhibited the growth of breast cancer cells in a P53-independent manner. Moreover, it induced apoptosis via the Fas/Fas L system and potentiated Fas-triggered apoptosis in MCF-7 cells. These findings may open interesting perspectives in human breast cancer treatment strategy.
Keywords: Apoptosis, breast cancer, cell growth, Fas, P53, sodium butyrate
Introduction
Chemotherapy and radiation are widely used for cancer treatment. Common features of many anti-cancer drugs and radiation are induction of DNA damage followed by the activation of the tumour suppressor protein P53 as a mediator of their cellular effects. The accumulation and activation of wild-type P53 results in at least two pathways, cell-cycle arrest and apoptosis. P53-dependent G1 arrest is mediated by direct transactivation of the p21WAF1 gene that encodes the inhibitor of cyclin-dependent kinases (Bates & Vousden, 1994). How p53 triggers apoptosis is not completely elucidated, but it seems to involve both transcription-dependent and -independent mechanisms (Sionov & Haupt, 1999; Sheikh & Fornace, 2000). Several recent publications reported that anti-cancer drugs may induce apoptosis via the Fas/Fas L system (Yu et al., 1999; Glick et al., 1999; Sun et al., 2000). Fas is a cell surface receptor comprising a type I integral membrane protein that expresses a cytoplasmic death domain and belongs to the tumour necrosis factor receptor superfamily (Nagata & Golstein, 1995). Activation of Fas by its ligand (Fas L) or a cross-linking antibody results in the oligomerization of its intracellular death domain and the recruitment of pro-caspase-8 or -10, leading to the activation of these caspases which activate downstream caspases, resulting in apoptotic cell death (Juo et al., 1998).
Butyric acid is produced during the fermentation of fibre by endogenous intestinal bacteria and is also present in fruits, vegetables and milk fat. The sodium salt of butyric acid, sodium butyrate, inhibits cell growth by favouring cell cycle arrest and promotes differentiation in normal as well as transformed cells (Barnard & Warwick, 1993). Moreover, sodium butyrate induces apoptosis in a number of cancer cells (Mandal & Kumar, 1996; Bernhard et al., 1999; Giuliano et al., 1999). Due to its growth-inhibiting and differentiation-inducing ability, butyrate was tested in the treatment of leukaemia and solid tumours, together with analogues that have better pharmacodynamic properties, alone or in combination with other anti-cancer drugs (Miller et al., 1987; Conley et al., 1998). In molecular terms, the action of butyrate is probably related to deacetylase inhibition, leading to hyperacetylation of chromatin components such as histones and nonhistone proteins and alterations in gene expression (Pazin & Kadonaga, 1997; Struhl, 1998). Nevertheless, neither genetically- nor pharmacologically-induced global histone hyperacetylation leads to general gene transcription (Pazin & Kadonaga, 1997). The genes responsible for inhibiting proliferation and inducing differentiation or death by butyrate remain elusive, although some promising candidates have been identified, e.g. the cycline-dependent kinase inhibitor p21WAF1 (Archer et al., 1998), c-myc (Heruth et al., 1993) and the anti-apoptotic bcl-2 gene (Mandal & Kumar, 1996). It has also been demonstrated that butyrate decreases the level of P53 in transformed cells but induces apoptosis independently of p53 (Janson et al., 1997). Bonnotte et al. (1998) described how butyrate potentiated Fas-dependent apoptosis induced by the exposing the cells to Fas L. In this work, we studied the effect of butyrate on the growth of breast cancer cells as well as its action mechanisms. In particular, we examined a possible causal role of P53 and Fas in butyrate-induced cell growth inhibition and apoptosis.
Methods
Cell culture
Breast cancer cell lines MCF-7, T47-D, BT-20, MDA-MB-231 were obtained from the American Type Cell Culture Collection. MCF-7ras cell line, established by transfecting v-Ha-Ras cDNA in MCF-7 cells (Sommers et al., 1990), was a gift from Prof M. Crépin (Laboratoire de Recherche Oncologie Cellulaire et Moléculaire Humaine, Université de Paris Nord, France). Cells were routinely grown in EMEM medium supplemented with 10% foetal calf serum (FCS), 5 UI ml−1 insulin, 100 UI ml−1 streptomycin, 100 μg ml−1 penicillin, and 45 μg ml−1 gentamicin.
Cell growth assays
Monolayer growth
Cells were plated in triplicate at a density of 45,000 cells/cm2 in 60 mm dishes. Cells were allowed to adhere overnight, then treated with 1 mM butyrate for 5 days. At the end of the treatment, cells were harvested with 0.25% trypsin and counted using an haematocytometer. Cell viability was assessed by Trypan blue exclusion test.
Three-dimensional culture in type I collagen gel
Collagen gel was prepared as described previously by Fauquette et al. (1997). Briefly, eight volumes of 2 mg ml−1 collagen solution were mixed with one volume of EMEM 10X and one volume of 22.2 g/L sodium bicarbonate. A volume of 500 μl fresh medium containing 2×105 cells was dispensed in 16 mm wells. After gel formation, 1 ml of fresh medium complemented with 10% FCS containing butyrate was added and changed every other day. Cells were treated with 1 mM butyrate for 5 days. At the end of the experiment, collagen gel was digested by 500 UI ml−1 of type XI collagenase and the number of cells was determined by haematocytometer count.
Anchorage-independent growth in soft agar
Cells were seeded at a density of 5000 cells/cm2 in 35 mm wells. The bottom layer was prepared with EMEM containing 10% FCS and 0.56% Bacto agar (Difco, U.S.A.) Cells were seeded on the upper layer in triplicate, in EMEM supplemented with 3% FCS containing 0.37% Bacto agar and butyrate. Cells were treated with 1 mM butyrate for 7 – 10 days before colonies of at least 20 cells were counted.
Cell cycle analysis
Cells were treated with 2.5 mM butyrate for 24 or 48 h. Cells (1×106) were trypsinized and fixed in ethanol (70%, −20°C, 30 min). Fixed cells were washed with PBS and incubated with propidium iodide (20 μg ml−1, 20°C, 45 min). The cell suspension was then filtered and analysed using a Coulter Epics XL/XL-MCl cytometer.
Determination of apoptotic cells
Hoechst staining
Apoptosis was determined by morphological analysis after Hoechst 33258 staining, as previously described (Toillon et al., 2000). A minimum of 500 – 1000 cells was examined for each case and the results expressed as the number of apoptotic cells over the total number of cells counted.
DNA laddering
Cells were seeded in 60 mm dishes. After 48 h of treatment with butyrate, apoptotic DNA fragmentation was detected, as previously described (Staley et al., 1997) using the ApoAlertTM LM-PCR Ladder Assay kit (Clontech, U.S.A.)
Subcellular fractionation
Pre-confluent cells were washed twice with PBS and collected in 1 ml extraction buffer (50 mM HEPES pH 7.0, 50 mM KCl, 5 mM MgCl2, 5 mM EDTA, 10 mg ml−1 leupeptin, and 1 mM PMSF). Cells were lysed by five cycles of freezing in liquid nitrogen and thawing at 37°C. The crude lysate was centrifuged (100,000×g, 1 h at 4°C) and the resulting supernatant was separated from the pellet. The pellet (membrane and organelles) was resuspended in 1 ml extraction buffer and the supernatant (cytosol) was concentrated through a 3 kDa molecular weight cut-off amicon membrane (Pall Filtron Corporation).
Western blot
Pre-confluent cell cultures were washed with PBS and lysed for 30 min at 4°C in lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1% NP40, 0.1% SDS, 1 mM PMSF, 1 mM orthovanadate, 1 mM Na4P2O7, 10 mg/ml aprotinin, and 10 mg ml−1 leupeptin). The lysate was sonicated, boiled, and clarified by centrifugation (13,000×g, 5 min at 4°C). The protein concentration of each sample was determined using a Biorad protein assay kit. Each sample was then loaded onto 5% stacking/12% running SDS polyacrylamide gel. Immunoblots were incubated with primary antibody (1 : 1000 dilution, overnight at 4°C). Detection was performed using a goat anti-mouse or a rabbit anti-goat secondary antibody (1 : 2000 dilution, 1.5 h at room temperature) and an ECL detection system (Amersham).
Transient transfections
MCF-7 and T47-D cells were transiently co-transfected with a pEGFp-C1 plasmid containing Green Fluorescent Protein (GFP) cDNA (Clontech) and wild-type p53 or antisense p53 expressing vector (Iotsova & Stehelin, 1995). Briefly, 2×105 cells were seeded into 35 mm dishes and the following day, cells were transfected with 0.5 μg GFP-plasmid, 0.5 μg plasmid of interest, and 10 μl transfection reagent (lipofectamine for T47-D cells, and lipofectine for MCF-7 cells) in 1 ml Opti-MEM (Life Technologies, Inc.) After transfection, cells were cultured for a further 18 h in FCS-containing medium and treated with butyrate for an additional 24 h. Cells were then fixed in 4% paraformaldehyde for 20 min at 4°C and stained with Hoechst for apoptosis detection. A random count of 500 GFP-positive cells was performed for each assay.
Materials
All cell culture reagents were obtained from BioWhittaker (France) except insulin which was obtained from Organon (France). Sodium butyrate, Hoechst 33258, Trypan blue, and electrophoresis reagents were from Sigma (U.S.A.) Caspase inhibitors, Fas antagonist and agonist antibodies were from R&D Systems (U.K.) Anti-Fas (B-10) antibody for Western blot, anti-P53 (DO-1), anti-Bcl-2 (100) and anti-actin (1 – 19) antibodies were from Santa Cruz Biotechnology (U.S.A.) Anti-Fas L antibody for Western blot (clone 13) was from Transduction Laboratories (U.S.A.) Anti-Bax (Ab-3) antibody was from Calbiochem (France). ECL reagents were obtained from Amersham Life Science (France).
Statistical analysis
Statistical significance was measured by Student's paired t-test. P for each data set is shown in the legend.
Results
Butyrate inhibits breast cancer cell growth under various culture conditions
In monolayer culture and collagen I gel, cell growth was inhibited in all the cell lines tested after 5 days of treatment with 1 mM butyrate (Figure 1). In soft agar culture, colony formation was strongly reduced by 1 mM butyrate in all the cell lines tested (Figure 1). Besides reducing the number of colonies, butyrate also reduced the size of colonies in these cells (data not shown). Under all cell culture conditions, cell growth inhibition was more pronounced with 2.5 mM butyrate than 1 mM (data not shown).
Figure 1.
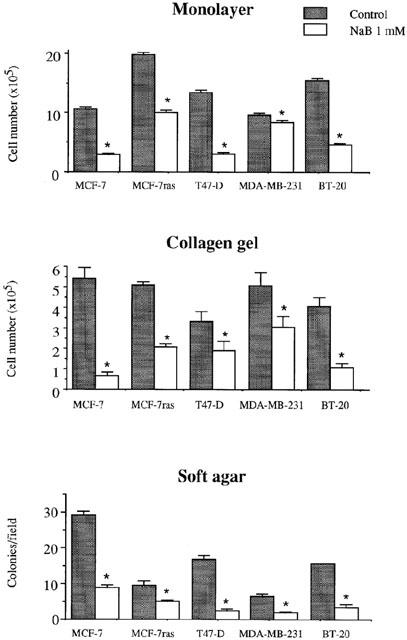
Effect of butyrate on the growth of breast cancer cell lines. Cells were treated with 1 mM butyrate in monolayer culture for 4 days, or in three-dimensional gel collagen culture for 5 days, or in soft agar culture for 7 – 13 days. The Figure shows the results of a triplicate assay in one experiment, which is representative of three independent experiments. *P<0.01.
Butyrate accumulates breast cancer cells in G1 and G2/M phases of the cell cycle
The distribution of breast cancer cells in different phases of the cell cycle was analysed after 24 and 48 h treatment with 2.5 mM butyrate (Figure 2). A decrease in the number of cells in the S phase was observed for all cell lines after 24 h treatment. After 48 h treatment, MCF-7, MCF-7ras, T47-D, and BT-20 cells were accumulated in G1 phase, whereas MDA-MB-231 cells were accumulated in G2/M phase.
Figure 2.
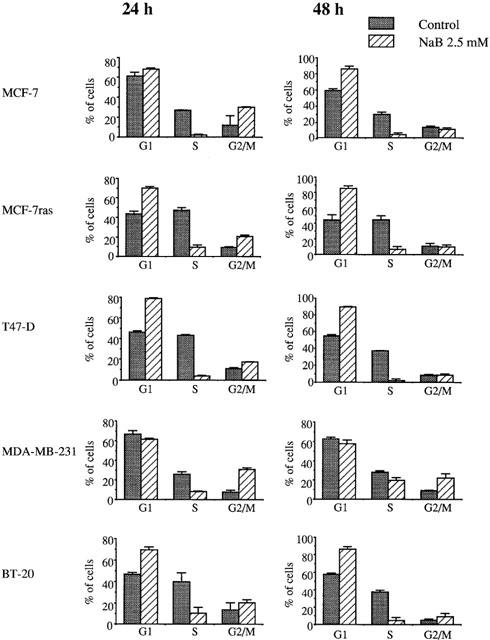
Cell-cycle histograms of breast cancer cell lines treated with butyrate. Cells were treated with 2.5 mM butyrate for 24 or 48 h, then analysed by flow cytometry of propidium iodide-stained nuclei.
Butyrate induces apoptosis of breast cancer cells
When cells were treated with butyrate for 24 h, MCF-7, MCF-7ras and BT-20 were induced into apoptosis in a dose-dependent manner, T47-D cells were only induced into apoptosis by 2.5 mM butyrate, and no apoptosis induction was observed in MDA-MB-231 cells (Figure 3). When cells were treated for 48 h, butyrate induced apoptosis in a dose-manner in all the cell lines tested except MDA-MB-231. Moreover, the percentages of apoptotic cells were higher after 48 than 24 h treatment (Figure 3). Analysis of DNA-fragmentation using a semi-quantitative ligation-mediated PCR assay showed a typical DNA ladder in MCF-7 and MCF-7ras cells treated with 2.5 mM butyrate for 48 h (Figure 4). However, no DNA ladder was detected in T47-D, MDA-MB-231, and BT-20 cells (Figure 4), suggesting that T47-D and BT-20 cells may undergo apoptosis without internucleosomal fragmentation of DNA.
Figure 3.
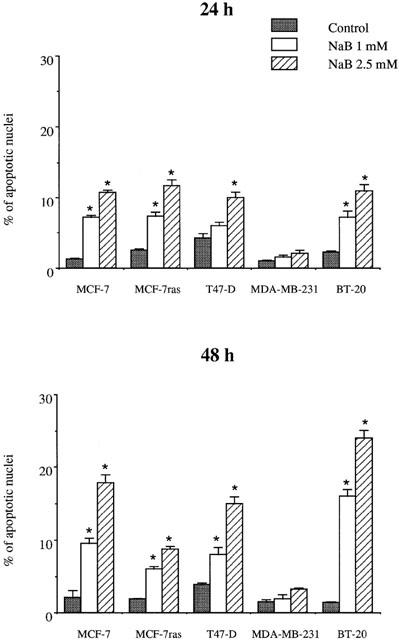
Effect of butyrate on apoptosis induction in breast cancer cell lines. Cells were treated with 1 or 2.5 mM butyrate for 24 or 48 h. Apoptosis was determined after Hoechst staining. The Figure shows the results of a triplicate assay in one experiment, which is representative of three independent experiments. *P<0.05.
Figure 4.
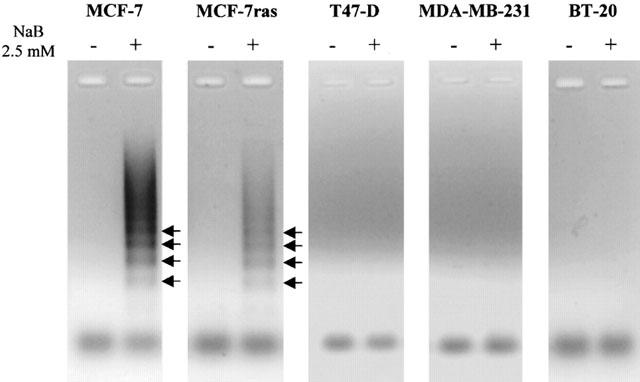
Analysis of DNA ladders in breast cancer cells. Cells were treated with 2.5 mM butyrate for 48 h. DNA fragmentation was displayed as described in Methods. Arrows show the formation of DNA ladders.
Butyrate modulates P53, Bcl-2 and Bax levels
As growth inhibition of breast cancer cells by butyrate is due to inhibition of proliferation and apoptosis induction (Figures 2 and 3), we performed immunoblots to determine the levels of different proteins controlling cell proliferation and/or apoptosis in MCF-7 and T47-D cells. Figure 5 shows that, in MCF-7 cells, butyrate increased P53 and Bax levels but decreased Bcl-2 levels, while, in T47-D cells, butyrate increased Bax levels but did not modify P53 and Bcl-2 levels.
Figure 5.
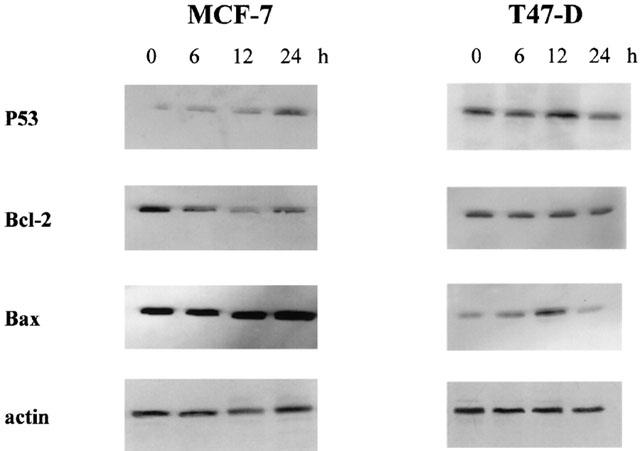
Western blot analysis of P53, Bcl-2, and Bax in MCF-7 and T47-D cells. Pre-confluent cells were treated with 2.5 mM butyrate for different periods of time. Proteins (20 μg) were electrophoresed and Western blots were performed as described in Methods. The experiment was repeated three times. The loading and transfer of equal amounts of protein were confirmed by immunodetection of actin.
P53 is not involved in butyrate-induced growth inhibition of breast cancer cells
As MCF-7 and T47-D express functional P53 (Sheikh et al., 1994), we next determined whether P53 was involved in butyrate-induced inhibition of proliferation and apoptosis. Cells were initially treated with 20 μM pifithrin-α, which specifically inhibits the transcriptional activity of P53 (Komarov et al., 1999). As shown in Figure 6, pifithrin-α did not modify cell growth inhibition (Figure 6A) or apoptosis induction (Figure 6B) when MCF-7 and T47-D cells were treated with 2.5 mM butyrate for 48 h. This suggested that the transcriptional activity of P53 was not involved in butyrate-induced growth inhibition. To further verify the role of P53 in butyrate-induced apoptosis, we transiently co-transfected cells with GFP cDNA and one of two p53 cDNA constructs: antisense or wild-type. As shown in Figure 7, transfection of MCF-7 and T47-D cells with all vectors increased basal apoptosis (by about 20%). However, when cells transfected with empty vector were treated with 2.5 mM butyrate for 24 h, a significant increase in apoptosis was observed compared to the control. In addition, neither wild-type nor antisense p53 modified butyrate-induced apoptosis when compared to empty vector-transfected cells.
Figure 6.
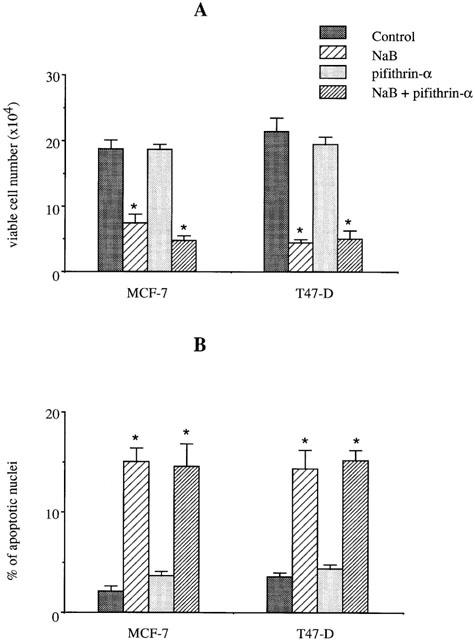
Effect of pifithrin-α on butyrate-triggered growth inhibition and apoptosis. MCF-7 and T47-D cells were treated with 2.5 mM butyrate in the presence or absence of 20 μg ml−1 pifithrin-α for 48 h. Viable cell number (A) and apoptosis (B) were determined as described in Methods. The Figure shows the results of a triplicate assay in one experiment, which is representative of three independent experiments. *P<0.01.
Figure 7.
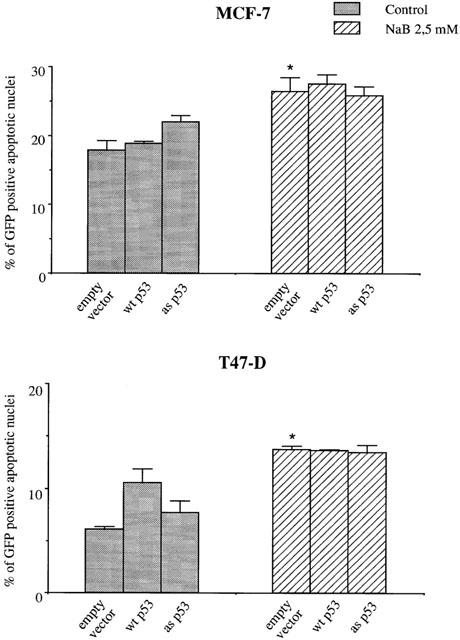
Effect of butyrate on the induction of apoptosis in cells transfected with p53. MCF-7 and T47-D cells were transiently co-transfected with a pL15TK plasmid containing wild-type (wt) or antisense (as) p53 cDNA and a pEGFp-C1 plasmid containing GFP cDNA (Clontech). Transfected cells were treated with 2.5 mM NaB for 24 h, then processed for apoptosis determination as described in Methods. The Figure shows the results of a triplicate assay in one experiment, which is representative of three independent experiments. *P<0.01.
Butyrate-induced apoptosis in MCF-7 cells is reduced by caspase inhibitors and involves Fas signalling
In current apoptosis models, an initial proapoptotic stress activates various caspases. In order to determine the transduction pathways involved in butyrate-triggered apoptosis, we treated MCF-7 cells with 2.5 mM butyrate in the presence or absence of cell-permeable caspase inhibitors for 48 h. As shown in Table 1, general caspase inhibitor as well as those of caspase-1, -2, -4, -9 -10 and -13 reduced butyrate-induced apoptosis by about half. To determine whether butyrate-induced apoptosis involved Fas signalling, we treated cells with anti-Fas agonist antibody and butyrate, alone or in combination, for different periods of time (Figure 8). Apoptosis increased after 20 h treatment with anti-Fas agonist and 24 h treatment with butyrate. When cells were treated with butyrate and anti-Fas agonist in combination, apoptosis was induced more rapidly, as a large percentage of apoptotic cells (about 15 – 20%) was already observed after 16 h treatment and about 50% of cells showed signs of apoptosis after 24 h treatment. Anti-Fas antagonist antibody had no effect on the basal apoptosis levels of MCF-7 cells, but it efficiently inhibited butyrate-triggered apoptosis (Figure 9), indicating that the basal apoptosis of breast cancer cells did not involve the Fas/Fas L system, while butyrate-triggered apoptosis did.
Table 1.
Effect of caspase inhibitors on NaB-induced apoptosis
Figure 8.
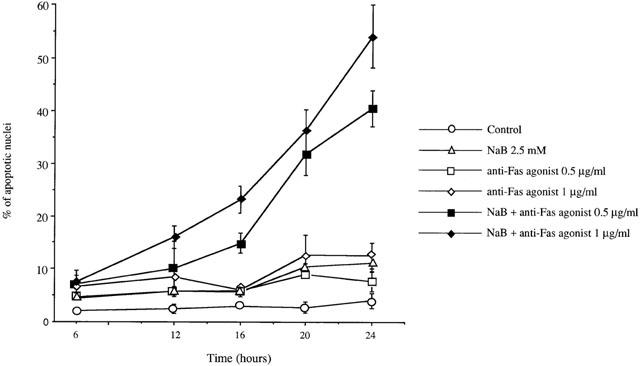
Butyrate potentiated Fas-induced apoptosis. MCF-7 cells were treated with 0.5 or 1 μg ml−1 anti-Fas agonist antibody and 2.5 mM butyrate, alone or in combination, for different periods of time. Apoptosis was determined after Hoechst staining. The Figure shows the results of a triplicate assay in one experiment, which is representative of three independent experiments.
Figure 9.
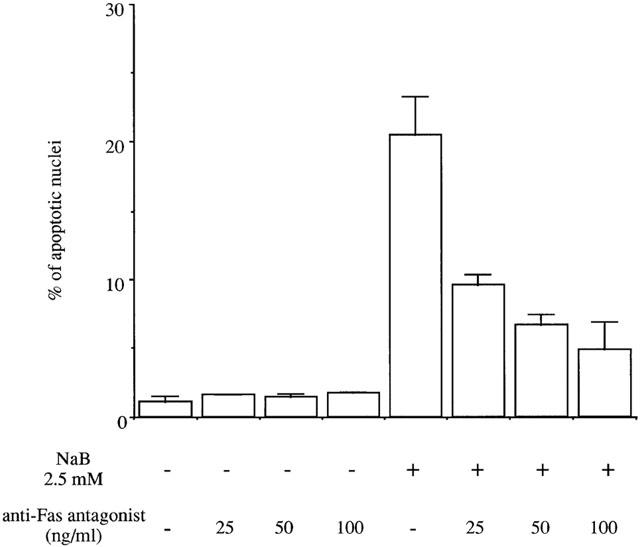
Butyrate-induced apoptosis was blocked by anti-Fas antagonist antibody. MCF-7 cells were treated with 2.5 mM butyrate in the presence or absence of 25, 50, or 100 ng ml−1 Fas antagonist antibody for 48 h. Apoptosis was determined after Hoechst staining. The Figure shows the results of a triplicate assay in one experiment, which is representative of three independent experiments.
Butyrate increases Fas and Fas L protein levels in MCF-7 cells
Since butyrate-induced apoptosis was modulated by anti-Fas antibodies, we determined whether butyrate could regulate Fas and Fas L levels. As shown in Figure 10 Fas levels in whole-cell extracts and membrane fractions increased after 16 h treatment with butyrate and the increase continued up to 24 h treatment. Concomitantly with the increase in membrane Fas, cytosolic Fas levels decreased with treatment time, suggesting a translocation of Fas from the cytosol to the membrane. Fas L levels were slightly increased by butyrate after 16 h treatment.
Figure 10.
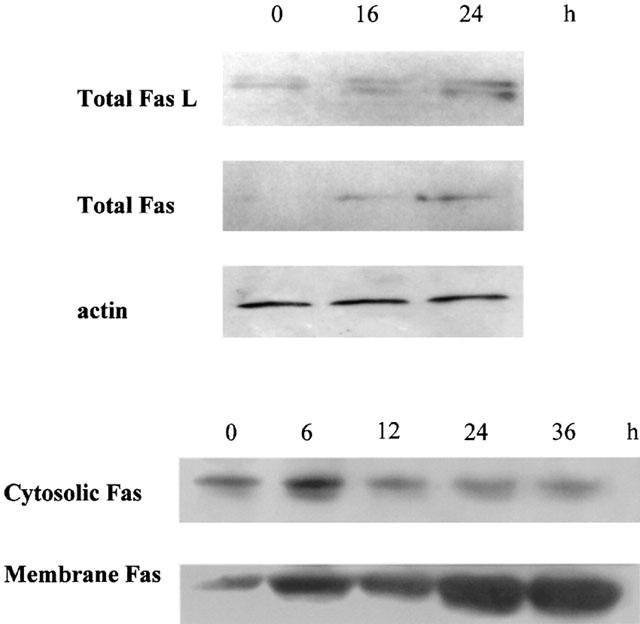
Butyrate increased Fas and Fas L levels. MCF-7 cells were treated with 2.5 mM butyrate for different periods of time. Proteins from whole-cell extracts, membrane, and cytosolic preparations (100 μg) were electrophoresed, and Western blots were performed as described in Methods. The experiment was repeated 4 times. The loading and transfer of equal amounts of protein were confirmed by immunodetection of actin.
Discussion
In this study, we have demonstrated that butyrate inhibits growth of all breast cancer cells analysed in monolayer culture, collagen gel, and soft agar. Three-dimensional collagen gel culture is closer to the in vivo situation than monolayer culture, and anchorage-independent growth in soft agar has been used extensively in clinical and experimental oncology as an in vitro indicator of malignancy. Our results indicate that butyrate has a wide spectrum of action on breast cancer cells. Moreover, butyrate induced cell cycle arrest in G1 and apoptosis in MCF-7, MCF-7ras, T47-D and BT-20 cells, and arrest in G2/M in MDA-MB-231 cells. These data demonstrate that butyrate is a potent growth inhibitor, not only for hormone-dependent MCF-7, MCF-7ras, and T47-D cells, but also for hormone-independent BT-20 and MDA-MB-231 cells. Therefore, butyrate could affect breast cancer cells, both in early (oestrogen-responsive) and advanced (oestrogen-resistant) stages of breast cancer development.
The P53 protein is a sequence-specific transcription factor that plays an important role in coupling DNA damage to the growth arrest and/or apoptotic response. However, we demonstrated that P53 was not involved in butyrate-induced growth inhibition of breast cancer cells in several ways: firstly, butyrate induced apoptosis and/or cell accumulation in the cell cycle of BT-20 and MDA-MB-231 cells which express a mutant, non-functional P53 (Sheikh et al., 1994); secondly, transient transfection with wild-type and antisense p53 did not modify butyrate-induced apoptosis in MCF-7 and T47-D cells, which express functional P53 (Sheikh et al., 1994) (Figures 6 and 7); finally, pifithrin-α, which inhibits the transcriptional activity of P53 (Komarov et al., 1999), did not modify cell growth or apoptosis in MCF-7 and T47-D treated with butyrate. These results are in agreement with the findings of Janson et al. (1997) who reported that butyrate induced P53-independent apoptosis in fibroblasts. The P53-independent growth inhibitory effect of butyrate is of major interest, as 15 to 60% of breast tumours contain p53 mutations (Bautista & Theillet, 1997; Hartman et al., 1997).
Fas is a cell surface receptor and belongs to the tumour necrosis factor receptor superfamily (Nagata & Golstein, 1995). It has recently been reported that Fas is implicated in the induction of apoptosis during mammary gland involution (Song et al., 2000). Defects in the Fas/Fas L apoptotic signalling pathway provide a survival advantage to cancer cells and may be implicated in tumorigenesis. Indeed, expression of Fas L by breast cancer cells is associated with the loss of Fas expression, thus eliminating the possibility of self-induced apoptosis. Moreover, Fas L-expressing tumour cells may be capable of avoiding host T cell-mediated immune control by eliminating activated anti-tumour T cells (Muschen et al., 2000; Mullauer et al., 2000). Significantly, constitutive down-regulation of Fas is involved in drug resistance (Landowski et al., 1997) and associated with a poor prognosis in breast cancer (Reimer et al., 2000). On the other hand, Fas is also implicated in anti-cancer drugs triggered apoptosis (Yu et al., 1999; Glick et al., 1999; Sun et al., 2000). In this study, we demonstrated that butyrate induced apoptosis in MCF-7 breast cancer cells via the Fas/Fas L system, as a functional knockout of the Fas system by Fas antagonist antibody efficiently reduced butyrate-induced apoptosis (Figure 9). Western blots analysis showed that butyrate increased both Fas and Fas L levels (Figure 9). Moreover, the increase in membrane Fas was due to a translocation from the cytosol to the membrane, as demonstrated by the concomitant decrease in cytosolic Fas levels (Figure 9). Yu et al. (1999) reported that Vitamin E succinate can convert Fas-resistant human breast cancer cells to the Fas-sensitive phenotype by translocating cytosolic Fas to the membrane. Our results indicate that Fas translocation is also involved in Fas-sensitive cells. Bonnotte et al. (1998) reported that butyrate potentiated Fas-dependent apoptosis in colon cancer cell lines, although it did not increase Fas receptor expression or modify the levels of Bcl-2, Bcl-xL, Bcl-xS, and Bax. We show here that butyrate increased Bax and decreased Bcl-2 levels in MCF-7 cells (Figure 5). The increased Bax/Bcl-2 ratio favours the pro-apoptotic action of Bax. Indeed, it has been reported that Bcl-2 may exert its anti-apoptotic activity partly by inhibiting the translocation of Bax from the cytosol to the mitochondria (Nomura et al., 1999; Murphy et al., 1999). It has also been reported that Fas-induced apoptosis is enhanced by mitochondrial insertion of Bax, which activates cytochrome c release and complete processing of caspase-7 like caspases in MCF-7 breast epithelial cells (Murphy et al., 1999). In accordance with these reports, it has been reported that overexpression of Bcl-2 protects cells from butyrate-induced apoptosis (Mandal & Kumar, 1996).
Overall, our results demonstrating that butyrate inhibited cell growth in a P53-independent manner and enhanced Fas-induced apoptosis may open up interesting prospects for the treatment of human breast cancer. As down-regulation of Fas is associated with a poor prognosis in breast cancer (Reimer et al., 2000), it remains to be determined whether butyrate treatment will prove useful in the fight against advanced breast cancer.
Acknowledgments
This work was supported by the Ligue National Contre le Cancer (Comité du Nord), the Association pour la Recherche sur le Cancer (ARC). We are grateful to Mrs Isabelle Pollet for her excellent technical assistance, as well as of the IFR 22 department.
Abbreviations
- Fas L
Fas Ligand
- FCS
foetal calf serum
- LM-PCR
ligation-mediated polymerase chain reaction
- MCF-7ras cell line
established by transfection of v-Ha-Ras cDNA in MCF-7 cells
- NaB
sodium butyrate
References
- ARCHER S.Y., MENG S., SHEI A., HODIN R.A. p21(WAF1) is required for butyrate-mediated growth inhibition of human colon cancer cells. Proc. Natl. Acad. Sci. U.S.A. 1998;12:6791–6796. doi: 10.1073/pnas.95.12.6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARNARD J.A., WARWICK G. Butyrate rapidly induces growth inhibition and differentiation in HT29 cells. Cell Growth Differ. 1993;4:495–501. [PubMed] [Google Scholar]
- BATES S., VOUSDEN K.H. p53 in signaling checkpoint arrest or apoptosis. Curr. Opinion Genet. Dev. 1994;6:1–7. doi: 10.1016/s0959-437x(96)90004-0. [DOI] [PubMed] [Google Scholar]
- BAUTISTA S., THEILLET CH. p53 mutations in breast cancer: incidence and relations to tumor aggressiveness and evolution of the disease. Path. Biol. 1997;45:882–892. [PubMed] [Google Scholar]
- BERNHARD D., AUSSERLECHNER M.J., TONKO M., LÖFFLER M., HARTMANN B.L., CSORDAS A., KOFLER R. Apoptosis induced by the histone deacetylase inhibitor sodium butyrate in human leukemic lymphoblasts. FASEB J. 1999;13:1991–2001. doi: 10.1096/fasebj.13.14.1991. [DOI] [PubMed] [Google Scholar]
- BONNOTTE B., FAVRE N., REVENEAU S., MICHEAU O., DROIN N., GARRIDO C., FONTANA A., CHAUFFERT B., SOLARY E., MARTIN F. Cancer cell sensitization to Fas-mediated apoptosis by sodium butyrate. Cell Death Differ. 1998;5:480–487. doi: 10.1038/sj.cdd.4400371. [DOI] [PubMed] [Google Scholar]
- CONLEY B.A., EGORIN M.J., TAIT N., ROSEN D.M., SAUSVILLE E.A., DOVER G., FRAM R.J., VAN ECHO D.A. Phase I study of the orally administrated butyrate prodrug, tributyrin, in patients with solid tumors. Clin. Cancer Res. 1998;4:629–634. [PubMed] [Google Scholar]
- FAUQUETTE W., DONG-LE BOURHIS X., DELANNOY-COURDENT A., BOILLY B., DESBIENS X. In vitro characterization of specific morphogenesis and invasiveness ability exhibited by various human mammary epithelial cells: involvement of the uPA/plasmin system. Biol. Cell. 1997;89:29–41. [Google Scholar]
- GIULIANO M., LAURICELLA M., CALVARUSO G., CARABILLO M., EMANUELE S., VENTO R., TESORIERE G. The apoptotic effects and synergistic intercation of sodium butyrate and MG132 in human retinoblastoma Y79 cells. Cancer Res. 1999;59:5586–5595. [PubMed] [Google Scholar]
- GLICK R.D., SWENDEMAN S.L., COFFEY D.C., RIFKIND R.A., MARKS P.A., RICHON V.M., LA QUALIA M.P. Hybrid polar histone deacetylase inhibitor induces apoptosis and CD95/CD95 ligand expression in human neuroblastoma. Cancer Res. 1999;59:4392–4399. [PubMed] [Google Scholar]
- HARTMAN A., BLASZYK H., KOVACH J.S., SOMMER S.S. The molecular epidemiology of p53 gene mutations in human breast cancer. TIG. 1997;13:27–33. doi: 10.1016/s0168-9525(96)10043-3. [DOI] [PubMed] [Google Scholar]
- HERUTH D.P., ZIRNSTEIN G.W., BRADLEY J.F., ROTHBERG P.G. Sodium butyrate causes an increase in the block to transcriptional elongation in the c-myc gene in SW837 rectal carcinoma cells. J. Biol. Chem. 1993;268:20466–20472. [PubMed] [Google Scholar]
- IOTSOVA V., STEHELIN D. Antisense p53 provokes changes in Hela cell growth and morphology. Eur. J. Cell Biol. 1995;68:122–132. [PubMed] [Google Scholar]
- JANSON W., BRANDNER G., SIEGEL J. Butyrate modulates DNA-damage-induced p53 response by induction of p53-independent differentiation and apoptosis. Oncogene. 1997;15:1395–1406. doi: 10.1038/sj.onc.1201304. [DOI] [PubMed] [Google Scholar]
- JUO P., KUO C.J., YUAN J., BLENIS J. Essential requirement for caspase-8/FLICE in the initiation of Fas-induced apoptotic cascade. Curr. Biol. 1998;8:1001–1008. doi: 10.1016/s0960-9822(07)00420-4. [DOI] [PubMed] [Google Scholar]
- KOMAROV P.G., KOMAROVA E.A., KONDRATOV R.V., CHRISTOV-TSELKOV K., COON J.S., CHERNOV M.V., GUDKOV A.V. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285:1733–1736. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- LANDOWSKI T.H., GLEASON-GUZMAN M.C., DALTON W.S. Selection for drug resistance results in resistance to Fas-mediated apoptosis. Blood. 1997;89:1854–1861. [PubMed] [Google Scholar]
- MANDAL M., KUMAR R. Bcl-2 expression regulates sodium butyrate-induced apoptosis in human MCF-7 breast cancer cells. Cell Death Differ. 1996;7:311–318. [PubMed] [Google Scholar]
- MILLER A.A., KURSCHEL E., OSIEKA R., SCHMIDT C.G. Clinical pharmacology of sodium butyrate in patients with acute leukemia. Euro. J. Cancer Clin. Oncol. 1987;23:1283–1287. doi: 10.1016/0277-5379(87)90109-x. [DOI] [PubMed] [Google Scholar]
- MULLAUER L., MOSBERGER I., GRUSCH M., RUDAS M., CHOTT A. Fas ligand is expressed in normal breast epithelial cells and is frequently up-regulated in breast cancer. J. Pathol. 2000;190:20–30. doi: 10.1002/(SICI)1096-9896(200001)190:1<20::AID-PATH497>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- MURPHY K.M., STREIPS U., LOCK R.B. Bax membrane insertion during Fas (CD95)-induced apoptosis precedes cytochrome c release and is inhibited by Bcl-2. Oncogene. 1999;18:5991–5999. doi: 10.1038/sj.onc.1203001. [DOI] [PubMed] [Google Scholar]
- MUSCHEN M., MOERS C., WARSKULAT U., EVEN J., NIEDERACAHER D., BECKMANN M.W. CD95 ligand expression as a mechanism of immune escape in breast cancer. Immunology. 2000;99:69–77. doi: 10.1046/j.1365-2567.2000.00921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAGATA S., GOLSTEIN P. The Fas death receptor. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- NOMURA M., SHIMIZU S., ITO T., NARITA M., MATSUDA H., TSUJIMOTO Y. Apoptotic cytosol facilitates Bax translocation to mitochondira that involves cytosolic factor regulated by Bcl-2. Cancer Res. 1999;59:5542–5548. [PubMed] [Google Scholar]
- PAZIN M.J., KADONAGA J.T. What's up and down with histone deacetylation and transcription. Cell. 1997;89:325–328. doi: 10.1016/s0092-8674(00)80211-1. [DOI] [PubMed] [Google Scholar]
- REIMER T., HERRNRING C., KOCZAN D., RICHTER D., GERBER B., KABELITZ D., FRIESE K., THIESEN H.-J. Fas L: Fas ratio-a prognostic factor in breast carcinomas. Cancer Res. 2000;60:822–828. [PubMed] [Google Scholar]
- SHEIKH M.S., FORNACE A.J., Jr Role of the p53 family members in apoptosis. J. Cell Physiol. 2000;182:171–181. doi: 10.1002/(SICI)1097-4652(200002)182:2<171::AID-JCP5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- SHEIKH M.S., LI X.S., CHEN J.C., SHAO Z.M., ORDONEZ J.V., FONTANA J.A. Mechanism of regulation of WAF1/Cip 1 gene expression in human breast carcinoma: role of p53-dependent and independent signal transduction pathways. Oncogene. 1994;9:3407–3415. [PubMed] [Google Scholar]
- SIONOV R.V., HAUPT Y. The cellular response to p53: the decision between life and death. Oncogene. 1999;18:6145–6157. doi: 10.1038/sj.onc.1203130. [DOI] [PubMed] [Google Scholar]
- SOMMERS C.L., PAPAGEORGE A., WILDING G., GELMANN E.P. Growth properties and tumorigenesis of MCF-7 cells transfected with isogenic mutants of rasH. Cancer Res. 1990;50:67–71. [PubMed] [Google Scholar]
- SONG J., SAPI E., BROWN W., NILSEN J., TARTARO K., KACINSKI B.M., CRAFT J., NAFTOLIN F., MOR G. Role of Fas and Fas ligand during mammary gland remodeling. J. Clin. Invest. 2000;106:1209–1210. doi: 10.1172/JCI10411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STALEY K., BLASCHKE A.J., CHUN J. Apoptotic DNA fragmentation is detected by a semi-quantitative ligation-mediated PCR of blunt DNA ends. Cell Death Differ. 1997;4:66–75. doi: 10.1038/sj.cdd.4400207. [DOI] [PubMed] [Google Scholar]
- STRUHL K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998;12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- SUN S.Y., YUE P., LOTAN R. Implication of multiple mechanisms in apoptosis induced by the synthetic retinoid CD437 in human prostate carcinoma cells. Oncogene. 2000;19:4513–4522. doi: 10.1038/sj.onc.1203810. [DOI] [PubMed] [Google Scholar]
- TOILLON R. A., ADRIAENSSENS E., WOUTERS D., LOTTIN S., BOILLY B., HONDERMARCK H., LE BOURHIS X. Apoptosis induction of MCF-7 cells by normal breast epithelial cells through P53 mediated pathway. Mol. Cell Biol. Res. Commun. 2000;3:338–344. doi: 10.1006/mcbr.2000.0236. [DOI] [PubMed] [Google Scholar]
- YU W., ISRAEL K., LIAO Q.Y., ALDAZ C.M., SANDERS B.G., KLINE K. Vitamin E succinate (VES) induces Fas sensitivity in human breast cancer cells: role for Mr 43,000 Fas in VES-triggered apoptosis. Cancer Res. 1999;59:953–961. [PubMed] [Google Scholar]