

Abstract
Agonists increase endothelial cell intracellular Ca2+, in part, by capacitative entry, which is triggered by the filling state of intracellular Ca2+ stores. It has been suggested that depletion of endoplasmic reticulum (ER) Ca2+ stores either leads to a physical coupling between the ER and a plasma membrane channel, or results in production of an intracellular messenger which affects the gating of membrane channels. As an axis involving the IP3 receptor has been implicated in a physical coupling mechanism the aim of this study was to examine the effects of the putative IP3 receptor antagonists/modulators, 2 aminoethoxydiphenyl borate (2APB) and xestospongin C, on endothelial cell Ca2+ entry.
Studies were conducted in fura 2 loaded cultured bovine aortic endothelial cells and endothelial cells isolated from rat heart.
2APB (30 – 300 μM) inhibited Ca2+ entry induced by both agonists (ATP 1 μM, bradykinin 0.1 μM) and receptor-independent mechanisms (thapsigargin 1 μM, ionomycin 0.5 and 5 μM). 2APB did not diminish endothelial cell ATP-induced production of IP3 nor effect in vitro binding of [3H]-IP3 to an adrenal cortex binding protein. Capacitative Ca2+ entry was also blocked by disruption of the actin cytoskeleton with cytochalasin (100 nM) while the initial Ca2+ release phase was unaffected.
Similarly to 2APB, xestospongin C (3 – 10 μM) inhibited ATP-induced Ca2+ release and capacitative Ca2+ entry. Further, xestospongin C inhibited capacitative Ca2+ entry induced by thapsigargin (1 μM) and ionomycin (0.5 μM).
The data are consistent with a mechanism of capacitative Ca2+ entry in vascular endothelial cells which requires (a) IP3 receptor binding and/or an event distal to the activation of the ER receptor and (b) a spatial relationship, dictated by the cytoskeleton, between Ca2+ release and entry pathways.
Keywords: Calcium, capacitative Ca2+ entry, Ca2+ stores, 2 aminoethoxydiphenyl borate, xestospongin C, inositol trisphosphate receptor, cytoskeleton
Introduction
The production of vasodilator factors such as nitric oxide (via constituitive NO synthase) and prostacyclin (via cyclo-oxygenase) by endothelial cells represent Ca2+-dependent processes (for example see references Martin & Michaelis, 1990; Lin et al., 2000; Mizuno et al., 2000). The increase in intracellular Ca2+ typically occurs following the activation of membrane receptors which initiate signal transduction pathways that lead to Ca2+ release from the endoplasmic reticulum (ER) and entry from the extracellular compartment.
In many cell types, including endothelial cells, release of intracellular Ca2+ has been shown to be coupled to subsequent Ca2+ entry by store depletion or capacitative Ca2+ entry (Putney, 1990; Schilling et al., 1992; Parekh & Penner, 1997; Fasolato & Nilius, 1998). Such coupling has been demonstrated in response to receptor-mediated stimuli (Putney, 1990; Schilling et al., 1992; Parekh & Penner, 1997; Fasolato & Nilius, 1998) and receptor-independent emptying of intracellular stores by agents such as thapsigargin and ionomycin (Putney, 1990; Schilling et al., 1992; Parekh & Penner, 1997; Fasolato & Nilius, 1998). While the exact mechanism by which the filling state of the ER is linked to Ca2+ entry is uncertain, it has been proposed that either a factor(s) modulating Ca2+ current across the cell membrane is released on emptying of the stores (Randriamampita & Tsien, 1993; Thomas & Hanley, 1995), or that store-depletion leads to a physical coupling between the ER and the plasma membrane (Irvine, 1990; Berridge, 1995). With respect to the former, evidence has been provided for the involvement of a number candidate signalling molecules including a small molecular weight phosphate-containing compound (Randriamampita & Tsien, 1993), a cytochrome P450 epoxygenase metabolite of arachidonic acid (Rzigalinski et al., 1999), and events requiring tyrosine phosphorylation (Fleming et al., 1996; Babnigg et al., 1997). In recent studies of clonal embryonic kidney cells Ma et al. (2000) have provided evidence supporting a direct coupling mechanism, and further, that this coupling requires the inositol trisphosphate (IP3) receptor or a related molecule (Ma et al., 2001). This conclusion was based on the observations that both rearrangement of the actin cytoskeleton and blockade of the IP3 receptor with 2 aminoethoxydiphenyl borate (2APB) uncoupled store depletion from Ca2+ entry. Similar results have been presented for other cell types, including human platelets, where an alternate IP3 receptor blocker, xestospongin C, inhibited both the association of IP3 receptors with the membrane Ca2+ channel protein Trp1, and capacitative Ca2+ entry (Rosado & Sage, 2001). However, cell or species specific differences may exist as Ribeiro et al. (1997) reported that NIH-3T3 cells do not require an intact cytoskeleton to demonstrate capacitative Ca2+ entry as thapsigargin-induced Ca2+ entry was unaffected by cytochalasin D treatment. Further, this group also reported differences in the regulation of capacitative Ca2+ entry between 3T3 and pancreatic acinar cells (Louzao et al., 1996). In addition to cell specific differences, per se, variation may occur as a result of the heterogeneity within the family of protein subunits which constitute the Ca2+ influx channels (Hofmann et al., 1999; Putney, 1999; Mery et al., 2001). In this regard some seven mammalian homologues of the Trp proteins have been identified (Putney, 1999).
The present study aimed to demonstrate capacitative Ca2+ entry pathways in bovine aortic and rat heart endothelial cells and determine specifically whether such Ca2+ entry is dependent on an axis involving the IP3 receptor-mediated mechanisms. Dependence on the IP3 receptor was examined using the cell permeable, small molecular weight, inhibitors 2APB as described by Maruyama et al. (1997) and xestospongin C (Gafni et al., 1997).
Methods
Cell culture
Bovine aortae, obtained from a local slaughterhouse, were placed in cold physiological salt solution and transported to the laboratory. Vessels were then trimmed of adherent tissue, washed, and filled with serum free DMEM (5.6 mM glucose) containing 0.2 mg collagenase and incubated for 20 mins at 37°C. Endothelial cells were detached from the vessel wall by gentle agitation and the resulting cell suspension centrifuged. The cell pellet was then resuspended in fresh DMEM containing 10% heat-inactivated foetal calf serum and incubated at 37°C in an atmosphere of 5% CO2. Cells were studied between passages 3 and 5.
For rat heart endothelial cells, hearts were removed from euthanized rats (protocol approved by the Animal Experimentation and Ethics Committee, RMIT University) and a polyethylene cannula exerted into the left ventricle. The cannulated preparation was connected to a re-circulating perfusion system and perfused with Joklik's medium containing heparin (1 u ml−1) and collagenase (0.7 mg ml−1) for 30 min, 37°C. Hearts were then mechanically disrupted and endothelial cells obtained by differential sieving and centrifugation (modified from Ford & Rovetto, 1987). Endothelial cells were then incubated in DMEM (5.6 mM glucose) with 20% foetal bovine serum at 37°C, 10% CO2.
Endothelial cells were characterized by positive staining for endothelial cell nitric oxide synthase and negative staining for α smooth muscle actin.
Measurement of intracellular Ca2+ (Ca2+i).
Following removal of endothelial cells from culture flasks (Ca2+ free solution containing 0.02% EDTA and 0.25% trypsin) cells (105 cells ml−1) were plated on glass coverslips which had been precoated with 50 μl of Engelberth Holm-Swarm (EHS) mouse sarcoma matrix (2.5 mg protein ml−1). Cells were allowed to adhere for 16 h (37°C, 5% CO2) prior to preparation for experiments. The coverslips were then transferred to a HEPES-buffered Kreb's solution containing the acetomethoxy ester of fura 2 (1 μM; 60 min). After loading cells were washed in fresh buffer to remove excess Ca2+ indicator.
Changes in intracellular Ca2+ related fluorescence were monitored using a video-based imaging system (Universal Imaging, PA, U.S.A.) coupled to an inverted microscope (20×Nikon Fluor objective lens, N.A. 0.75). As an index of changes in Ca2+i, the ratio of emitted fluorescence (510 nm) intensities was calculated following excitation at 340 and 380 nm. Fluorescence excitation (340 and 380 nm) was obtained by passing light from a 75 W Xenon source through a computer-controlled filter wheel. In all experiments the responses in various protocols were averaged across 8 – 10 cells/coverslip. Preliminary experiments verified that similar responses were obtained in single cells and groups of cells.
Protocols
Initial studies established the concentration – response relationships for the inhibitory effect of 2APB (10 – 300 μM) on ATP (1 μM)-induced increases in intracellular Ca2+. The effects of 2APB were examined both in the presence of extracellular Ca2+ and in separate experiments during re-addition of Ca2+ following exposure to ATP in the absence of extracellular Ca2+. Additional experiments were performed using bradykinin to mobilize intracellular Ca2+ to determine whether the processes of capacitative Ca2+ entry were common for different modes of receptor activation.
Following establishment of the inhibitory concentrations of 2APB, the effect of 100 μM 2APB on thapsigargin (1 μM) and ionomycin (0.5 and 5 μM)-induced Ca2+ entry was determined.
To support studies conducted with 2APB, additional experiments were performed with the IP3 receptor antagonist xestospongin C (Gafni et al., 1997). Initial studies examined the concentration-dependent effects of xestospongin C (3 – 10 μM) on ATP (1 μM)-induced Ca2+ responses. The responses to xestospongin C were studied both in the presence of extracellular Ca2+ and during Ca2+ re-addition following ATP stimulation in the absence of extracellular Ca2+. Further studies examined the effect of xestospongin C (6 and 10 μM) on thapsigargin (1 μM) and ionomycin (0.5 μM)-stimulated Ca2+ entry.
An additional set of studies were performed to examine whether cytoskeletal disruption dissociated agonist-induced Ca2+ release from capacitative Ca2+ entry, and whether this manoeuvre paralleled the effects of 2APB. Cells were prepared on EHS coated glass coverslips and loaded with fura 2 as described above. The cells were then treated with 100 nM cytochalasin D (60 min, 37°C) and ATP-induced changes in Ca2+i determined as above. Disruption of the actin cytoskeleton was confirmed by FITC-phalloidin staining and confocal microscopy.
To determine whether 2APB exerted effects on production of IP3, as opposed to an effect distal to generation of the second messenger, ATP-induced changes in cellular IP3 were determined using a radioreceptor assay (Amersham). In brief, cells were plated on EHS-coated glass coverslips, as above, and placed in six-well culture plates containing DMEM. After the 16 h adherence period cells were washed and placed in Krebs buffer and stimulated with ATP (1 μM) in the presence and absence of 2APB (30 and 100 μM). In preparation for assay, IP3 was extracted from cells using ice-cold 20% perchloric acid followed by neutralization in 60 mM HEPES, 1.5 M KOH solution. IP3 production was measured at baseline and 10 s after the addition of ATP. This timepoint was chosen as preliminary studies have shown that in our endothelial cell preparations, IP3 production in response to purinergic stimulation, is maximal at 10 s (data not shown) which is comparable with published studies (e.g. Purkiss et al., 1994).
Statistical methods
Changes in Ca2+i were assessed as changes in the 340 : 380 nm fluorescence ratio at baseline (i.e. in the absence of agonist/drug treatment). Baseline levels were designated as 100% and responses normalized to this value. Group data is shown as mean±s.e.mean. Statistical differences between treatments has been determined by analysis of variance (ANOVA) with appropriate post hoc tests. Simple comparison of the means of two groups was determined using the Student t-test. Statistical significance was accepted at the P<0.05 level.
Chemicals and reagents
2APB (Aldrich Chemical Co., Milwaukee, WI, U.S.A.) was prepared as a 0.5 M stock solution in dimethysulphoxide; subsequent dilutions were made in physiological salt solution. ATP (Sigma Chemical Co., St Louis, MO, U.S.A.) and bradykinin (Sigma) were dissolved in physiological salt solution. Thapsigargin (Sapphire Bioscience, New South Wales, Australia) was dissolved in DMSO and subsequent dilutions made in physiological salt solution. Ionomycin (Sigma) was dissolved in chloroform and further diluted in physiological salt solution. Xestospongin C (Calbiochem) was prepared in DMSO as a 5.6 mM stock solution with subsequent dilution in physiological salt solution.
Results
Effects of 2APB on endothelial cell capacitative Ca2+ entry
The effect of 2APB, in bovine aortic endothelial cells, on ATP-induced changes in Ca2+i are detailed in Figure 1. Figure 1a illustrates that in the presence of extracellular Ca2+ (2.5 mM) ATP (1 μM) stimulation results in a biphasic change in Ca2+i, with a rapid increase followed by a decline to a steady-state level which remains significantly above basal levels. 2APB alone had no effect on basal fluorescence but caused a concentration-dependent inhibition of both phases of Ca2+ mobilization (30 – 300 μM; Figure 1a) with 100 μM 2APB abolishing the secondary phase (Figure 1a,b). To illustrate capacitative Ca2+ entry, cells were exposed to ATP in the absence of extracellular Ca2+, after which Ca2+ was returned to the superfusate in the continued presence of the agonist (Figure 1b). 2APB (100 μM) added prior to ATP both inhibited the initial Ca2+ release and prevented the influx of Ca2+ following the return of the cation (Figure 1b). Similarly, when 2APB was added immediately prior to the re-addition of Ca2+, capacitative Ca2+ entry was inhibited consistent with the capacitive entry mechanism per se being inhibited in this condition rather than solely being a consequence of attenuated IP3-mediated store release (Figure 1c).
Figure 1.
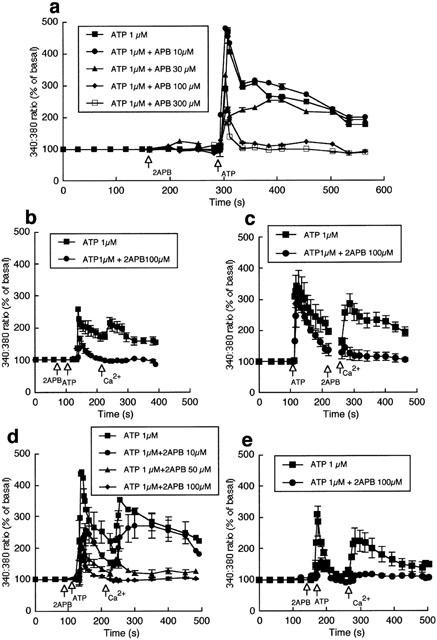
Effects of 2APB on ATP-induced changes in intracellular Ca2+. Studies shown in (a – d) were performed in bovine aortic endothelial cells and those in (e) in rat heart endothelial cells. (a) Shows the concentration-dependent effects of 2APB on ATP-induced changes in Ca2+i in the presence of extracellular Ca2+ (n=10). 2APB inhibits both the initial release of Ca2+and the subsequent plateau phase. (b) Illustrates the effect of 100 μM 2APB on ATP-induced Ca2+ release in the absence of extracellular Ca2+ and the effect of subsequent re-addition of Ca2+ (1 mM) to the superfusate (n=6). (c) Demonstrates the effect of 2APB specifically on the Ca2+ entry phase; addition of 2APB after ATP-induced Ca2+ release and immediately prior to the re-addition of extracellular Ca2+ prevented capacitative Ca2+ entry (n=6). (d) Shows the concentration dependence of the effects of 2APB (10 – 100 μM) on the ATP (1 μM)-induced Ca2+ release and influx components (n=5). (e) Demonstrates that 2APB similarly inhibits ATP (1 μM)-induced Ca2+ release and influx in cultured rat heart endothelial cells (n=6). Results are presented as mean±s.e.mean; n=number of coverslips. For clarity, representative error bars are shown.
To determine whether 2APB inhibits the Ca2+ release and entry components in bovine endothelial cells, the effects of 10, 50 and 100 μM 2APB on ATP (1 μM)-induced Ca2+ mobilization were compared (Figure 1d). While both components showed a 2APB concentration – dependent inhibition it appears that a residual release component persisted in the presence of 100 μM 2APB, whereas this concentration of 2APB abolished the influx component.
Endothelial cells cultured from rat heart showed a qualitatively similar response to ATP, with an initial Ca2+i release phase followed by Ca2+ entry (Figure 1e). As observed with the bovine cells, 2APB inhibited both phases of Ca2+ mobilization (Figure 1e).
Similar results were obtained when bradykinin (0.1 μM) was used to mobilize Ca2+i. For example, 100 μM 2APB decreased the initial Ca2+ release peak from 268±69% of baseline to 107±5% (P<0.05) and the secondary Ca2+ entry phase from 224±47% to 108±4% (P<0.05; n=5). This suggests that the effects of 2APB are not specific to ATP-receptor mediated events.
To illustrate the effects of 2APB on receptor-independent mechanisms of stimulating capacitative Ca2+ entry, separate preparations of bovine endothelial cells were treated with ionomycin (0.5 and 5 μM; Figure 2) or thapsigargin (1 μM; Figure 3). Ionomycin added to cells in the absence of extracellular Ca2+ resulted in a release of intracellular Ca2+, which was followed by an influx of Ca2+ on re-addition of the cation to the superfusate (Figure 2a,b). Concentration-dependent effects of ionomycin on both phases of Ca2+ mobilization were observed (Figure 2a,b). Addition of 2APB (100 μM) prior to ionomycin treatment prevented subsequent Ca2+ influx (Figure 2a,b). When 2APB was added to ionomycin-treated cells during the Ca2+ influx component there was a rapid decrease in intracellular Ca2+, consistent with 2APB inhibiting the Ca2+ entry process (Figure 2c). Washout of 2APB restored ionomycin-induced Ca2+ influx (data not shown).
Figure 2.
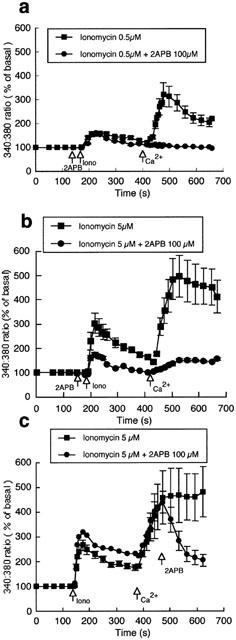
Effect of 2APB (100 μM) on ionomycin (0.5, n=6 and 5 μM, n=18)-induced changes in intracellular Ca2+ (a and b). (c (n=6)) Illustrates that addition of 2APB during the phase of capacitative Ca2+ entry inhibits entry causing a decline in Ca2+i. Results are presented as mean±s.e.mean. For clarity, representative error bars are shown.
Figure 3.
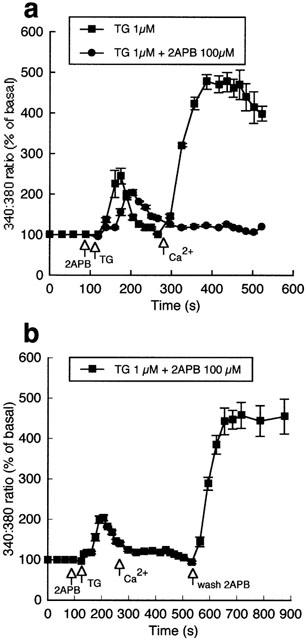
Effect of 2APB (100 μM) on thapsigargin (1 μM)-induced changes in intracellular Ca2+. In the absence of extracellular Ca2+ thapsigargin induced a transient increase in Ca2+i; subsequent addition of Ca2+ to the superfusate resulted in a large increase in Ca2+i (a; n=5). (b) Illustrates that 2APB while not preventing the initial thapsigargin-induced increase in Ca2+ prevented subsequent capacitative Ca2+ entry (n=5). Washout of 2APB restored entry of Ca2+. Results are presented as mean±s.e.mean. For clarity, representative error bars are shown.
Exposure of bovine aortic endothelial cells to the ER Ca2+ ATPase inhibitor thapsigargin resulted in an increase in Ca2+i and Ca2+ influx on re-addition of Ca2+ to the superfusate (Figure 3a). As in the case of ionomycin, 2APB significantly inhibited the thapsigargin-induced Ca2+ influx component (Figure 3a,b). Figure 3b further illustrates that washout of 2APB, in the presence of Ca2+ and thapsigargin, restores Ca2+ influx.
Effect of xestospongin C on endothelial cell Ca2+ responses to ATP and capacitative Ca2+ entry
The effects of xestospongin C, in bovine aortic endothelial cells, on ATP-induced changes in Ca2+i are shown in Figure 4. Xestospongin C alone had no effect on basal fluorescence but appeared to cause a concentration-dependent inhibition of both phases of Ca2+ mobilization (3 – 6 μM; Figure 4a). To specifically examine capacitative, Ca2+ entry, cells were exposed to ATP in the absence of extracellular Ca2+, after which Ca2+ was returned to the superfusate in the continued presence of the agonist (Figure 4b). Xestospongin C added prior to ATP both inhibited the initial Ca2+ release and prevented the influx of Ca2+ following the return of the cation (Figure 4b). For example, in the control state re-addition of Ca2+ caused a peak increase in fluorescence of 354±23% (n=7), while in the presence of xestospongin C (10 μM) the change in fluorescence was reduced to 172±23% (n=3; P<0.05).
Figure 4.
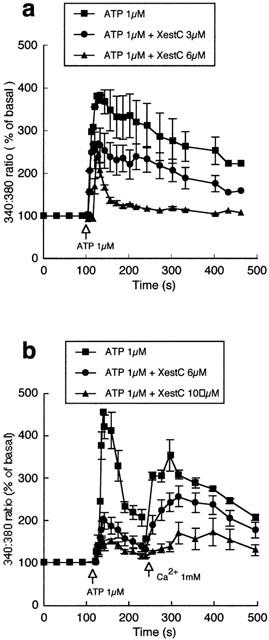
Effects of xestospongin C on intracellular Ca2+ in bovine endothelial cells. (a) Shows the concentration-dependent effects of xestospongin C on ATP-induced changes in intracellular Ca2+. Studies were performed in the presence of extracellular Ca2+. (b) Illustrates the effect of xestospongin C on ATP-induced Ca2+ release in the absence of extracellular Ca2+ and the effect of subsequent re-addition of Ca2+ (1 mM) to the superfusate. Results are presented as mean±s.e.mean. For clarity, representative error bars are shown.
To further confirm an inhibitory effect of xestospongin C on capacitative Ca2+ entry, the response to Ca2+ re-addition following thapsigargin (1 μM) treatment was compared in the absence and presence of the IP3 receptor antagonist. Xestospongin C (6 and 10 μM) significantly inhibited Ca2+ entry on re-addition of extracellular Ca2+ (Figure 5a). Similarly, Ca2+ entry induced by ionomycin (0.5 μM) was significantly attenuated by pretreatment with xestospongin C (10 μM) (Figure 5b). To specifically demonstrate an effect on the Ca2+ entry component, xestospongin C (10 μM) was added to ionomycin (0.5 μM) treated cells at the time when Ca2+ was returned to the superfusate (Figure 5c). As with xestospongin C pretreated cells, Ca2+ entry was significantly (P<0.05) attenuated. Additional experiments demonstrated that xestospongin C treatment also reversed Ca2+ entry induced by thapsigargin (1 μM) (data not shown). Collectively these data indicate that xestospongin C inhibits the capacitative Ca2+ entry phase in bovine endothelial cells.
Figure 5.
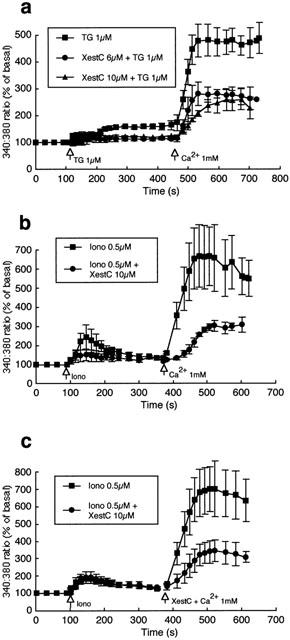
Effects of xestospongin C on thapsigargin and ionomycin-induced Ca2+ entry in bovine endothelial cells. (a) Illustrates the inhibitory effect of xestospongin C (6 and 10 μM; n=5 and 4 respectively) compared to control (n=7) on capacitative Ca2+ entry induced by exposure of endothelial cells to thapsigargin (1 μM). (b) Shows the inhibitory effect of xestospongin C (10 μM) pretreatment on ionomycin (5 μM; n=3)-induced Ca2+ entry compared with control (n=3). (c) Illustrates that xestospongin C (10 μM) added at the time of Ca2+ addition to the superfusate also attenuates capacitative Ca2+ entry (n=4 for each condition). Results are presented as mean±s.e.mean. For clarity, representative error bars are shown.
Effect of cytoskeletal disruption on endothelial cell capacitative Ca2+ entry
Cytochalasin D treatment, per se, had no apparent significant effect on baseline intracellular Ca2+, as indicated by fluorescence ratio values measured in 0 mM extracellular Ca2+ conditions, prior to the addition of ATP. Control, 0.42±0.03; 100 nM cytochalasin 0.47±0.05; 1 μM cytochalasin 0.51±0.05 (n=6, ANOVA, P<0.331, n.s.)
Exposure of cells to cytochalasin D (100 nM and 1 μM) had no apparent effect on ATP-induced Ca2+ release, while significantly inhibiting subsequent capacitative Ca2+ entry (Figure 6). Effectiveness of cytochalasin in disrupting the actin cytoskeleton was confirmed by confocal microscopy of FITC-phalloidin labelled endothelial cells (data not shown). Consistent with published studies (Rosado & Sage, 2001), cytochalasin treatment appeared to cause retraction of the cytoskeleton from the plasma membrane with a clumping of phalloidin-staining material within deeper regions of the cells. In addition, cytochalasin appeared to cause concentration-dependent changes in cell morphology evident as an increased occurrence of cytoplasmic protrusions.
Figure 6.
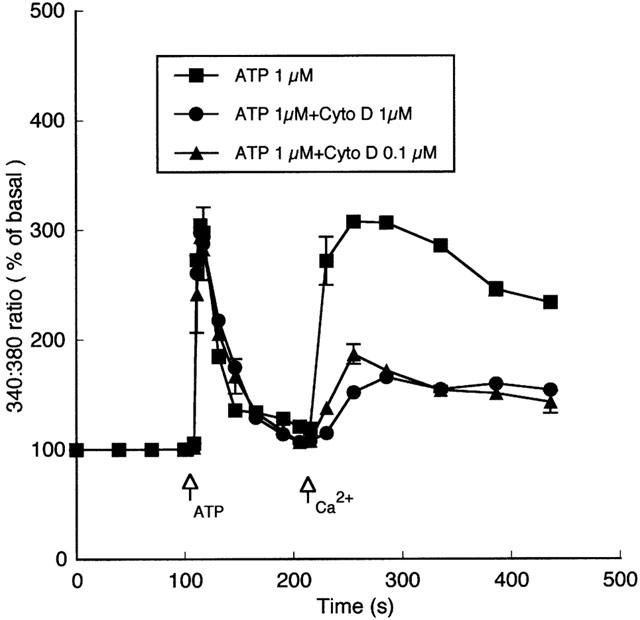
Effects of cytoskeletal disruption on ATP-induced changes in intracellular Ca2+. Cells were exposed to cytochalasin for 1 h at 37°C in a tissue culture incubator. As treatment did not cause a significant change in baseline flourescence ratio between groups, data are presented as normalized to per cent change from baseline prior to ATP stimulation. Exposure of cells to cytochalasin D (100 nM or 1 μM) had no apparent effect on the initial ATP-induced Ca2+ release while significantly inhibiting the capacitative Ca2+ entry phase (n=6 for each condition). Results are presented as mean±s.e.mean. For clarity, representative error bars are shown.
Effects of 2APB on ATP-induced inositol trisphosphate production
ATP (1 μM) caused a significant increase in IP3 production; 2.95±2.22 pmol/well baseline compared to 9.86±3.44 pmol/well after 10 s of ATP exposure (P<0.05 compared to baseline). In the presence of 2APB (100 μM) ATP caused a similarly significant increase in IP3 production; baseline 3.63±3.63 pmol well−1 compared to 9.92±2.97 pmol well−1 after 10 s of ATP exposure (P<0.05 compared to baseline). Results are presented as mean±s.e.mean for six coverslips from two independent experiments.
To verify, under cell free conditions, that 2APB did not inhibit IP3 binding, concentration response curves for IP3 and 2APB in displacing [3H]-IP3 from bovine adrenal cortex were prepared. 2APB (3 – 100 μM) did not result in a measurable alteration in the binding of [3H]-IP3 to the binding protein (data not shown).
Discussion
The results of the present study are consistent with a mechanism of capacitative Ca2+ entry in vascular endothelial cells which requires IP3 receptor binding or an event distal to the activation of the ER receptor. This conclusion is based on the observation that 2APB and xestospongin C, putative IP3 receptor antagonists or modulators, inhibit capacitative Ca2+ entry induced by either agonists (ATP, bradykinin) or receptor-independent Ca2+ mobilization (via ionomycin or thapsigargin). Control experiments demonstrated that 2APB did not lead to a reduction in IP3 production or [3H]-IP3 binding. Further, the finding of similar effects of 2APB on Ca2+ mobilization in endothelial cells from both bovine aorta and rat heart suggest that the findings are consistent across species and possibly between vascular sites.
Consistent with a number of previous studies (for example Lynch et al., 1992; Vaca & Kunze, 1994; Wang & Van Breemen, 1997) exposure of endothelial cells to ATP or bradykinin resulted in a biphasic change in intracellular Ca2+; an initial rapid increase that is a function of ER release and a sustained plateau that is, in part, dependent on Ca2+ entry from the extracellular space. As endothelial cells lack voltage gated Ca2+ channels, entry of this cation is considered to primarily occur through receptor/ligand gated channels and mechanisms related to the filling state of the ER, that is capacitative Ca2+ entry (Barritt, 1999; Lin et al., 2000; Sedova et al., 2000). The existence of the latter in the present studies was suggested by the influx of Ca2+ that occurred when the cation was returned to the superfusate of cells initially exposed to the agonists in the absence of extracellular Ca2+. Further, when the ER Ca2+ store was depleted by the ionophore, ionomycin, or the Ca2+ ATPase inhibitor, thapsigargin, Ca2+ entry was stimulated. As these latter compounds act on the filling state of the ER the data is consistent with a capacitative Ca2+ entry mechanism.
Two principal mechanisms have been proposed for the coupling of the ER filling state to Ca2+ entry; [1] that store depletion causes the release of a factor which acts to alter the gating properties of channels within the cell membrane (Randriamampita & Tsien, 1993; Thomas & Hanley, 1995) and [2] that store depletion results in a conformational change in an ER element which forms a direct or physical communication with the plasma membrane to allow Ca2+ entry (Irvine, 1990; Berridge, 1995). Recent studies of Ma et al. (2000) have been used to support a model involving a physical association between the IP3 receptor on the ER and a Ca2+ entry channel on the plasma membrane (Berridge et al., 2000). The involvement of the IP3 receptor was suggested from studies using both 2APB and xestospongin, while the physical association between the two compartments was suggested by studies altering the actin cytoskeleton. As in the present study, disruption of the cytoskeleton with cytochalasin D, in a cell line derived from pulmonary artery endothelium, was shown to inhibit capacitative Ca2+ entry without an effect on the initial agonist-induced release of Ca2+ (Holda & Blatter, 1997). Further support for a dynamic role of the actin cytoskeleton is provided by the studies of Rosado & Sage (2001) in human platelets, showing that both stabilization of the actin cytoskeleton with jasplakinolide and disruption by cytochalasin prevent capacitative Ca2+ entry.
The exact role that the cytoskeleton appears to play in capacitative Ca2+ entry appears to vary between cell types. In contrast to the findings of the present study, and those described above (Holda & Blatter, 1997; Rosado & Sage, 2001), Riberio et al. (1997) reported that in NIH-3T3 cells cytochalasin treatment specifically affects the agonist-induced release of Ca2+ while having no effect on capacitative Ca2+ entry. The differences, particularly in regard to the Ca2+ release phase, may relate to the extent of cytochalasin treatment as Riberio et al. (1997) used a 10 μM concentration which markedly changed cell morphology while in the present study 100 nM and 1 μM concentrations of cytochalasin were employed. The lower concentrations were chosen to avoid gross changes in cell shape and detachment of the cells from the underlying matrix. In a study examining the role of capacitative Ca2+ entry in regulation of adenylyl cyclase isoforms, of C6-2B rat glioma cells, Fagan et al. (1998) have suggested that regulation of the cyclase requires an intimate association with the Ca2+ entry pathway; although this relationship could not be altered by agents known to disrupt the cytoskeleton (cystochalasin, nocodozole, colchicine). Similarly to the studies of Riberio et al. (1997) cytoskeletal disruption did not impair capacitative Ca2+ entry in response to thapsigargin treatment. In studies of embryonic kidney cells (HEK293) Ma et al. (2000) reported that condensation of cortical actin by treatment with the phosphatase inhibitor calyculin A impaired capacitative Ca2+ entry while not affecting IP3-mediated Ca2+ release. Formation of a dense cortical actin layer was considered to impair the physical interaction between the plasma membrane and endoplasmic reticulum. This group, however, has previously reported that disassembly of the actin cytoskeleton with cytochalasin D did not impair coupling between Ca2+ release and Ca2+ entry (Patterson et al., 1999). In addition to these studies of other investigators, we did not find cytochalasin treatment to markedly inhibit capacitative Ca2+ entry in arteriolar smooth muscle (Potocnik & Hill, 2001). Thus, collectively the available data suggest that dependence of capacitative Ca2+ entry on a functional cytoskeleton may vary between cell types. Conceivably the physical relationship between the endoplasmic reticulum and Ca2+ entry channels is dependent on different structural elements in differing cell types. Alternatively contrasting results may reflect other factors such as differences in the Ca2+ entry channels in the varying cell types (Putney, 1999) or variation in the amount/distribution of cytoskeletal elements.
The results of the present studies add to a growing body of evidence supporting a modulatory effect of 2APB on IP3 receptor-mediated processes (Maruyama et al., 1997; Ascher-Landsberg et al., 1999; Gysembergh et al., 1999; Ma et al., 2000). With respect to ATP-induced Ca2+ mobilization 2APB concentration-dependently attenuated both the initial release phase and subsequent Ca2+ entry. 2APB did not, however, appear to have any effect on ATP-induced IP3 production or basal Ca2+ levels as assessed by changes in the 340 : 380 nm fluorescence ratio. In contrast to the latter observation Gysembergh et al. (1999) reported an effect of 2APB (1 – 10 μM) on rabbit cardiac myocytes, while Maruyama et al. (1997) found that concentrations greater than 90 μM 2APB were required to increase baseline Ca2+i in a rat cerebral microsomal preparation and greater than 200 μM 2APB was required to increase basal Ca2+i in human platelets. Whether these differences reflect tissue specific or methodological differences is currently uncertain.
Recent studies have suggested that 2APB may exert a direct inhibitory effect on capacitative entry at sites other than the IP3 receptor. Consistent with this, 2APB has been shown to inhibit Ca2+ entry in DT 40 B cells in which the IP3 receptor has been deleted (Broad et al., 2001), and in excised membrane patches from rat basophilic leukaemia cells (Braun et al., 2001). In the present study, however, both putative IP3 receptor antagonists, 2APB and xestospongin, inhibited endothelial cell capacitative Ca2+ entry, suggesting that in this cell type the IP3 receptor may indeed be a component of the capacitative Ca2+ entry pathway. Consistent with these results, Rosado & Sage (2001) have recently shown that xestospongin inhibits capacitative Ca2+ entry in human platelets and the association between IP3 receptors and Trp1. No evidence currently exists for xestospongin exerting an effect directly at the level of Trp channels, although it has been suggested that in solution 2APB may dimerise thereby resembling the molecular shape of xestospongin C (Van Rossum et al., 2000; Liu & Ambudkar, 2001). The apparently discrepant results may also relate to the particular Ca2+ channel proteins (for example mammalian homologues of Trp channels (Putney, 1999)) or IP3 receptor subtypes expressed in different cell types. With respect to the latter it has been shown in A7r5 cells that specific IP3 receptor subtypes (namely the IP3R1 subtype) are associated with capacitative Ca2+ entry, while the IP3R3 subtype appears unrelated to this mode of Ca2+ entry (Wang et al., 2001).
In more recent studies Ma et al. (2001) have suggested that 2APB, while being an IP3 receptor antagonist, may also interact with a regulatory protein that exerts an action over both IP3 receptors and capacitative Ca2+ entry channels. Such an effect could be consistent with both the data from the present studies and those suggesting an inhibitory effect of 2APB proximal to the ER in this signalling pathway.
That 2APB inhibited store-depletion mediated Ca2+ entry in the present studies was demonstrated firstly by the addition of 2APB to ATP-stimulated endothelial cells following the initial release peak (Figure 1) and secondly by inhibition of Ca2+ entry following exposure of cells to ionomycin (Figure 2) and thapsigargin (Figure 3). In the latter case Ca2+ depletion of the ER occurs independently of plasma membrane receptor activation. Somewhat surprisingly, 2APB reduced the initial Ca2+ release caused by 5 μM ionomycin while not affecting the initial increase in Ca2+i induced by either 0.5 μM ionomycin or thapsigargin (1 μM). Conceivably the higher concentration of the ionophore, as well as depleting intracellular Ca2+ stores, exerts an additional effect at the level of the ER membrane. Alternatively, at the higher concentration of ionomycin unidentified effects are evident; perhaps related to the greater release of Ca2+ seen under this condition (relative to 0.5 μM ionomycin). Regardless of this effect, the data presented are consistent with 2APB inhibiting store depletion mediated Ca2+ entry in endothelial cells.
An alternate explanation of the data relates to the functional relationship between ryanodine release channels of the ER and plasma membrane KCa channels. Ca2+ release from the ER into the subplasmalemmal space has been shown to activate KCa channels, resulting in membrane hyperpolarization (Frieden & Graier, 2000). This change in membrane potential subsequently increases the driving force for Ca2+ entry and has been implicated in agonist-induced production of a number of Ca2+-dependent vasoactive compounds (Luckhoff & Busse, 1990a). Support of this pathway has also been derived from the observation that K+ channel activators lead to hyperpolarization and Ca2+ entry in endothelial cells (Luckhoff & Busse, 1990b). Analogous to the data obtained in the present studies disruption of the normal relationship between the superficial ER and the plasma membrane with nocodozole prevented ryanodine-induced activation of KCa channels in a human umbilical vein endothelial cell line (Frieden & Graier, 2000). While it is possible that such a mechanism would have been disrupted in the current studies, by exposure of cells to cytochalasin, it appears unlikely that both the 2APB (see also Maruyama et al., 1997) and xestospongin data could be explained simply by a ryanodine receptor-Ca2+ entry pathway. Further, if the endothelial cells studied exhibit both IP3 receptor-dependent mechanisms and Ca2+-induced Ca2+ release (CICR) (Mozhayeva, 1996) the effectiveness of 2APB and xestospongin C in preventing Ca2+ entry suggests that the IP3 receptor dependent processes occur proximally to CICR.
In summary, the data presented demonstrate the effectiveness of 2APB and xestospongin C, putative modulators of the IP3 receptor, in inhibiting agonist-induced Ca2+ entry in vascular endothelial cells. Further, the fact that cytoskeletal disruption with cytochalasin specifically inhibited the Ca2+ influx phase, as opposed to the release component, adds support to the requirement of a physical coupling, or spatial proximity, for the coupling of store depletion to Ca2+ entry in endothelial cells. Differences in the effect of agents such as cytochalasin on Ca2+ entry, however, suggest that variation in the exact coupling mechanism may exist between cell types. Given the ability of the endothelial cell in responding to physiological forces such as shear stress (Davies et al., 1997) and the fact that the cytoskeleton is implicated in mechanotransduction (Ingber, 1997), these results may be of direct relevance to physiological mechanisms of endothelial cell autacoid production.
Acknowledgments
Work described in these studies was supported by grants from the National Health and Medical Research Council of Australia, National Heart Foundation and the Edward Dunlop Foundation. Sincere thanks are given to Dr Simon Potocnik for assistance with confocal microscopy studies verifying the effects of cytochalasin on endothelial cells. Appreciation is extended to Marjorie Dunlop, PhD, University of Melbourne, for constructive comments prior to submission of the manuscript.
Abbreviations
- 2APB
2 aminoethoxydiphenyl borate
- ATP
adenosine triphosphate
- CICR
Ca2+-induced Ca2+ release
- DMEM
Dulbecco's modified essential medium
- DMSO
dimethyl sulphoxide
- EHS
Engelberth Holm-Swarm
- ER
endoplasmic reticulum
- IP3
inositol trisphosphate
- Trp
transient receptor potential
References
- ASCHER-LANDSBERG J., SAUNDERS T., ELOVITZ M., PHILLIPPE M. The effects of 2-aminoethoxydiphenyl borate, a novel inositol 1,4,5-trisphosphate receptor modulator on myometrial contractions. Biochem. Biophys. Res. Comm. 1999;264:979–982. doi: 10.1006/bbrc.1999.1602. [DOI] [PubMed] [Google Scholar]
- BABNIGG G., BOWERSOX S.R., VILLEREAL S.R. The role of pp60c-src in the regulation of calcium entry via store-operated calcium channels. J. Biol. Chem. 1997;272:29434–29437. doi: 10.1074/jbc.272.47.29434. [DOI] [PubMed] [Google Scholar]
- BARRITT G.J. Receptor-activated Ca2+ inflow in animal cells: a variety of pathways tailored to meet different Ca2+ signalling requirements. Biochem. J. 1999;337:153–169. [PMC free article] [PubMed] [Google Scholar]
- BERRIDGE M.J. Capacitative calcium entry. Biochem. J. 1995;312:1–11. doi: 10.1042/bj3120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERRIDGE M.J., LIPP P., BOOTMAN M.D. The calcium entry pas de deux. Science. 2000;287:1064–1065. doi: 10.1126/science.287.5458.1604. [DOI] [PubMed] [Google Scholar]
- BRAUN F-J., BROAD L.M., ARMSTRONG D.L., PUTNEY J.W., Jr Stable activation of single Ca2+ release-activated Ca2+ channels in divalent cation-free solutions. J. Biol. Chem. 2001;276:1063–1070. doi: 10.1074/jbc.M008348200. [DOI] [PubMed] [Google Scholar]
- BROAD L.M., BRAUN F-J., LIEVREMONT J-P., BIRD G.St.J., KUROSAKI T., PUTNEY J.W., Jr Role of the phospholipase C-inositol 1,4,5-trisphosphate pathway in calcium release-activated calcium current and capacitative calcium entry. J. Biol. Chem. 2001;276:15945–15952. doi: 10.1074/jbc.M011571200. [DOI] [PubMed] [Google Scholar]
- DAVIES P.F., BARBEE K.A., VOLIN M.V., ROBOTEWSKYJ A., CHEN J., JOSEPH L., GRIEM M.L., WERNICK M.N., JACOBS E., POLACEK D.C., DEPAOLA N., BARAKAT A.I. Spatial relationships in early signaling events of flow-mediated endothelial mechanotransduction. Annu. Rev. Physiol. 1997;59:527–549. doi: 10.1146/annurev.physiol.59.1.527. [DOI] [PubMed] [Google Scholar]
- FAGAN K.A., MONS N., COOPER D.M.F. Dependence of the Ca2+-inhibitable adenylyl cyclase of C6-2B glioma cells on capacitative Ca2+ entry. J. Biol. Chem. 1998;273:9297–9305. doi: 10.1074/jbc.273.15.9297. [DOI] [PubMed] [Google Scholar]
- FASOLATO C., NILIUS B. Store depletion triggers the calcium release-activated calcium current (ICRAC) in macrovascular endothelial cells: a comparison with Jurkat and embyonic kidney cell lines. Pflügers Arch. 1998;436:69–74. doi: 10.1007/s004240050605. [DOI] [PubMed] [Google Scholar]
- FLEMING I., FISSLTHALER B., BUSSE R. Interdependence of calcium signaling and protein tyrosine phosphorylation in human endothelial cells. J. Biol. Chem. 1996;271:11009–11015. doi: 10.1074/jbc.271.18.11009. [DOI] [PubMed] [Google Scholar]
- FRIEDEN M., GRAIER W.F. Subplasmalemmal ryanodine-sensitive Ca2+ release contributes to Ca2+-dependent K+ channel activation in a human umbilical vein endothelial cell line. J. Physiol. 2000;524:715–724. doi: 10.1111/j.1469-7793.2000.00715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FORD D.A., ROVETTO M.J. Rat cardiac myocyte adenosine transport and metabolism. Am. J. Physiol. 1987;252:H54–H63. doi: 10.1152/ajpheart.1987.252.1.H54. [DOI] [PubMed] [Google Scholar]
- GAFNI J., MUNSCH J.A., LAM T.H., CATLIN M.C., COSTA L.G., MOLINSKI T.F., PESSAH I.N. Xestospongins: potent membrane permeable blockers of the inositol 1,4,5-trisphosphate receptor. Neuron. 1997;19:723–733. doi: 10.1016/s0896-6273(00)80384-0. [DOI] [PubMed] [Google Scholar]
- GYSEMBERGH H., LEMAIRE S., PIOT C., SPORTOUCH C., RICHARD S., KLONER R.A., PRZYKLENK K. Pharmacological manipulation of Ins(1,4,5)P3 signaling mimics preconditioning in rabbit heart. Am. J. Physiol. 1999;277:H2458–H2469. doi: 10.1152/ajpheart.1999.277.6.H2458. [DOI] [PubMed] [Google Scholar]
- HOFMANN T., OBUKHOV A.G., SCHAEFER M., HARTENECK C., GUDERMANN T., SCHULTZ G. Diect activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- HOLDA J.R., BLATTER L.A. Capacitative calcium entry is inhibited in vascular endothelial cells by disruption of cytoskeletal microfilaments. FEBS Lett. 1997;403:191–196. doi: 10.1016/s0014-5793(97)00051-3. [DOI] [PubMed] [Google Scholar]
- INGBER D.E. Tensegrity: the architectural basis of cellular mechanotransduction. Annu. Rev. Physiol. 1997;59:575–599. doi: 10.1146/annurev.physiol.59.1.575. [DOI] [PubMed] [Google Scholar]
- IRVINE R.F. “Quantal” Ca2+ release and the control of Ca2+ entry by inositol phosphates – a possible mechanism. FEBS Lett. 1990;263:5–9. doi: 10.1016/0014-5793(90)80692-c. [DOI] [PubMed] [Google Scholar]
- LIN S., FAGAN K.A., LI K-X., SHAUL P.W., COOPER D.M.F., RODMAN D.M. Sustained endothelial nitric oxide synthase activation requires capacitative Ca2+ entry. J. Biol. Chem. 2000;275:17979–17985. doi: 10.1074/jbc.275.24.17979. [DOI] [PubMed] [Google Scholar]
- LIU X., AMBUDKAR I.S. Characteristics of a store-operated calcium-permeable channel, SOCC: sarcoendoplasmic reticulum calcium pump function controls channel gating. J. Biol. Chem. 2001;276:29891–29898. doi: 10.1074/jbc.M103283200. [DOI] [PubMed] [Google Scholar]
- LOUZAO M.C., RIBEIRO C.M.P., BIRD G.St.J., PUTNEY J.W., Jr Cell type-specific modes of feedback regulation of capacitative calcium entry. J. Biol. Chem. 1996;271:14807–14813. doi: 10.1074/jbc.271.25.14807. [DOI] [PubMed] [Google Scholar]
- LUCKHOFF A., BUSSE R. Calcium influx into endothelial cells and formation of endothelium-derived relaxing factor is controlled by membrane potential. Pflügers Arch. 1990a;416:305–311. doi: 10.1007/BF00392067. [DOI] [PubMed] [Google Scholar]
- LUCKHOFF A., BUSSE R. Activators of potassium channels enhance calcium influx into endothelial cells as a consequence of potassium currents. Naunyn Schmiedebergs Arch. Pharmacol. 1990b;342:94–99. doi: 10.1007/BF00178979. [DOI] [PubMed] [Google Scholar]
- LYNCH M., GILLESPIE J.I., GREENWELL J.R., JOHNSON C. Intracellular calcium ‘signatures' evoked by different agonists in isolated bovine aortic endothelial cells. Cell Calcium. 1992;13:227–233. doi: 10.1016/0143-4160(92)90011-g. [DOI] [PubMed] [Google Scholar]
- MA H-T., PATTERSON R.L., VAN ROSSUM D.B., BIRNBAUMER L., MIKOSHIBA K., GILL D.L. Requirement of the inositol trisphosphate receptor for activation of store-operated Ca2+ channels. Science. 2000;287:1647–1651. doi: 10.1126/science.287.5458.1647. [DOI] [PubMed] [Google Scholar]
- MA H.-T., VENKATACHALAM K., LI H.-S., MONTELL C., KUROSAKI T., PATTERSON R.L., GILL D.L. Assessment of the role of the inositol 1,4,5-trisphosphate receptor in the activation of transient receptor potential channels and store-operated Ca2+ entry channels. J. Biol. Chem. 2001;276:18888–18896. doi: 10.1074/jbc.M100944200. [DOI] [PubMed] [Google Scholar]
- MARTIN T.W., MICHAELIS K.C. Ca2+-dependent synthesis of prostaglandin I2 and mobilization of arachidonic acid from phospholipids in cultured endothelial cells permeabilized with saponin. Biochim. Biophys. Acta. 1990;1054:159–168. doi: 10.1016/0167-4889(90)90237-8. [DOI] [PubMed] [Google Scholar]
- MARUYAMA T., KANAJI T., NAKADE S., KANNO T., MIKOSHIBA K. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of ins (1,4,5)P3-induced Ca2+ release. J. Biochem (Tokyo) 1997;122:498–505. doi: 10.1093/oxfordjournals.jbchem.a021780. [DOI] [PubMed] [Google Scholar]
- MERY L., MAGNINO F., SCHMIDT K., KRAUSE K.H., DUFOUR J.F. Alternative splice variants of hTrp4 differentially interact with the C-terminal portion of the inositol 1,4,5-trisphosphate receptors. FEBS Lett. 2001;487:377–383. doi: 10.1016/s0014-5793(00)02362-0. [DOI] [PubMed] [Google Scholar]
- MIZUNO O., KOBAYASHI S., HIRANO K., NISHIMURA J., KUBO C., KANAIDE H. Stimulus-specific alteration of the relationship between cytosolic Ca2+ transients and nitric oxide production in endothelial cells ex vivo. Br. J. Pharmacol. 2000;130:1140–1146. doi: 10.1038/sj.bjp.0703420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOZHAYEVA M.G. [Ca2+]i elevation evoked by Ca2+ readdition to the medium after agonist-induced Ca2+ release can involve both IP3-, and ryanodine-sensitive Ca2+ release. Pflügers Arch. 1996;433:180–187. doi: 10.1007/s004240050265. [DOI] [PubMed] [Google Scholar]
- PAREKH A.B., PENNER R. Store depletion and calcium influx. Physiol. Rev. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- PATTERSON R.L., VAN ROSSUM D.B., GILL D.L. Store-operated Ca2+ entry: evidence for a secretion-like coupling model. Cell. 1999;98:487–499. doi: 10.1016/s0092-8674(00)81977-7. [DOI] [PubMed] [Google Scholar]
- POTOCNIK S.J., HILL M.A. Pharmacological evidence for capacitative Ca2+ entry in cannulated and pressurized skeletal muscle arterioles. Brit. J. Pharmacol. 2001;134:247–256. doi: 10.1038/sj.bjp.0704270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PURKISS J.R., WILKINSON G.F., BOARDER M.R. Differential regulation of inositol 1,4,5-trisphosphate by co-existing P2Y-purinoceptors and nucleotide receptors on bovine aortic endothelial cells. Br. J. Pharmacol. 1994;111:723–728. doi: 10.1111/j.1476-5381.1994.tb14797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PUTNEY J.W., Jr Capacitative calcium entry revisited. Cell Calcium. 1990;11:611–624. doi: 10.1016/0143-4160(90)90016-n. [DOI] [PubMed] [Google Scholar]
- PUTNEY J.W., Jr TRP, inositol 1,4,5-trisphosphate receptors, and capacitative calcium entry. Proc. Natl Acad. Sci. 1999;96:14669–14671. doi: 10.1073/pnas.96.26.14669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RANDRIAMAMPITA C., TSIEN R.Y. Emptying of intracellular Ca2+ stores releases a novel small messenger that stimulates Ca2+ influx. Nature. 1993;364:809–814. doi: 10.1038/364809a0. [DOI] [PubMed] [Google Scholar]
- RIBERIO C.M.P., REECE J., PUTNEY J.W., Jr Role of the cytoskeleton in calcium signaling in NIH 3T3 cells: An intact cytoskeleton is required for agonist-induced [Ca2+]i signaling, but not for capacitative calcium entry. J. Biol. Chem. 1997;272:26555–26561. doi: 10.1074/jbc.272.42.26555. [DOI] [PubMed] [Google Scholar]
- ROSADO J.A., SAGE S.O. Activation of store-mediated calcium entry by secretion-like coupling between the inositol 1,4,5-trisphosphate receptor type II and human transient receptor potential (hTrp1) channels in human platelets. Biochem. J. 2001;256:191–198. doi: 10.1042/0264-6021:3560191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RZIGALINSKI B.A., WILLOUGHBY K.A., HOFFMAN S.W., FALCK J.R., ELLIS E.F. Calcium influx factor, further evidence it is 5,6,-epoxyeicosatrienoic acid. J. Biol. Chem. 1999;274:175–182. doi: 10.1074/jbc.274.1.175. [DOI] [PubMed] [Google Scholar]
- SCHILLING W.P., CABELLO O.A., RAJAN L. Depletion of the inositol 1,4,5-trisphosphate-sensitive intracellular Ca2+ store in vascular endothelial cells activates the agonist-sensitive Ca2+ influx pathway. Biochem. J. 1992;284:521–530. doi: 10.1042/bj2840521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEDOVA M., KLISHIN A., HUSER J., BLATTER L.A. Capacitative Ca2+ entry is graded with degree of intracellular Ca2+ store depletion in bovine vascular endothelial cells. J. Physiol. 2000;523:549–559. doi: 10.1111/j.1469-7793.2000.t01-3-00549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOMAS D., HANLEY M.R. Evaluation of calcium influx factors from stimulated Jurkat T-lymphocytes by microinjection into Xenopus oocytes. J. Biol. Chem. 1995;270:6429–6432. doi: 10.1074/jbc.270.12.6429. [DOI] [PubMed] [Google Scholar]
- VACA L., KUNZE D.L. Depletion of intracellular Ca2+ stores activates a Ca2+ selective channel in vascular endothelium. Am. J. Physiol. 1994;267:C950–C955. doi: 10.1152/ajpcell.1994.267.4.C920. [DOI] [PubMed] [Google Scholar]
- VAN ROSSUM D.B., PATTERSON R.L., MA H.-T., GILL D.L. Ca2+ entry mediated by store depletion, s-nitrosylation, and TRP3 channels. J. Biol. Chem. 2000;275:28562–28568. doi: 10.1074/jbc.M003147200. [DOI] [PubMed] [Google Scholar]
- WANG X., VAN BREEMEN C. Multiple mechanisms of activating Ca2+ entry in freshly prepared rabbit aortic endothelial cells. J. Vasc. Res. 1997;34:196–207. doi: 10.1159/000159223. [DOI] [PubMed] [Google Scholar]
- WANG Y., CHEN J., WANG Y., TAYLOR C.W., HIRATA Y., HAGIWARA H., MIKOSHIBA K., TOYO-OKA T., OMATA M., SAKAKI Y. Crucial role of type 1, but not type 3, inositol 1,4,5-trisphosphate (IP3) receptors in IP3-induced Ca2+ release, capacitative Ca2+ entry, and proliferation of A7r5 vascular smooth muscle cells. Circ. Res. 2001;88:202–209. doi: 10.1161/01.res.88.2.202. [DOI] [PubMed] [Google Scholar]