

Abstract
A histidine residue in the N-terminal extracellular region of α1,2,3,5 subunits of the human GABAA receptor, which is replaced by an arginine in α4 and α6 subunits, is a major determinant for high affinity binding of classical benzodiazepine (BZ)-site ligands. The effect of mutating this histidine at position 105 in the α5 subunit to an arginine (α5H105R) on BZ-site pharmacology has been investigated using radioligand binding on HEK293 and L(tk-) cells and two electrode voltage clamp recording on Xenopus oocytes in which GABAA receptors of subtypes α5, α5H105R, α4 and α6 were co-expressed with β3γ2s.
The classical BZs, diazepam and flunitrazepam (full agonists on the α5 receptor) showed negligible affinity and therefore negligible efficacy on α5H105R receptors. The β-carbolines DMCM and βCCE (inverse agonists on the α5 receptor) retained some affinity but did not exhibit inverse agonist efficacy at α5H105R receptors. Therefore, the α5H105R mutation confers an α4/α6-like pharmacology to the classical BZs and β-carbolines.
Ro15-4513, flumazenil, bretazenil and FG8094, which share a common imidazobenzodiazepine core structure, retained high affinity and were higher efficacy agonists on α5H105R receptors than would be predicted from an α4/α6 pharmacological profile. This effect was antagonized by DMCM, which competes for the BZ-site and therefore is likely to be mediated via the BZ-site.
These data indicate that the conserved histidine residue in the α subunit is not only a key determinant in the affinity of BZ-site ligands on α5 containing GABAA receptors, but also influences ligand efficacy.
Keywords: Benzodiazepine, GABAA, α subunit, mutagenesis, diazepam
Introduction
γ-aminobutyric acid (GABA) is the major inhibitory neurotransmitter in the mammalian central nervous system. The GABAA receptor is a member of the ligand-gated ion channel superfamily (Schofield et al., 1987). It is formed by the pentameric arrangement of heterogeneous membrane spanning protein subunits which form a central chloride ion channel (for reviews see MacDonald & Olsen, 1994; Rabow et al., 1995; Sieghart, 1995; Whiting et al., 1995). The flow of chloride ions through the GABAA receptor channel is influenced by numerous ligands in addition to GABA. For example, benzodiazepines (BZ) are able to act through a discrete binding site on the receptor to allosterically modulate the action of GABA (MacDonald & Barker, 1978; Squires & Braestrup, 1977). To date nineteen GABAA subunits have been isolated and cloned: α1 – 6, β1 – 4, γ1 – 3, ρ1 – 3, δ, ε, π and θ (Barnard et al., 1998). Current evidence indicates that the stoichiometry of the principal GABAA subtypes include two α subunits, two β subunits and one γ subunit (Chang et al., 1996). GABA binding sites are formed at the interface between the α and β subunits (Levitan et al., 1988) and the BZ-site is formed at the interface between an α and γ subunit (Stephenson et al., 1990).
A critical role of a histidine residue in BZ pharmacology was first implicated when chemical modification by an amino reagent altered binding of a BZ-site ligand in rat cortical membranes (Burch & Ticku, 1981; Maksay & Ticku, 1984). Studies on GABAA receptor subunit mutants (mainly α1) implicate a number of residues in BZ-site ligand binding (Amin et al., 1997; Buhr et al., 1996; 1997a, 1997b; Casula et al., 2001; Schaerer et al., 1998; Wingrove et al., 1997). A histidine residue in the N-terminal extracellular region of α1,2,3 and 5 subunits (position 105 in α5) of the human GABAA receptor, which is represented by an arginine in α4 and α6 subunits (position 106 in α4 and 100 in α6), has been further identified as a major determinant for high affinity binding of conventional BZ-site ligands (Davies et al., 1998; Wieland et al., 1992). The BZ agonist diazepam has high affinity for α1, α2, α3 and α5 containing GABAA receptors whereas α4 and α6 subtypes are diazepam insensitive (Hadingham et al., 1996; Wafford et al., 1996). Mutation of the conserved histidine (in the α1-, α2-, α3-, α5-containing receptors) to an arginine abolished binding of diazepam (Wieland et al., 1992). Conversely, mutation of the arginine residue at position 100 in the diazepam-insensitive α6 subunit to a histidine bestowed diazepam sensitivity on the receptor (Wieland et al., 1992).
The purpose of this study was to characterize the functional consequences of mutating the histidine residue at position 105, to an arginine, on the BZ-site pharmacology of α5 receptors. Specifically, we tested the hypothesis that this mutation confers an α4/α6 receptor-like pharmacology on α5 receptors. Both the binding affinity and the functional efficacy of a range of BZ-site compounds were investigated using radioligand binding and two electrode voltage clamp electrophysiology.
Methods
Ligand binding
The binding affinity of various BZ-site ligands was determined using a conventional radioligand binding assay (as described by Wingrove et al., 1997). HEK293 (Human Embryonic Kidney) cells were plated out at 4×106 cells/10 cm dish in DMEM supplemented with 10% foetal calf serum. After 24 h incubation at 37°C in an atmosphere of 95% O2/5% CO2, the cells were transfected with subunit cDNA (2 μg per subunit per plate) and the vector pAdVAntage (6 μg per plate) (Promega, Madison, WI, U.S.A.), to increase cDNA expression, using calcium phosphate precipitation (Chen & Okayama, 1988). After incubation at 37°C in an atmosphere of air supplemented with 3 – 5% CO2 for 48 – 72 h, the cells were harvested into phosphate buffered saline, pelleted and resuspended in assay buffer (KH2PO4, 10 mM; KCl, 100 mM; pH 7.4) then homogenized through a 27-gauge needle. In some studies, L (tk−) cells (murine fibroblasts) stably transfected with GABAA receptor subunits, as previously described (Hadingham et al., 1992; 1993), were utilized, however the affinity of BZ-site ligands in these two expression systems was highly similar (unpublished observations) and therefore these data were pooled.
The radiolabelled BZ-site ligand [3H]-Ro15-4513 was incubated with the cells in a saturation assay to determine the KD. Non-specific binding was obtained by incubation in the presence of 10 μM Ro15-4513. The assay was terminated after 90 min incubation at 4°C by filtration onto GF/B filters (Brandel, Montreal, Quebec, Canada) using a Tomtec cell harvester (Receptor Technologies Ltd., Oxon, U.K.) The filters were washed three times with 5 ml ice-cold assay buffer and dried before using liquid scintillation counting (LS6500 Scintillation System, Beckman Instruments Inc., Fullerton, CA, U.S.A.) to detect filter retained radioactivity. Displacement assays using sub-KD concentrations of [3H]-Ro15-4513 to determine the Ki of the unlabelled ligands were performed in a similar manner. The inhibition constant (Ki) was calculated using the Cheng-Prusoff equation (Cheng & Prusoff, 1973; Ki=IC50/(1+[Radioligand]/Ki=IC50/(1+[Radioligand]/KD)).
Data points were fitted by nonlinear regression analysis (Prism version 3.01; Graphpad) to give the values of KD from the saturation assay and the IC50 from the radioligand displacement assay (one site competition). The Ki and the KD values were calculated from at least three independent experiments and expressed as mean±standard error of the mean. The 2-tail unpaired Student's t-test was used to test significance (P<0.05).
Efficacy determination
The efficacies of the ligands for the BZ-site of the GABAA receptor (diazepam, flunitrazepam, βCCE, DMCM, Ro15-4513, flumazenil, bretazenil and FG 8094 were determined using the two electrode voltage clamp technique on oocytes expressing recombinant GABAA receptors (as described by Ebert et al., 1997).
Briefly, oocytes were surgically removed from a female Xenopus laevis anaesthetized in a 0.4% solution of ethyl m-aminobenzoate. The oocytes (stage V and VI) were manually defolliculated in isolation medium (mM: NaCl, 108; KCl, 2; HEPES, 1; EDTA, 0.1; pH 7.9 with NaOH) followed by 5 min of mild collagenase treatment. Oocytes were then transferred to Modified Barth's Solution (mM: MBS; NaCl, 88; KCl, 1; HEPES, 10; NaHCO3, 2.4; CaCl, 0.91; MgSO4 (7H2O), 0.82; Ca(NO3)2(4H2O), 0.33; pH 7.5 with NaOH) for cDNA injection.
The cDNAs encoding the α, β and γ GABAA subunits, used in this investigation were obtained as described by Hadingham et al. (1993; 1995; 1996). The subunit cDNAs (engineered into the expression vector pCDM8 or pcDNA Amp) were diluted in injection buffer (mM: NaCl 88, KCl 1, HEPES 15, in sterile d.H2O, adjusted to pH 7.0 with 1 M NaOH, sterile filtered through 0.2 μM filter) to a total cDNA concentration of 20 ng μl−1 or 6.7 ng μl per subunit, ∼15 nl of this mix was injected directly into the nucleus of the oocyte.
Oocytes were stored for up to 4 days following injection in incubation media (1 ml each of penicillin (10,000 units ml−1), streptomycin (10 mg ml−1) and gentamycin (50 mg ml−1) made up with 22 mg Na+pyruvate in l litre MBS). For recording, the oocytes were then impaled by the two electrodes, one electrode to voltage clamp the cell at −60 mV, and the other electrode to measure the change in current following drug application. GABA was applied until the peak of the response was observed, usually 30 s or less. At least 3 min wash time was allowed between each GABA application to prevent desensitization. Concentration response curves were calculated using a non-linear squares fitting program to the equation: f(x)=Bmax/(1+(EC50/x)n) where x is the drug concentration, EC50 is the concentration of drug eliciting a half maximal response and n is the Hill coefficient. The effects of GABA receptor modulators were examined on control GABA EC20 responses with a pre-application time of 30 s. Only oocytes with an EC20 within a fixed range were selected (1 – 6 μM for α5β3γ2s and α6β3γ2s; 1 – 12 μM for α5H105Rβ3γ2s) to minimize the chance of high (GABA) affinity doublet receptors with no γ subunit (no BZ-site) expressed by the oocyte. The efficacy of GABA receptor modulators was determined as the percentage change in the current activated by the EC20 concentration of GABA using the equation: Efficacy (%)=((current in presence of modulator/control current)×100)−100.
Data represent values of n⩾3 that are expressed as the mean±standard error of the mean. The 2-tailed unpaired Student's t-test was used to test significance (P<0.05).
Materials
All salts utilized for the MBS and injection buffers were obtained from BDH and were of AnalaR grade or better. The BZ-site ligands diazepam, flunitrazepam, ethyl-β-carboline-3-carboxylate (βCCE), dimethoxy-4-ethyl-β-carboline-3-carboxylate (DMCM), Ro15-4513, flumazenil, bretazenil and FG 8094 were all purchased from Sigma Chemical Company. All BZ-site ligands were dissolved in dimethylsulphoxide (DMSO), made up to a stock concentration of 2 or 10 mM and stored in a freezer (−20°C) until use. The radiolabelled BZ-site ligand [3H]-Ro15-4513 was obtained from New England Nuclear Research Products, Boston, MA, U.S.A.
Results
Ligand binding affinity
Radioligand binding assays were performed to investigate the effect of the α5H105R mutation on binding affinity of BZ-site ligands. Saturation binding experiments were conducted to calculate the mean values for the KD of the [3H]-Ro15-4513 radioligand on α5, α5H105R and α6 containing GABAA receptors. Ro 15-4513 showed the greatest affinity for the α5 receptor with a KD of 1.2 nM (pKD=8.92±0.04), there was a significant reduction in affinity for the α5H105R receptor which had a KD of 8.8 nM (pKD=8.08±0.09) (P=0.02). The affinity was significantly lower again for the α6 receptor with a KD of 20.9 nM (pKD=7.68±0.08) (P=0.04). A KD of 3.4±0.6 nM has previously been reported for the α4 receptor in a stably transfected Ltk cell line (Sur et al., 1999).
Mean Ki values for a variety of BZ-site ligands of different structural classes were determined by displacement of specific [3H]-Ro15-4513 binding for α5, α5H105R, α4 and α6 containing GABAA receptors (Table 1). The classic 1,4-benzodiazepines diazepam and flunitrazepam which showed high affinity for the α5 receptor (Ki of 20.7 and 2.19 nM respectively), showed a dramatic loss of affinity on mutation of the histidine residue (Ki on α5H105R >10,000 nM) consistent with an α4 or α6-like pharmacological profile (Ki also >10,000 nM).
Table 1.
Relative binding affinity of BZ-site ligands for human α5, α5H105R, α4 and α6 recombinant GABAA receptors
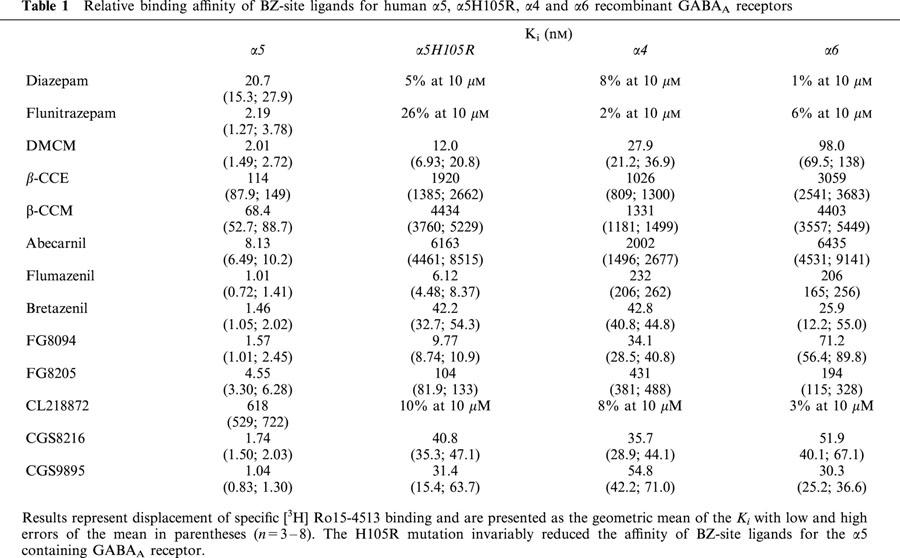
Of the four β-carboline ligands examined, DMCM, β-CCE, β-CCM and abecarnil, all exhibited reduced affinity at α5H105R receptors and Ki values were consistent with an α4- or α6-like profile (Table 1). Affinities were also determined for a subset of 1,4-benzodiazepines which differ from classical ligands in that they possess an imidazobenzodiazepine core. Flumazenil, Ro15-4513, bretazenil, FG8094 and FG8205 all exhibit high affinity binding to α5 receptors (1.01, 1.20, 1.46, 1.57 and 4.55 nM, respectively) and three of these compounds, flumazenil, Ro-15-4513 and FG8094 retained appreciable affinity following point-mutation of histidine 105 to arginine (α5H105R affinities 6.12, 8.80 and 9.77 nM respectively). Three additional compounds from alternative structural classes, CL218872, CGS8216 and CGS9895, all displayed a loss of affinity at the mutated receptor (α5 Ki values 618, 1.74, 1.04; α5H105 >10,000, 40.8, 31.4 nM), consistent with an α4/α6 profile.
Effect of α5H105R mutation on GABA potency
Full sequential concentration response curves to GABA (0.3 – 100 μM) were constructed on α5, α5H105R and α6 containing GABAA receptors expressed in Xenopus oocytes (Figure 1). All curves shared a sigmoidal relationship with similar Hill slopes (1.1±0.1; 1.3±0.1; 1.4±0.3 for α5, α5H105R and α6 respectively, Figure 1B). GABA had comparable potency on α5 containing GABAA receptors (EC50=5.1 μM; pEC50=5.29±0.06) and α6 containing GABAA receptors ( EC50 = 1.1 μM; pEC50 = 5.97 ± 0.04 ) (P<0.0001). The insertion of the α5H105R mutation reduced the potency of GABA (EC50=13 μM; pEC50=4.88±0.05) when compared to the α5 containing GABAA receptor (P<0.0001).
Figure 1.
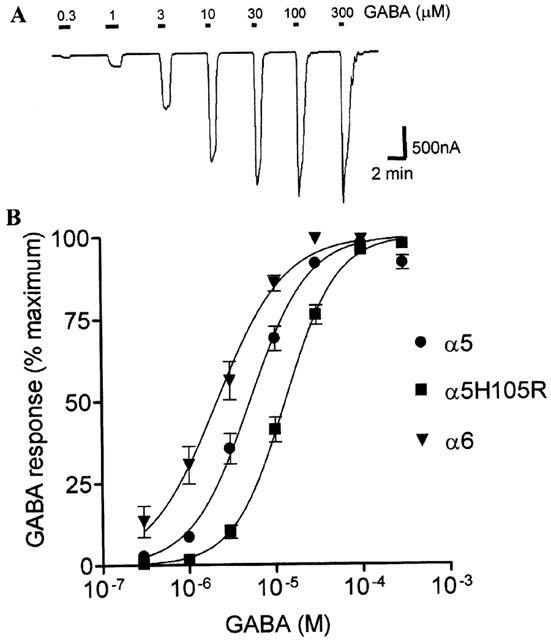
Potency of GABA on human α5, α5H105R and α6 containing GABAA receptors. (A) Example of a two electrode voltage clamp recording of membrane current from Xenopus oocytes expressing αxβ3γ2s GABAA receptors, showing responses to sequential bath application of increasing concentrations of GABA. (B) Graph showing the concentration dependent response to GABA (0.3 – 300 μM) on α5, α5H105R and α6 containing GABAA receptors. Responses were normalized to the maximum GABA response for each oocyte and the mean±s.e.mean from n⩾3 oocytes is shown. Curves on all subunits showed a characteristic monophasic sigmoidal profile with similar Hill slopes.
Effect of α5H105R mutation on BZ efficacy
The ability of selected BZs to modulate an EC20 dose of GABA was then investigated (Figure 2A). The classical BZs, diazepam and flunitrazepam, both significantly potentiated the response to GABA being full agonists on the α5 receptor (+116.4±12.5% and +147.2±1.7% respectively), whereas they were inactive on both α4 and α6 receptors (Figure 2B). Mutation of the histidine residue in α5 (α5H105R) also rendered diazepam and flunitrazepam inactive (+2±2% and −5±3% respectively), consistent with a α4/α6 pharmacological profile (Figure 2B).
Figure 2.
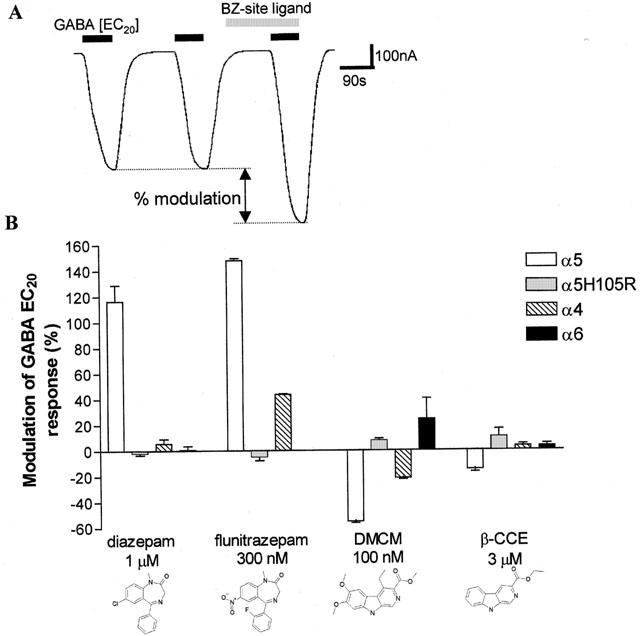
Relative efficacy of conventional BZ-site ligands on human α5, α5H105R, α4 and α6 containing GABAA receptors. (A) Typical trace showing the consistent change in membrane current induced by an EC20 concentration of GABA and modulation of this response upon bath application of a BZ-site ligand. The efficacy of the BZ-site ligand is determined as the percentage modulation in the current activated by an EC20 concentration of GABA using the equation: modulation of GABA EC20 response (%)=((GABA EC20 induced current in presence of the BZ-site ligand/GABA EC20 induced current prior to BZ-site ligand application)×100)−100. A positive change is indicative of a BZ-site agonist, a negative change of an inverse agonist and no change indicates an antagonist. In the example shown, the ligand produced a small potentiation of the GABA response, consistent with partial agonist activity at the BZ-site. In subsequent figures, the efficacy of BZ-site ligands has been determined as above and then is presented as the mean±s.e.mean of data from n⩾3 oocytes. Only one BZ-site ligand was tested per oocyte. (B) Bar graph showing the efficacy of classical BZs (diazepam and flunitrazepam) and β-carbolines (DMCM and β-CCE) on human α5, α5H105R, α4 and α6 containing GABAA receptors expressed in Xenopus oocytes. All ligands were tested at a concentration 100 times their binding affinity on the α5 containing GABAA receptor.
The β-carbolines, DMCM and βCCE significantly diminished the response to GABA being inverse agonists (−55±1% and −15±2% respectively) at α5 receptors. On mutation of the histidine residue in α5 (α5H105R) the inverse agonism was significantly reduced (+8±2% and +10±6% respectively, P<0.05), more consistent with α4 (−22±1% and +3±2% respectively) and α6 (+21±13% and +3±2% respectively) pharmacology (Figure 2B).
The compounds Ro15-4513, flumazenil and bretazenil, an inverse agonist (−15±2%), an antagonist (−5±6%) and a weak agonist (+13±3%) on α5 receptors, all showed significantly greater agonism on the mutant receptor (+89±2%, +72±4% and +22±4% respectively) than predicted from their weak agonism or antagonism on α4 (+27±4%; +5±1% and +9±1% respectively) or α6 (+3±9%; +13±2% and +13±6% respectively) receptors (P<0.05, Figure 3A).
Figure 3.
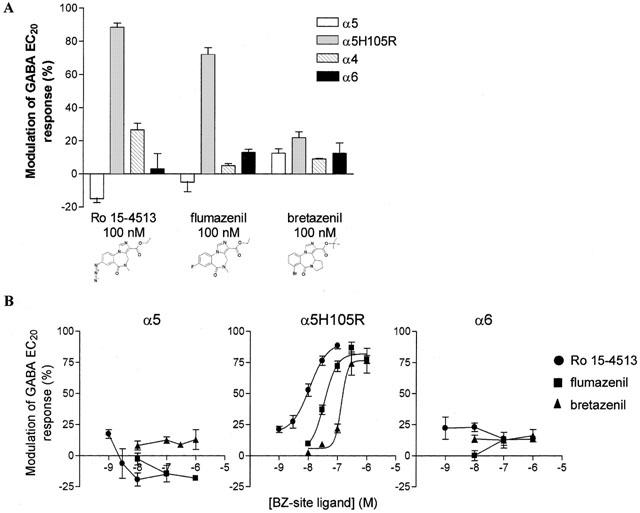
Relative efficacy of BZ-site ligands containing an imidazobenzodiazepine core on human α5, α5H105R and α6 containing GABAA receptors. (A) Bar graph showing the percentage modulation of GABA EC20 responses (mean±s.e.mean from n⩾3 oocytes) by Ro15-4513, flumazenil and bretazenil on human α5, α5H105R, α4 and α6 containing GABAA receptors expressed in Xenopus oocytes (determined as described in Figure 2A). All ligands were tested at a concentration 100 times their binding affinity on the α5 containing GABAA receptor. All three ligands showed partial agonist activity at the α5H105R containing GABAA receptor. (B) To determine the concentration dependence of responses to these BZ-site ligands, the effect of increasing concentrations of a BZ-site ligand (in the range 10−9 – 10−6 M) on sequential applications of an EC20 concentration of GABA were determined in different oocytes. Again only one BZ-site ligand was tested at one concentration per oocyte as described in Figure 2A. The graphs show the concentration dependent response (mean±s.e.mean from n⩾3 oocytes) to these BZ-site ligands on α5, α5H105R and α6 containing GABAA receptors. Monophasic sigmoidal concentration-response curves for Ro15-4513, flumazenil and bretazenil were observed on α5H105R containing GABAA receptors, whereas these ligands had little effect on α5 and α6 GABAA receptors throughout the concentration range tested.
To investigate the unpredicted agonism seen with Ro15-4513, flumazenil and bretazenil further, different concentrations of these compounds (10−9 – 10−6 M) were investigated and concentration response curves were constructed. Ro15-4513, flumazenil and bretazenil were relatively inactive on α5 and α6 containing GABAA receptors. However, all three compounds displayed partial agonism on the α5H105R receptor (Ro15-4513, EC50=11 nM, pEC50=7.95±0.09; flumazenil, EC50=35 nM pEC50=7.45±0.07; and bretazenil, EC50=135 nM, pEC50=6.87±0.11), Figure 3B). Curves to all three compounds on the α5H105R mutant showed monophasic sigmoidal relationships indicating that the compounds are binding to a single site.
Response to Ro15-4513, flumazenil and bretazenil in the presence of a ligand which competes for the BZ-site
To determine whether Ro15-4513, flumazenil and bretazenil are acting via the BZ-site, a competition study with ligand binding to the BZ-site was conducted (Figure 4). The conventional antagonist, flumazenil could not be used due to its agonist effect on the mutant receptor (+72±4%). Therefore, DMCM was selected as it has a lack of efficacy on the mutant receptor (+8±2%) without an accompanying loss of affinity (Ki=12 nM). DMCM was applied at an equal concentration (100 nM) to the agonists which was ∼100 times its affinity on the wild-type α5 receptor. In each case the agonist response (Ro15-4513, 89±2% flumazenil, 72±4%; bretazenil, 22±4%) was significantly inhibited to approximately 50% (Ro15-4513, by 44% to 50±5%; flumazenil, by 52% to 34±6%; bretazenil, by 55% to 10±1%, P<0.05) in the presence of DMCM. DMCM was also tested at a higher concentration (1 μM) with Ro15-4513, which caused greater inhibition (by 64% to 32±5%) indicating that DMCM inhibits binding of this compound in a concentration dependent manner.
Figure 4.
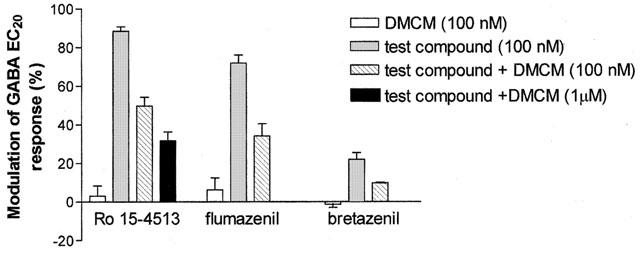
Determination of the site of action of imidazobenzodiazepines on human α5H105R containing GABAA receptors. The bar graph shows the percentage modulation of GABA EC20 responses (mean±s.e.mean from n⩾3 oocytes) by Ro15-4513, flumazenil and bretazenil on human α5H105R containing GABAA receptors expressed in Xenopus oocytes (determined as described in Figure 2A, in the presence and absence of a ligand which competes for the BZ-site (DMCM, Ki =12 nM). Again only one test compound was studied per oocyte; the effect of test compounds in the presence and absence of DMCM was also determined in different oocytes as the effects of such compounds are often irreversible. DMCM was applied at an equal concentration (100 nM) to the agonists which was ∼100 times its affinity on the wild-type α5 receptor and also at a higher concentration (1 μM) with Ro15-4513.
Ro15-4513, flumazenil and bretazenil all share a common imidazobenzodiazepine core with a carboxylic acid at position-3 (Figure 3). To test the hypothesis that the presence of this core structure increases agonist properties to ligands on the α5H105R receptor, another related compound FG8094, which shares the imidazobenzodiazepine core structure, was tested. Consistent with our hypothesis, FG8094 (an inverse agonist on α5-containing receptors, −16.4±4.3%) produced a partial agonist response on the α5H105R containing GABAA receptor (+54.2±14.6%).
Discussion and conclusions
The pharmacology of the BZ-site of GABAA receptors has been extensively studied by several groups. The main approaches used to investigate the structural requirements include site-directed mutagenesis of recombinant receptors and molecular modelling (Benson et al., 1998; Boileau & Czajkowski, 1999; Casula et al., 2001; Davies et al., 1998; Dunn et al., 1999; Renard et al., 1999; Sigel & Buhr, 1997; Sigel et al., 1998; Wingrove et al., 1997; Wisden & Stephens, 1999; Zhang et al., 1995). Characterization of the pharmacophore of the BZ-site has been complicated by the existence of multiple receptor isoforms, the diverse range of structures that are able to bind to the BZ-site and allosterically modulate the actions of GABA, and by the observations that efficacy cannot be predicted from binding affinity.
It is possible that site-directed mutagenesis of the histidine at position 105 in the α5 subunit to an arginine may exert its effects not at the level of the BZ-site but may produce a conformational change in the receptor itself, thereby disrupting the GABA binding site. If this were the case, a large change in GABA potency would be expected. Although the potency of GABA for the α5H105R receptor (compared to the α5 wild-type receptor) was reduced, the reduction in EC50 was only equivalent to a shift of half a log unit. Considering the potency of GABA for a given receptor subtype can vary by up to one log unit with time after cDNA injection (α5 containing GABAA receptors, day 1 EC50=14 μM, pEC50=4.86±0.08 cf. Day 4 EC50=3 μM, pEC50=5.52±0.11), this change in potency is not indicative of a significant alteration in the conformation of the GABA binding site of the receptor. The reason for the difference observed may be explained by the process of only selecting the oocytes with a GABA EC20 value within the specified range for each receptor subtype, and these ranges can differ slightly between subunits.
The affinity and maximum efficacies of the BZs measured in oocytes were similar to those using binding techniques, suggesting that a different surrounding membrane environment and possible variations in the post-translational modifications provided by different cell types are not critical factors for the interactions of these drugs at their binding sites (Pritchett & Seeburg, 1990). The hypothesis that mutating this conserved histidine residue to an arginine confers pharmacological properties more consistent with an α4/α6 isoform holds for the classical BZs and β-carbolines. Diazepam and flunitrazepam showed no affinity and negligible efficacy for the α5H105R receptor, consistent with previous studies (Wieland et al., 1992); the lack of efficacy of these ligands can most likely be attributed to their lack of affinity for the receptor. Similarly, DMCM and βCCE exhibited both a loss of binding affinity and a loss of partial inverse agonist efficacy at the α5H105R receptor, consistent with the expected pharmacology of α4/α6 receptors.
The compounds Ro15-4513, flumazenil, bretazenil and FG8094, which share a common imidazobenzodiazepine core, were all higher efficacy agonists on the α5H105R receptor than would have been predicted based on a simple switch from an α5 to an α4/α6 pharmacology. Data from previous studies with Ro15-4513 and bretazenil on α1H101R, α2H101R, α3H126R and α5H105R containing GABAA receptors (Benson et al., 1998) support this.
This cannot be attributed to an increase in affinity of these ligands, since they show similar or lower affinity for the α5H105R receptor than the α5 wild-type receptor. Another explanation for the unpredicted agonism with these compounds could be that they bind to another region on the mutant GABAA receptor revealed by the disruption of the BZ-site by point mutation. However, there are three reasons why this is unlikely. Firstly, the binding studies show that [3H]-Ro15-4513 binds competitively to the same site as DMCM and βCCE on the α5H105R receptor. Sceondly, the concentration response curves for these compounds show no evidence of a biphasic response which would be seen when a compound binds to two discrete binding sites producing different effects. Finally, DMCM was able to inhibit the effect of these compounds in a concentration dependent manner. Therefore it is likely that Ro15-4513, flumazenil and bretazenil elicit their agonist effect via an interaction with the BZ-site on the mutant receptor. So, even though the ligands studied bind to the same general site, the consequence on ion channel opening is not the same.
Whilst it might have been anticipated that Ro15-4513 would retain some affinity for the α5H105R receptor, given that this has been the ligand generally used to characterize diazepam-insensitive GABAA receptors, perhaps more surprising was the fact that flumazenil, FG8094 and DMCM retained significant affinity at the mutant receptor. This would not have been predicted based on their affinities for α4 and α6-containing receptors and suggests that His105 is not a key determinant for high affinity binding of these ligands, implicating the importance of other residues. This theory of ligand-receptor interaction means certain residues in the BZ-site may be involved purely in the binding of the ligand to the BZ-site acting as a form of anchorage holding the ligand in the correct alignment. While other residues within the binding site do not interact with the ligand directly but facilitate the change in conformation of the receptor upon the binding of the allosteric modulator (Sigel et al., 1998).
Photoaffinity labelling experiments have implicated the pendant phenyl ring of the classical BZs (e.g. diazepam) in binding to the imidazo ring of this conserved histidine residue (probably through a π – π interaction; McKernan et al., 1998). It has also been demonstrated that the conserved histidine residue is not required for the high affinity binding of flumazenil, Ro15-4513 and FG8094 as they still bind to the BZ-site in the presence of photoincorporated flunitrazepam (McKernan et al., 1998).
The primary objective of this study was to test the hypothesis that the α5H105R mutation confers α4/α6-containing receptor-like properties on to the α5-containing receptor. This investigation showed that the problem is more complex than first thought. The reason for this complexity is that the conserved histidine residue is directly involved in the binding of certain compounds to the BZ-site (e.g. classical BZs and some β-carbolines) yet for other compounds containing an imidazobenzodiazepine core (e.g. flumazenil, Ro15-4513, FG8094) the residue is not required for high affinity binding to the BZ-site but does influence the equilibrium between different conformations of the receptor.
These data support the hypothesis that GABA receptor subtypes contain common and highly conserved structural determinants that influence both the affinity and efficacy of BZ-site ligands and that can be exploited to investigate the pharmacological significance of specific GABAA receptor subtypes.
Abbreviations
- βCCE
ethyl-β-carboline-3-carboxylate
- BZ
benzodiazepine
- DMCM
dimethoxy-4-ethyl-β-carboline-3-carboxylate
- GABA
γ-aminobutyric acid
References
- AMIN J., BROOKS-KAYAL A.R., WEISS D.S. Two tyrosine residues on the α subunit are crucial for benzodiazepine binding and allosteric modulation of γ-aminobutyric acidA receptors. Mol. Pharmacol. 1997;51:833–841. doi: 10.1124/mol.51.5.833. [DOI] [PubMed] [Google Scholar]
- BARNARD E.A., SKOLNICK P., OLSEN R.W., MOHLER H., SIEGHART W., BIGGIO G., BRAESTRUP C., BATESON A.N., LANGER S.Z. International Union of Pharmacology. XV. Subtypes of γ-aminobutyric acidA receptors. Classification on the basis of subunit structure and receptor function. Pharmacol. Rev. 1998;50:294–313. [PubMed] [Google Scholar]
- BENSON J.A., LÖW K., KEIST R., MOHLER H., RUDOLPH U. Pharmacology of recombinant γ-Aminobutyric AcidA receptors rendered diazepam-insensitive by point-mutated α-subunits. FEBS Letters. 1998;431:400–404. doi: 10.1016/s0014-5793(98)00803-5. [DOI] [PubMed] [Google Scholar]
- BOILEAU A.J., CZAJKOWSKI C. Identification of transduction elements for benzodiazepine modulation of the GABAA receptor: Three residues are required for allosteric coupling. J. Neurosci. 1999;19:10213–10220. doi: 10.1523/JNEUROSCI.19-23-10213.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUHR A., BAUR R., MALHERBE P., SIGEL E. Point mutations of the α1β2γ2 GABAA receptor affecting modulation of the channel by ligands of the benzodiazepine binding site. Mol. Pharmacol. 1996;49:1080–1084. [PubMed] [Google Scholar]
- BUHR A., BAUR R., SIGEL E. Subtle changes in residue 77 of the γ subunit of α1β2γ2 GABAA receptors drastically alter the affinity for ligands of the benzodiazepine binding site. J. Biol. Chem. 1997a;272:11799–11804. doi: 10.1074/jbc.272.18.11799. [DOI] [PubMed] [Google Scholar]
- BUHR A., SCHAERER M.T., BAUR R., SIGEL E. Residues at positions 206 and 209 of the α1 subunit of γ-aminobutyric acidA receptors influence affinities for benzodiazepine binding site ligands. Mol. Pharmacol. 1997b;52:676–682. doi: 10.1124/mol.52.4.676. [DOI] [PubMed] [Google Scholar]
- BURCH T.P., TICKU M.K. Histidine modification with diethyl pyrocarbonate shows heterogeneity of benzodiazepine receptors. Proc. Natl. Acad. Sci. U.S.A. 1981;78:3945–3949. doi: 10.1073/pnas.78.6.3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CASULA M.A., BROMIDGE F.A., PILLAI G.V., WINGROVE P.B., MARTIN K., MAUBACH K., SEABROOK G.R., WHITING P.J., HADINGHAM K.L. Identification of amino acid residues responsible for the alpha5 subunit binding selectivity of L-655,708, a benzodiazepine binding site ligand at the GABA(A) receptor. J. Neurochem. 2001;77:445–451. doi: 10.1046/j.1471-4159.2001.00289.x. [DOI] [PubMed] [Google Scholar]
- CHANG Y., WANG R., BAROT S., WEISS D.S. Stoichiometry of a recombinant GABAA receptor. J. Neurosci. 1996;16:5415–5424. doi: 10.1523/JNEUROSCI.16-17-05415.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN C.A., OKAYAMA H. Calcium phosphate-mediated gene transfer. A highly efficient transfection system for stably transforming cells with plasmid DNA. BioTechniques. 1988;6:632–638. [PubMed] [Google Scholar]
- CHENG Y.C., PRUSOFF W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (IC50) of an enzymatic reaction. BioChem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- DAVIES M., BATESON A.N., DUNN S.M.J. Structural requirements for ligand interactions at the benzodiazepine recognition site of the GABAA receptor. J. Neurochemistry. 1998;70:2188–2194. doi: 10.1046/j.1471-4159.1998.70052188.x. [DOI] [PubMed] [Google Scholar]
- DUNN S.M.J., DAVIES M., MUNTONI A.L., LAMBERT J.J. Mutagenesis of the rat α1 subunit of the γ-Aminobutyric acidA receptor reveals the importance of residue 101 in determining the allosteric effects of benzodiazepine site ligands. Molecular Pharmacology. 1999;56:768–774. [PubMed] [Google Scholar]
- EBERT B., THOMPSON S.A., SAOUNATSOU K., MCKERNAN R., KROGSGAARD-LARSEN P., WAFFORD K.A. Differences in agonist/antagonist binding affinity and receptor transduction using recombinant human γ-aminobutyric acid type A receptors. Mol. Pharmacol. 1997;52:1150–1156. [PubMed] [Google Scholar]
- HADINGHAM K.L., GARRETT E.M., WAFFORD K.A., BAIN C., HEAVENS R.P., SIRINATHSINGHJI D.J., WHITING P.J. Cloning of cDNAs encoding the human γ-aminobutyric acid type A receptor α6 subunit and characterization of the pharmacology of α6-containing receptors. Mol. Pharmacol. 1996;49:253–259. [PubMed] [Google Scholar]
- HADINGHAM K.L., HARKNESS P.C., MCKERNAN R.M., QUIRK K., LE BOURDELLES B., HORNE A.L., KEMP J.A., BARNARD E.A., RAGAN C.I., WHITING P.J. Stable expression of mammalian type A γ-aminobutyric acid receptors in mouse cells: demonstration of functional assembly of benzodiazepine-responsive sites. Proc. Natl. Acad. Sci. U.S.A. 1992;89:6378–6382. doi: 10.1073/pnas.89.14.6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HADINGHAM K.L., WAFFORD K.A., THOMPSOM S.A., PALMER D.J., WHITING P.J. Expression and pharmacology of human GABAA receptors containing gamma 3 subunits. Eur. J. Pharmacol. 1995;291:301–309. doi: 10.1016/0922-4106(95)90070-5. [DOI] [PubMed] [Google Scholar]
- HADINGHAM K.L., WINGROVE P.B., WAFFORD K.A., BIAN C., KEMP J.A., PALMER K.J., WILSON A.W., WILCOX A.S., SIKELA J.M., RAGAN C.I., WHITING P.J. Role of the β subunit in determining the pharmacology of human γ-aminobutyric acid type A receptors. Mol. Pharmacol. 1993;44:1211–1218. [PubMed] [Google Scholar]
- LEVITAN E.S., SCHOFIELD P.R., BURT D.R., RHEE L.M., WISDEN W., KOHLER M., FUJITA N., RODRIGUEZ H.F., STEPHENSON A., DARLISON M.G., BARNARD E.A., SEEBURG P.H. Structural and functional basis for GABA-A receptor heterogeneity. Nature. 1988;335:76–79. doi: 10.1038/335076a0. [DOI] [PubMed] [Google Scholar]
- MACDONALD R., BARKER J.L. Benzodiazepines specifically modulate GABA-mediated postsynaptic inhibition in cultured mammalian neurones. Nature. 1978;271:563–564. doi: 10.1038/271563a0. [DOI] [PubMed] [Google Scholar]
- MACDONALD R.L., OLSEN R.W. GABAA receptor channels. Annu. Rev. Neuroscience. 1994;17:569–602. doi: 10.1146/annurev.ne.17.030194.003033. [DOI] [PubMed] [Google Scholar]
- MAKSAY G., TICKU M.K. Characterization of gamma-aminobutyric acid benzodiazepine receptor complexes by protection against inactivation by group-specific reagents. J. Neurochem. 1984;42:1715–1727. doi: 10.1111/j.1471-4159.1984.tb12763.x. [DOI] [PubMed] [Google Scholar]
- MCKERNAN R.M., FARRAR S., COLLINS I., EMMS F., ASUNI A., QUIRK K., BROUGHTON H. Photoaffinity labelling of the Benzodiazepine Binding Site of α1β3γ2 γ-Aminobutyric AcidA receptors with flunitrazepam identifies a subset of ligands that interact directly with His102 of the α subunit and predicts orientation of these within the benzodiazepine pharmacore. Mol. Pharmacol. 1998;54:33–43. doi: 10.1124/mol.54.1.33. [DOI] [PubMed] [Google Scholar]
- PRITCHETT D.B., SEEBURG P.H. γ-aminobutyric acidA receptor α5-subunit creates novel type II benzodiazepine receptor pharmacology. J. Neurochem. 1990;54:1802–1804. doi: 10.1111/j.1471-4159.1990.tb01237.x. [DOI] [PubMed] [Google Scholar]
- RABOW L.E., RUSSEK S.J., FARB D.H. From ion currents to genomic analysis: recent advances in GABAA receptor research. Synapse. 1995;21:189–274. doi: 10.1002/syn.890210302. [DOI] [PubMed] [Google Scholar]
- RENARD S., OLIVER A., GRANGER P., AVENET P., GRAHAM D., SEVRIN M., GEORGE P., BESNARD F. Structural elements of the γ-Aminobutyric acid type A receptor conferring subtype selectivity for benzodiazepine site ligands. J. Biological Chemistry. 1999;274:13370–13374. doi: 10.1074/jbc.274.19.13370. [DOI] [PubMed] [Google Scholar]
- SCHAERER M.T., BUHR A., BAUR R., SIGEL E. Amino acid residue 200 on the α1 subunit of GABAA receptors affects the interaction with selected benzodiazepine binding site ligands. Eur. J. Pharmacol. 1998;354:283–287. doi: 10.1016/s0014-2999(98)00456-7. [DOI] [PubMed] [Google Scholar]
- SCHOFIELD P.R., DARLINSON M.G., FUJITA N., BURT D.R., STEPHENSON F.A., RODRIGUEZ H., RHEE LM., RAMACHANDRAN J., REALE V., GLENCORSE T.A., SEEBURG P.H., BARNARD E.A. Sequence and functional expression of the GABA-A receptor shows a ligand-gated receptor superfamily. Nature. 1987;328:221–227. doi: 10.1038/328221a0. [DOI] [PubMed] [Google Scholar]
- SIEGHART W. Structure and pharmacology of γ-Aminobutyric acidA receptor subtypes. Pharmacol. Rev. 1995;47:181–234. [PubMed] [Google Scholar]
- SIGEL E., BUHR A. The benzodiazepine binding site of GABAA receptors. Trends Pharmacol. Sci. 1997;18:425–429. doi: 10.1016/s0165-6147(97)01118-8. [DOI] [PubMed] [Google Scholar]
- SIGEL E., SCHAERER M.T., BUHR A., BAUR R. The benzodiazepine binding pocket of recombinant α1β2γ2 γ-Aminobutyric AcidA receptors: relative orientation of ligands and amino acid side chains. Mol. Pharmacol. 1998;54:1097–1105. doi: 10.1124/mol.54.6.1097. [DOI] [PubMed] [Google Scholar]
- SQUIRES R.F., BRAESTRUP C. Benzodiazepine receptors in rat brain. Nature. 1977;266:732–734. doi: 10.1038/266732a0. [DOI] [PubMed] [Google Scholar]
- STEPHENSON F.A., DUGGAN M.J., POLLARD S. The γ2 subunit is an integral component of the γ-aminobutyric acidA receptor but the α1 polypeptide is the principle site of the agonist benzodiazepine photoaffinity labelling reaction. J. Biol. Chem. 1990;265:21160–21165. [PubMed] [Google Scholar]
- SUR C., FARRAR S.J., KERBY J., WHITING P.J., ATACK J.R., MCKERNAN R.M. Preferential coassembly of α4 and δ subunits of the GABAA receptor in rat thalamus. Mol. Pharmacol. 1999;56:110–115. doi: 10.1124/mol.56.1.110. [DOI] [PubMed] [Google Scholar]
- WAFFORD K.A., THOMPSON S.A., THOMAS D., SIKELA J., WILCOX A.S., WHITING P.J. Functional characterization of human γ-aminobutyric acidA receptors containing the α4 subunit. Mol. Pharmacol. 1996;50:670–678. [PubMed] [Google Scholar]
- WHITING P.J., MCKERNAN R.M., WAFFORD K.A. Structure and pharmacology of vertebrate GABAA receptor subtypes. Int. Re. Neurobiol. 1995;38:95–138. doi: 10.1016/s0074-7742(08)60525-5. [DOI] [PubMed] [Google Scholar]
- WIELAND H.A., LÜDDENS H., SEEBURG P.H. A single histidine in GABAA receptors is essential for benzodiazepine agonist binding. J. Biol. Chem. 1992;267:1426–1429. [PubMed] [Google Scholar]
- WINGROVE P.B., THOMPSON S.A., WAFFORD K.A., WHITING P.J. Key amino acids in the γ subunit of the γ-aminobutyric acidA receptor that determine ligand binding and modulation at the benzodiazepine site. Mol. Pharmacol. 1997;52:874–881. doi: 10.1124/mol.52.5.874. [DOI] [PubMed] [Google Scholar]
- WISDEN W., STEPHENS D.N. Towards better benzodiazepines. Nature. 1999;401:751–752. doi: 10.1038/44482. [DOI] [PubMed] [Google Scholar]
- ZHANG W., KOEHLER K.F., ZHANG P., COOK J.M. Development of a comprehensive pharmacore model for the benzodiazepine receptor. Drug Design Dis. 1995;12:192–248. [PubMed] [Google Scholar]