

Abstract
Redox and ROS regulation of MAPK-mediated TNF-α biosynthesis is not well characterized. It was hypothesized that the involvement of the MAPK pathway in regulating LPS-mediated TNF-α secretion is redox-dependent, NF-κB-sensitive and attenuated by N-acetyl-L-cysteine (NAC) and other antioxidants.
In alveolar epithelial cells, LPS induced a time- and dose-dependent phosphorylation of MAPKp38. This was associated with the activation of MAPK-activated protein kinase, which phosphorylated the small heat-shock protein, Hsp27.
MAPKp38 inhibition (SB-203580) abrogated LPS-induced TNF-α production. MAPKERK blockade (PD-98059) attenuated TNF-α secretion, an effect synergistically amplified in the presence of SB-203580.
Regulation of NF-κB by selective inhibitors revealed that this pathway is partially involved in regulating LPS-mediated TNF-α secretion. Whereas the proteasome inhibitor, MG-132, had no effect on LPS-mediated TNF-α production, CAPE, sulfasalazine and SN-50, a cell-permeant NF-κB inhibitor, attenuated but did not abrogate TNF-α biosynthesis.
LPS up-regulated ROS, an effect abrogated by 4′-hydroxy-3′-methoxy-acetophenone and NAC, which reduced TNF-α secretion, induced the accumulation of GSH, reduced the concentration of GSSG, and blockaded the phosphorylation/activation of MAPKp38 pathway.
ROS induced MAPKp38 phosphorylation and selective antioxidants, including the permeant GSH precursor, γ-GCE, reduced ROS-dependent MAPKp38 phosphorylation.
These results indicate that the MAPK pathway and MAPK-mediated regulation of TNF-α production is redox-dependent, GSH-mediated and requires, at least in part, a NF-κB/ROS-sensitive mechanism.
Keywords: Antioxidant, cytokines, endotoxin, immunopharmacology, pathophysiology, p38 MAPK, TNF-α
Introduction
Many extracellular stimuli, including pro-inflammatory cytokines and other inflammatory mediators (Herlaar & Brown, 1999), elicit specific cellular responses through the activation of mitogen-activated protein kinase (MAPK) signalling pathways (Marshall, 1995; Garrington & Johnson, 1999). MAPKs are proline-targeted serine-threonine kinases that transduce environmental stimuli to the nucleus and they themselves are activated by upstream MAPK kinases (MAPKKs) on both threonine and tyrosine residues within an ‘activation loop' (Su & Karin, 1996; Garrington & Johnson, 1999). Once activated, MAPKs can phosphorylate and activate other kinases or nuclear proteins, including potential transcription factors, such as NF-κB, and substrates (Stein et al., 1997). The novel mammalian reactivating protein kinase (p38/RK) MAPKs and ERK are stress-activated protein kinases (SAPK) that mediate responses to cellular stresses such as u.v. irradiation, osmotic imbalance, heat shock, DNA damage, bacterial products such as lipopolysaccharide (LPS), and inflammatory signals (Lee et al., 1994; Wang et al., 1997). Furthermore, inflammatory mediators, such as cytokines, including tumour necrosis factor (TNF)-α, activate the MAPK pathway in several cell types (Raingeaud et al., 1995; Moriguchi et al., 1997; Zu et al., 1998). Of note, MAP kinase has been recently implicated in regulating pro-inflammatory cytokine biosynthesis (Ballard-Croft et al., 2001; Lee et al., 1994) and transcription (Rutault et al., 2001).
The tripeptide L-γ-glutamyl-L-cysteinyl-glycine, or glutathione (GSH), a ubiquitous thiol, plays a major role in maintaining intracellular reduction-oxidation (redox) equilibrium in the lung (Meister, 1988; Hayes & McLellan, 1999; Rahman, 1999; 2000; Haddad & Land, 2000a; Haddad et al., 2000). The cysteinyl moiety of GSH provides the reactive thiol as a functional element responsible for the diverse properties of glutathione, including (i) an antioxidant potential mediated by the peroxidase coupled reaction, (ii) regulation of cellular sulphydryl status and redox equilibrium, (iii) governing pathways in neuro-immune-endocrine interactions as an immunopharmacological thiol, and (iv) regulation of the expression/activation of redox-sensitive transcription factors induced by stress-evoked responses (Dröge et al., 1994; Hayes & McLellan, 1999; Haddad et al., 2000). The pivotal role of redox cycle in maintaining the integrity of the biological system in the face of oxidative stress and other challenges is, therefore, of particular relevance. The immunopharmacological potential assigned to glutathione (Thompson et al., 1985; Dröge et al., 1994), furthermore, stems from established observations. Interleukin (IL)-1 induced responses in mesangial cells, for instance, occurred through modulating redox equilibrium (Rovin et al., 1997). In addition, reactive oxygen species (ROS) signalling pathways regulating the transcription of IL-4 (Jeannin et al., 1995), IL-6, IL-8 (Gosset et al., 1999) and TNF-α (Neuschwander-Tetri et al., 1996; Gosset et al., 1999) were mediated through a thiol-dependent mechanism. Interestingly, antioxidants (Matsumoto et al., 1998; Reimund et al., 1998; Barrett et al., 1999; Haddad et al., 2001a) and glutathione precursors (Jeannin et al., 1995; Gosset et al., 1999; Pena et al., 1999; Haddad et al., 2001c) have been shown to down-regulate cytokine synthesis, activation and downstream processes. In this respect, N-acetyl-L-cysteine (NAC), an antioxidant and a GSH precursor (Bernard, 1991; Haddad et al., 2000), has been shown to ameliorate cytokine transcription and synthesis (Matsumoto et al., 1998; Tsuji et al., 1999; Hashimoto et al., 2001), and suppress ROS-mediated lung injury (Bernard, 1991).
Redox regulation of MAPKp38- and MAPKERK-mediated cytokine transcription and biosynthesis has not been well characterized. It has been shown previously that thioredoxin (TRX), a redox control protein, negatively regulated MAPKp38 kinase activation and MAPKp38-mediated IL-6 expression (Hashimoto et al., 1999a, 1999b). In addition, Hashimoto et al. (2001) recently reported that NAC attenuated TNF-α-induced MAPKp38 activation and the downstream IL-8 dependent pathway. One mechanism involved in the generation of ROS in a variety of cells is the oxidative burst, a process catalysed by the multicomplex enzyme NADPH oxidase; MAPKp38 has been implicated in regulating this complex and subsequently coupled intracellular redox signalling with the oxidative burst-dependent regulation of pro-inflammatory signals (Ridley et al., 1997; Nick et al., 1999; Partrick et al., 2000). However, redox regulation of LPS-induced MAPK-mediated TNF-α biosynthesis in the alveolar epithelium has yet to be ascertained and the underlying signalling mechanism was unravelled. Therefore, the aim of the present study was to investigate the role that glutathione and antioxidants play in regulating MAPK phosphorylation/activation, and to determine whether ROS and redox equilibrium are involved in MAPK-dependent regulation of TNF-α biosynthesis.
Methods
Chemicals and reagents
Unless otherwise indicated, chemicals of the highest analytical grade were purchased from Sigma-Aldrich (Dorset, England, U.K.). The pyridinyl imidazole SB-203580, a specific inhibitor of MAPKp38 (Lee et al., 1994) and PD-98059, a specific inhibitor of MAPKERK, were obtained from Calbiochem-Novabiochem Corporation (England, U.K.); SB-203580 and PD-98059 were reconstituted in dimethyl sulphoxide (DMSO), where the final concentration of DMSO was determined to be ⩽0.01%. γ-Glutamylcysteinyl-ethyl ester (γ-GCE), a cell-permeant GSH analogue, was purchased from Calbiochem-Novabiochem Corporation and reconstituted in sterile deionized water and stored at 4°C. Phospho-MAPKp38 (Thr180/Tyr182) and MAPKp38 antibodies were purchased from New England Biolabs Incorporation (England, U.K.). Recombinant heat-shock protein 27 (Hsp27) was obtained from Calbiochem-Novabiochem Corporation and kept stored at −70°C.
Primary cultures of alveolar epithelia
Foetal alveolar type II (fATII) epithelial cells were isolated from lungs of foetuses, essentially as reported elsewhere (Haddad & Land, 2000a, 2000b). Briefly, foetal rats were removed from pregnant Sprague-Dawley rats by caesarean section at day 19 of gestation (term=22 days), the lungs excised, teased free from heart and upper airway tissue, and were finely minced then washed free of erythrocytes using sterile, chilled Mg2+- and Ca2+-free Hanks' balanced salt solution (HBSS; 0.5 ml foetus−1). The cleaned lung tissue was resuspended in 1 ml foetus−1 HBSS containing trypsin (0.1 mg ml−1), collagenase (0.06 mg ml−1) and DNase I (0.012% w v−1), and was agitated at 37°C for 20 min. The solution was then centrifuged at 100 ×g for 2 min to remove undispersed tissue, the supernatant was saved to a fresh sterile tube and an equal volume of Dulbecco's modified Eagle medium (DMEM) with 10% (v v−1) foetal calf serum (FCS) was added to the supernatant. After passing the supernatant through a 120 μm pore sterile mesh, the filtrate was centrifuged at 420 ×g for 5 min, the pellet re-suspended in 20 mls DMEM/FCS and the cells were placed into a T-150 culture flash for 1 h at 37°C to enable fibroblasts and non-epithelial cells to adhere. Unattached cells were washed three times by centrifugation at 420×g for 5 min each and then seeded onto 24 mm diameter Transwell-clear permeable supports (Costar; 0.4 μm pore size) at a density of 5×106 cells per filter and were allowed to adhere overnight at 152 Torr (≈21% O2/5% CO2). DMEM/FCS was exchanged for 4 mls of serum free PC-1 media (Biowhittaker, MD, U.S.A.) pre-equilibrated to pO2=152 Torr and 37°C 24 h later and cells were maintained at this pO2 until the experiment. In each case, and under conditions of independent treatment and pre-treatments, the adenylate energy charge, an index of cell viability and competence, remained ⩾0.7 and transepithelial monolayer resistance was monitored constantly at 250–350 Ω cm2 or more (Haddad & Land, 2000a; Haddad et al., 2000).
Drug treatment and measurement of pro-inflammatory cytokine TNF-α by ELISA
Epithelial cells were pre-treated for 2 h with NAC (1–50 mM), washed twice in pre-equilibrated PC-1 medium and subsequently challenged with LPS (1 μg ml−1) for 24 h (LPS was derived from Escherichia coli, serotype 026 : B6). Cell-free supernatants were assayed for pro-inflammatory (R&D Systems, U.K.) cytokine biosynthesis by two-site, solid phase, sandwich enzyme-linked immunosorbent assay (ELISA), essentially as recounted previously (Haddad et al., 2001a, 2001c, 2001d). Briefly, rabbit immunoaffinity purified polyclonal anti-rat TNF-α (2 μg ml−1) antibody was used to coat high-binding microtitre plates (MaxiSorp, Nunc, U.K.) in bicarbonate buffer (0.1 M NaHCO3 and 0.1 M NaCl, pH 8.2) (Safieh-Garabedian et al., 1997); Haddad et al., 2001a, 2001c). After blocking in 3% bovine serum albumin (BSA), recombinant (standard) and biotinylated (recognition) immunoaffinity purified sheep anti-rat cytokine antibodies were employed for secondary detection. The colour was developed using streptavidin-poly-HRP (Amersham Life Sciences, U.K.) coupled with 3,3′,5,5′-tetramethyl-benzidine dihydrochloride (TMB) and 1 mM H2O2. The optical density was read at 450 nm against a background filter measuring at 595 nm, where the inter- and intra-assay coefficients of variations were reported at ⩽10%. Results were extracted from the linear regression of the positive slope and cytokine concentration was expressed in pg ml−1.
Measurement of intracellular levels of reduced (GSH) and oxidized (GSSG) glutathione
Reduced (GSH) glutathione concentrations were determined spectrophotometrically (Haddad & Land, 2000a; Haddad et al., 2000) in neutralized perchloric acid (PCA; 7%) extracts by following the glyoxylase-catalysed production of S-lactyl-GSH at 240 nm in a 1-ml volume containing 790 μl phosphate buffer (25 mM KH2PO4; 25 mM K2HPO4; pH 6.8), 150 μl 1% BSA, 10 μl sample, 10 μl glyoxylase-I (1 mg ml−1), and 40 μl methylglyoxal (0.1 M). Oxidized glutathione (GSSG) was determined in the same cuvette by addition of 1 mg ml−1 glutathione reductase and 8 μl of 12 mM β-NADPH and then by following the change in absorbance at 340 nm (Haddad & Land, 2000a; Haddad et al., 2000). Drift inherent to the assay was controlled by subtracting the absorbance change observed over the same time period from control cuvettes containing the same reaction components but with a matched sample volume of deionized water. Protein content of each PCA precipitate was re-dissolved in 1 M NaOH and determined enabling results to be expressed as μmoles mg−1 protein.
Preparation of subcellular extracts for Western analysis of p38 MAP kinase activation/phosphorylation
Analysis of p38/RK phosphorylation
Total protein extraction was performed by homogenizing primary cell tissue in a suitable volume of a buffer (1 : 40 w v−1) containing 20 mM HEPES (pH 7.5), 1.5 mM MgCl2, 0.2 mM EDTA and 0.1 M NaCl (Haddad & Land, 2000a; Haddad et al., 2000). Before extraction 5 mM dithiothreitol (DTT), 1 mM phenylmethylsulphonyl fluoride (PMSF), and 1.2 mM sodium orthovanadate (Na3VO4) were added to the buffer. The cellular debris was pelleted by centrifugation at 10,000×g for 30 min at 4°C, and the collected supernatant was mixed with an equal volume of the same extracting buffer but containing in addition 40% (v v−1) glycerol. Threonine and tyrosine phosphorylation of p38 MAPK was analysed according to instructions given in commercially available kits (New England Biolabs, Inc., Beverly, MA, U.S.A.). The kit employs specific anti-phospho-p38 MAPK antibodies against Thr180/Tyr182 sites that do not cross-react with phosphorylated threonine/tyrosine of extracellular signal-regulated kinase (ERK) 1/2 or c-Jun-NH2-terminal kinase (JNK). Analysis of Thr180/Tyr182 phosphorylation of p38 MAPK was performed as follows: Extracted proteins (20–25 μg) were resolved over sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS–PAGE; 7.5%) gels at RT, blotted onto nitrocellulose membrane, and non-specific binding sites were subsequently blocked. The membrane was probed with specific antibody to phosphorylated threonine and tyrosine of p38 MAPK for primary detection. Anti-rabbit Ig-biotinylated antibody (Amersham Life Science, U.K.) was employed for secondary detection followed by the addition of streptavidin-HRP conjugate and visualized on film by chemiluminescence. Phosphorylation-independent state of p38 MAPK using a specific antibody was used as an internal reference for semi-quantitative loading in parallel lanes for each variable. Western blots were scanned by NIH MagiScanII and subsequently quantitated by UN-Scan-IT automated digitizing system (Version 5.1; 32-bit), and the ratio of the density of the band to that of the non-phosphorylated form was performed.
Analysis of Hsp27 phosphorylation by the upstream activated MAPKAP-K2 kinase
Active phosphorylated p38 MAPK regulates a kinase cascade that may be followed by determining the terminal phosphorylation of Hsp27 by MAPKAP-K2. After treatment with LPS for the indicated doses and time points, cells were ruptured in 250 μl of lysis buffer (mM): HEPES (pH 7.4) 20, EGTA 2, β-glycerophosphate 50, Na3VO4 1, NaF 5, 1% (v v−1) Triton X-100; 10% (v v−1) glycerol; DTT 1, PMSF 1, 10 μg/ml leupeptin; and 10 μg/ml aprotinin, for 30 min on ice. Cell debris was removed by centrifugation at 10,000×g for 10 min at 4°C. The kinase activity of MAPKAPK-2, which is specifically regulated by the phosphorylation of the upstream p38 MAPK, was assayed with recombinant heat-shock protein 27 (Hsp27) as a substrate. Briefly, 1 μg of Hsp27 was added to a microcentrifuge tube containing 10 μl (≈10 μg) of cellular extract. Kinase reaction was initiated by the addition of 10 μl of γ-32P-labelled Mg2+/ATP solution (10 mM MgCl2/1 mM ATP/2 μCi of [γ-32P]-ATP) and performed at 36°C for 20 min. The final concentration of Mg2+/ATP in the assay was adjusted to a 2.5 μM and 0.25 μM, respectively. Reactions were terminated by the addition of 5 μl Laemmli sample buffer and subseqeuntly boiled (95°C) for 5 min. Samples were separated by SDS–PAGE (17.5% (w v−1) gel). Gels were blotted onto a Whatmann paper and dried for 2 h prior to exposure to autoradiography with a phosphorimager, followed by specific quantitation of the corresponding bands.
Selective inhibition of p38 MAP kinase and LPS-induced release of pro-inflammatory cytokines
Cells were pre-incubated for 2 h with SB-203580 (0, 0.01, 0.1, 1, 10, 100 μM) (Calbiochem, U.K.), a selective inhibitor of MAPK p38/RK. This was followed by exposure to LPS (1 μg ml−1) for 24 h and cell-free supernatants were collected for cytokine analysis (TNF-α) by ELISA (Haddad et al., 2001a, 2001b, 2001c), as recounted above.
Selective inhibition of p42/44 MAP ERK kinase and LPS-induced release of pro-inflammatory cytokines
Cells were pre-incubated for 2 h with PD-98059 (0, 0.01, 0.1, 1, 10, 100 μM) (Calbiochem, U.K.), a selective inhibitor of MAPK p42/44/ERK. This was followed by exposure to LPS (1 μg ml−1) for 24 h and cell-free supernatants were collected for cytokine analysis (TNF-α) by ELISA (Haddad et al., 2001a, 2001b; 2001c), as recounted above. Separately, cells were pre-treated (2 h) with a combination of SB-203580 and PD-98059, exposed to LPS (1 μg ml−1) for 24 h and subsequently assayed for TNF-α.
Selective inhibition of the nuclear factor-κB signalling pathway and the regulation of LPS-induced release of TNF-α
Cytokines have been demonstrated to induce NF-κB, whose activation has been implicated in mediating biological responses, including the expression of genes encoding cytokines (Baldwin, 1996). Cells were pre-treated (2 h) with various inhibitors then monolayers washed twice in pre-equilibrated PC-1 medium and subsequently challenged with LPS (1 μg ml−1) for 24 h. In order to determine whether LPS-induced release of TNF-α is regulated, at least in part, by NF-κB, we used carbobenzoxy-L-leucyl-L-leucyl-L-leucinal (MG-132; Calbiochem, U.K.), a potent, reversible proteasome inhibitor (Ki=4 nM), reported to inhibit NF-κB activation (Baldwin, 1996). Cells were pre-treated with MG-132 (0, 1, 10, 50 μM) for 2 h before exposure to LPS for further 24 h. Sulfasalazine (SSA; 0, 0.1, 1, 10 mM) and caffeic acid phenylethyl ester (CAPE; 0, 1, 10, 100 μM; Calbiochem, U.K.), which are potent specific inhibitors of NF-κB, were also incorporated. TNF-α release was assayed (24 h) following challenge. Separately, cells were pre-treated for 2 h with SN-50 (0, 1, 10, 20 μM; Calbiochem, U.K.), a specific permeating inhibitor of NF-κB nuclear translocation, followed by exposure to LPS (1 μg ml−1) for 24 h. This peptide contains the nuclear localization sequence (NLS) for the p50 NF-κB subunit and the amino-terminal sequence of Kaposi fibroblast growth factor to promote cell permeability. An inactive mutant control for SN-50 peptide (SN-50 M; 20 μM) corresponding to the same peptide sequence with substitutions of Lys363 for Asn and Arg364 for Gly in the NLS region has been used to confirm the specificity and selectivity of SN-50.
Assessment of intracellular reactive oxygen species (ROS) accumulation with LPS
For the determination of hydrogen peroxide (H2O2), cell supernatants post addition of LPS (1 μg ml−1) were collected and centrifuged at 2000×g for 5 min at 4°C, and then treated with 1 M NaOH. The change in absorbency at 600 nm was monitored against phenol red solution (PRS) containing (in mM): NaCl 140, K3PO4 10, glucose 5.5, phenol red 0.28, and U ml−1 horseradish peroxidase 20 (Haddad et al., 2001a, 2001c). Standard curves using PRS and H2O2 were prepared (0–100 μM), and results extrapolated from the linear regression were converted to nmoles/mg protein. For the quantification of superoxide anion (O2−.) release, the supernatant was discarded after 24 h of culture, and cells were then reincubated with LPS in the presence of 80 μM ferricytochrome c (fc) suspended in HBSS. The amount of O2−. released was determined by measuring the absorgency at 550 nm against blanks containing fc and superoxide dismutase (SOD; 300 u ml−1). All experiments were performed in duplicates and data are presented as the amount of O2−. released based on nmoles of reduced fc/min/mg protein (Haddad et al., 2001a, 2001c). The relatively non-specific measure of ·OH production, which reacts with dihydrorhodamine (DHR), thereby yielding water and a tertiary rather stable free radical, allows this radical to undergo rearrangement of the π electrons, leading to formation of fluorescent rhodamine. Cells were cultured at 105 well−1 in flat-bottomed microtitre plates at normoxia. Wells were washed with sterile, pre-equilibrated saline, then culturing was continued in Kreb's/Ringer solution containing 50 μM DHR and LPS as indicated. Fluorescence was measured at excitation/emission wavelengths of 485/535 nm. Negative/positive controls were included to monitor the specificity of the Fenton reaction (Haddad et al., 2001a, 2001c). The ·OH level measured of the control (no LPS) was calibrated to 100%, and variables were plotted against this level as logarithmic fluorescence units.
Exposure to ROS-generating systems and the role of antioxidants in MAPKp38 signalling
The rate of induced production of O2−. by xanthine (X) in the presence of xanthine oxidase (XO) was measured as the reduction of fc, as detailed previously (Haddad et al., 2001a, 2001c). X (100 μM) and XO (0–2 mU ml−1) were incubated in microtiter plates coated with 105 cells well−1 with 100 μl of reaction solution containing 50 μM fc in HBSS. The degree of fc reduction was measured at 550 nm. The standardized reaction allows production of O2− at an average rate of ≈8.10±0.09 nmoles min−1 ml−1 (XO=2 mu ml−1). Separately, cells were incubated in the presence of X (100 μM) and XO (2 mu ml−1) or H2O2 (100 μM) for 15 min at 37°C. Subcellular extracts were prepared for assessing MAPKp38 phosphorylation/activation as recounted above. Independently, in order to investigate the possibility of ROS acting as signalling messengers in MAPKp38 signalling, selective antioxidants against O2−·, ·OH and H2O2 radicals were employed. This choice was based on the antioxidant potency and specificity: Dimethyl sulphoxide (DMSO) is a prototypical scavenger of ·OH (Parker et al., 1985; Wasil et al., 1987, 1,3-dimethyl-2-thiourea (DMTU) scavenges H2O2 and ·OH (Fox, 1984; Parker et al., 1985; Wasil et al., 1987), and 4′-hydroxy-3′-methoxy-acetophenone (HMAP) is an NADPH-oxidase inhibitor which may also block ROS production by mitochondria (Lapperre et al., 1999). Epithelial cells were pre-treated for 2 h with DMSO (1%), DMTU (10 mM) and HMAP (10 mM), before exposure to LPS (1 μg ml−1) for 15 min. Extracts were then prepared for the assessment of MAPKp38 phosphorylation/activation. Separately, in order to determine the level of participation of mitochondrial ROS, cells were pre-treated with diphenylene iodonium (DPI; 10 μM), a non-specific inhibitor of flavin-dependent O2−. producing enzymes, including not only mitochondrial but also the classical NADPH oxidase (Chandel et al., 1998), or KCN (10 mM), an inhibitor of mitochondrial respiratory chain, followed by exposure to LPS (1 μg ml−1) for 15 min. In addition, γ-GCE (100 μM), a cell-permeant precursor of GSH, was added to monolayers 2 h before exposure to LPS (1 μg ml−1) for 15 min, followed by assessment of MAPKp38 phosphorylation/activation.
Statistical analysis and data presentation
Data are the means and the error bars the s.e.mean. Statistical evaluation of the difference in mean separation was performed by one-way analysis of variance (ANOVA), followed by post hoc Tukey's test, and the a priori level of significance at 95% confidence level was considered at P⩽0.05.
Results
The excitatory role of LPS in mediating the phosphorylation of MAPK p38/RK
The induction of threonine/tyrosine phosphorylation of p38/RK MAP kinase reflects the activation state of this kinase in regulating the downstream pathway involving MAPKAP-K2. In addition, the role of LPS in mediating the phosphorylation of p38/RK MAPK is not well characterized in the foetal alveolar epithelium. Consequently, we examined the phosphorylation of this kinase in response to LPS in controlled time- and dose-dependent experiments. As shown in Figure 1A, exposure of alveolar epithelial cells to LPS (1 μg ml−1) induced, in a time-dependent manner, the phosphorylation of p38/RK kinase (p-p38) on threonine/tyrosine residues as determined by a selective antibody probed on an SDS–PAGE analysis gel (see Methods). The positive control (+ve control) contained protein extracts prepared from C-6 glioma cells stimulated with anisomycin to induce the phosphorylation of threonine and tyrosine residues of p38/RK kinase. The phosphorylation state-independent p38/RK MAP kinase is shown in the lower panel of Figure 1A to verify semi-quantitative loading for gel analysis per loading lane. The amount of p38/RK phosphorylation was quantified by an image analyser to indicate that the peak of p38/RK phosphorylation due to LPS stimulation is somewhere around the 15-min time point, as shown in the histogram (Figure 1B). This reference time point (15 min) was subsequently adopted in further experiments. The phosphorylation of p38/RK MAP kinase became immediately active within minutes post addition of LPS (significantly active at 2 min), continued to increase up until 30 min, thereafter declining, still significantly different at 60 min, but its activity was lost between 90–120 min (Figure 1A,B). Figure 1C shows the dose-dependent analysis of LPS stimulation (0–1000 ng ml−1) at 15 min, indicating a maximum induction of p38/RK phosphorylation at 100–1000 ng ml−1 concentration range (Figure 1D). The steady, phosphorylation-independent (p38) was also quantitated at various doses of LPS (Figure 1C) to account for equal loading per lane. The specificity of SB-203580 was confirmed by the fact that this compound selectively prevented LPS-mediated phosphorylation of MAPKp38 without affecting the phosphorylation-dependent state of either JNK or ERK kinase (Data not shown).
Figure 1.
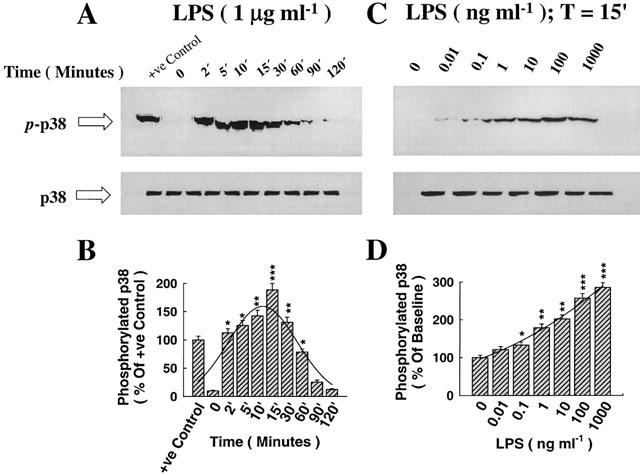
The immunoregulatory role of lipopolysaccharide (LPS) in augmenting the phosphorylation and activation of p38 MAPK. Foetal alveolar epithelial cells were stimulated with LPS (1 μg ml−1) for the desired time points as indicated. Lysates were separated by a 7.5% SDS–PAGE, transferred to membranes and probed with specific antibodies recognizing the phosphorylated and steady state forms of p38 MAPK. (A) LPS induced, in a time-dependent manner, the phosphorylation of p38, with maximum induction at between 10–15 min (p-p38 indicates the phosphorylated form and p38 that of the steady-state form). The positive control lane contained loading of anisomycin extract for the recognition of the specificity of phosphorylated p38 bands. (B) Histogram analysis of p38 phosphorylation for the time-response curve and Pseudo-Voight curve fitting showing the peak at 15 min. (C) Dose-response curve showing the effect of LPS (15 min) on p38 phosphorylation and steady-state forms, with maximum induction at a concentration of 1000 ng ml−1. (D) Histogram analysis of p38 phosphorylation for the dose-response curve and Pseudo-Voigt curve fitting showing gradual and maximum induction with LPS. *P<0.05, **P<0.01, *** P<0.001, as compared with control (LPS=0 ng ml−1). n=3, which represents the number of independent experiments with separate cell preparations.
The effect of LPS on the activation of MAPKAP-K2, the downstream kinase controlled by p38/RK MAP kinase, as assessed by the phosphorylation of its substrate, Hsp27
The activity of MAPKAP-K2 was evaluated by the phosphorylation state of its substrate, Hsp27. A representative SDS–PAGE Western gel showing the effect of LPS (1 μg ml−1) on the amount of γ-32[ATP] bound to Hsp27 indicating the degree of its phosphorylation (p-Hsp27) is shown in Figure 2A, exhibiting the time response curve. As shown in Figure 2A, the peak time for Hsp27 phosphorylation consistently occurred at a later time (30–60 min) than the activation of p38/RK MAP kinase (15 min). The time peak was determined by histogram analysis of the phosphorylated band, as shown in Figure 2B. The dose-response curve showing the effect of LPS at 45 min on Hsp27 phosphorylation is shown in Figure 2C. There was gradual increase in the activity of MAPKAP-K2 as assessed by Hsp27 phosphorylation on LPS exposure, peaking at between 10–100 ng ml−1, thereafter declining but still statistically significant at 10,000 ng ml−1 (Figure 2D).
Figure 2.
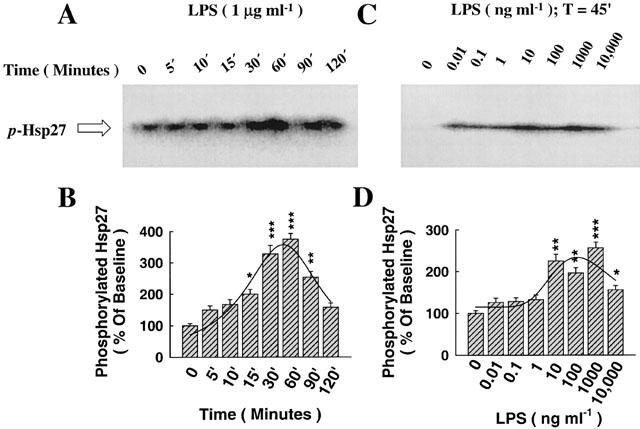
The regulation by LPS of the activity of MAPKAP-K2, the downstream kinase activated by p38 phosphorylation, through the phosphorylation of its substrate, Hsp27. (A) LPS induced, in a time-dependent manner, the phosphorylation of Hsp27 (p-Hsp27), an index of the activity of MAPKAP-K2, with peak analysis at 30–60 min. (B) Histogram analysis of Hsp27 phosphorylation for the time-response curve and Pseudo-Voigt curve fitting showing the peak at ≈45 min. (C) Dose-response curve showing the effect of LPS (45 min) on Hsp27 phosphorylation, with maximum induction at a concentration of 1000 ng ml−1, thereafter declining. (D) Histogram analysis of Hsp27 phosphorylation for the dose-response curve and Pseudo-Voigt curve fitting showing gradual and maximum induction with LPS. *P<0.05, **P<0.01, ***P<0.001, as compared with control (LPS=0 ng ml−1). n=3, which represents the number of independent experiments with separate cell preparations.
The role of p38/RK signalling pathway and N-acetyl-L-cysteine (NAC) pre-treatment in mediating the effect of LPS on TNF-α biosynthesis
The activity, but not the activation, of p38/RK MAP kinase is selectively blocked by the pyridinyl imidazole compound SB-203580, thereby blockading the downstream pathways associated with the activation of MAPKAP-K2, Hsp27 phosphorylation and pro-inflammatory cytokine transcription and biosynthesis (Garrington & Johnson, 1999). As shown in Figure 3A, exposure of alveolar epithelial cells to medium alone for 24 h had no effect on the release of TNF-α into the supernatant. In addition, pre-treatment with SB-203580 (100 μM) for 1 h and further exposure to medium alone had no apparent effect on TNF-α secretion. In contrast, administration of LPS (1 μg ml−1) for 24 h enhanced the secretion of TNF-α into the supernatant (Figure 3A). Pre-treatment with SB-203580, followed by exposure to LPS blockaded, in a dose-dependent manner, LPS-dependent biosynthesis of TNF-α, as shown in Figure 3A. At all concentrations of SB-203580 used, there were no signs of cytotoxicity associated with the treatment, nor were there any decreases in the transepithelial resistance of monolayers (Data not shown). Figure 3B shows the dose-dependent inhibition of LPS-induced TNF-α biosynthesis due to pre-treatment (2 h) with N-acetyl-L-cysteine (NAC), an antioxidant and a precursor of cysteine, the rate-limiting amino acid in the biosynthesis of glutathione (GSH) (Meister, 1988; Rahman, 1999; Haddad & Land, 2000a; Haddad et al., 2000).
Figure 3.
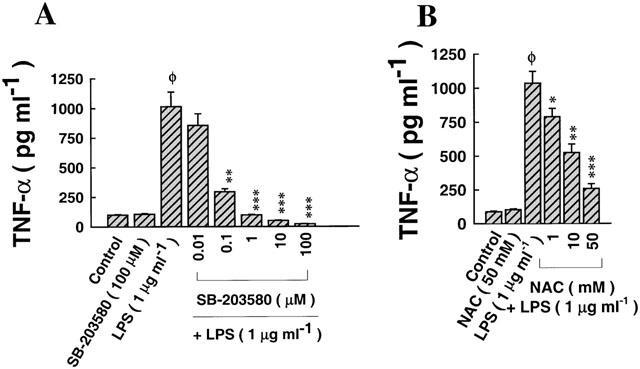
The effect of selective inhibition of p38 MAPK and NAC pre-treatment on LPS-induced TNF-α biosynthesis. (A) Pre-treatment with SB-203580 (2 h), a selective inhibitor of p38 MAPK, prior to exposure to LPS (1 μg ml−1), reduced LPS-induced TNF-α production in a dose-dependent manner. LPS upregulated the secretion of TNF-α approximately 4 fold relative to cells incubated in medium alone (control). Pre-treatment with SB-203580 (100 μM) on its own did not affect TNF-α synthesis. SB-203580 reduced LPS-induced TNF-α secretion at doses ⩾0.1 μM, with maximum inhibition at 100 μM (IC50=0.054±0.005 μM). (B) Pre-treatment with NAC (2 h) before stimulating with LPS reduced, in a dose-dependent manner, the induction of TNF-α, effective at doses ⩾1 mM, with maximum suppression at 50 mM (IC50=26.07±2.25 mM). ΦP<0.05, as compared with control; *P<0.05, **P<0.01, ***P<0.001, as compared with LPS (1 μg ml−1). n=4, which represents the number of independent experiments run in duplicate with separate cell preparations.
The role of p42/44/ERK signalling pathway and its synergism with p38/RK in mediating the effect of LPS on TNF-α biosynthesis
The effect of PD-98059, a selective inhibitor of MAPKERK pathway, on LPS-mediated TNF-α secretion is shown in Figure 4A. Exposure of alveolar epithelial cells to medium alone for 24 h had no effect on the release of TNF-α into the supernatant. In addition, pre-treatment with PD-98059 (100 μM) for 1 h and further exposure to medium alone had no apparent effect on TNF-α secretion. In contrast, administration of LPS (1 μg ml−1) for 24 h enhanced the secretion of TNF-α into the supernatant (Figure 4A). Pre-treatment with PD-98059, followed by exposure to LPS blockaded, in a dose-dependent manner, LPS-dependent biosynthesis of TNF-α, but to a lesser extent than SB-203580 (Figures 3A, 4A). At all concentrations of PD-98059 used, there were no signs of cytotoxicity associated with the treatment, nor was there any decrease in the transepithelial resistance of monolayers (Data not shown). A combination of SB-203580 and PD-98059 exhibited a mild synergistic effect, especially at a dose of 0.01 μM, which was ineffective with either inhibitor used on its own (Figure 4B).
Figure 4.
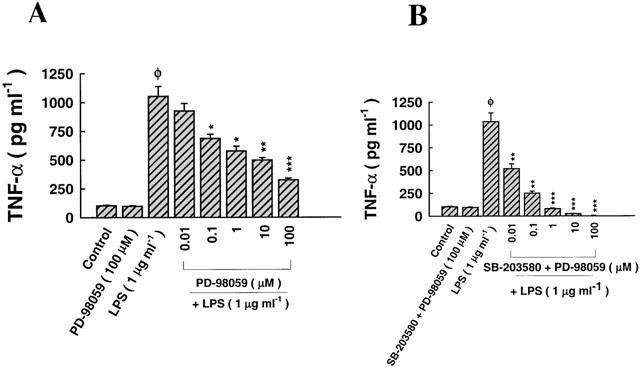
The effect of selective inhibition of p42/44 MAPK on LPS-induced TNF-α biosynthesis. (A) Pre-treatment with PD-98059 (2 h), a selective inhibitor of MAPKERK, prior to exposure to LPS (1 μg ml−1), reduced LPS-induced TNF-α production in a dose-dependent manner. LPS upregulated the secretion of TNF-α approximately 4 fold relative to cells incubated in medium alone (control). Pre-treatment with PD-98059 (100 μM) on its own did not affect TNF-α synthesis. PD-98059 reduced LPS-induced TNF-α secretion at doses ⩾0.1 μM, with maximum inhibition at 100 μM (IC50=5.12±0.47 μM). (B) Pre-treatment with a combination of SB-203580 and PD-98059 synergistically reduced, in a dose-dependent manner, the induction of TNF-α, effective at doses ⩾0.01 μM, with maximum and complete suppression at 100 μM (IC50=0.023±0.003 μM). ΦP<0.05, as compared with control; *P<0.05, **P<0.01, ***P<0.001, as compared with LPS (1 μg ml−1). n=3, which represents the number of independent experiments run in duplicate with separate cell preparations.
The effect of proteasome inhibitor and selective inhibition of NF-κB in mediating the effect of LPS on TNF-α biosynthesis
As shown in Figure 5A, exposure of epithelial cells to medium alone for 24 h had no effect on the secretion of TNF-α. Similarly, pre-treatment of monolayers with MG-132 (50 μM), a specific inhibitor of the proteasome complex, reportedly known to blockade the NF-κB pathway, followed by exposure to medium alone for 24 h had no effect on TNF-α production either (Figure 5A). Exposure to LPS (1 μg ml−1) for 24 h induced TNF-α biosynthesis ≈3–4 fold relative to medium alone measured at the same time point. Pre-treatment with MG-132 prior to challenge with LPS did not affect or reduce LPS-dependent release of TNF-α (Figure 5A). As shown in Figure 5B, pre-treatment of monolayers with CAPE (100 μM), a specific inhibitor of NF-κB, followed by exposure to medium alone for 24 h had no effect on TNF-α production either (Figure 5B). Pre-treatment with CAPE prior to challenge with LPS reduced LPS-dependent release of TNF-α at a minimum effective dose ⩾10 μM (Figure 5B). Figure 5C shows that pre-treatment of monolayers with SSA (10 mM), a potent and specific inhibitor of NF-κB, followed by exposure to medium alone for 24 h had no effect on TNF-α production either (Figure 5C). Pre-treatment with SSA prior to challenge with LPS reduced LPS-dependent release of TNF-α at the highest dose used in this study (10 mM) (Figure 5C). As shown in Figure 5D, pre-treatment of monolayers with SN-50 (20 μM), a potent and specific permeating inhibitor of NF-κB, followed by exposure to medium alone for 24 h had no effect on TNF-α production either (Figure 5D. Pre-treatment with SN-50 prior to challenge with LPS reduced, in a dose-dependent manner, LPS-dependent release of TNF-α (Figure 5D). Mutation of the SN-50 peptide (SN-50M; 20 μM) reversed the inhibitory effect of this peptide on LPS-mediated TNF-α biosynthesis (Figure 5D). The 50% minimum inhibitory effective concentration (IC50) of NF-κB inhibitors on LPS-mediated TNF-α biosynthesis is given in Table 1.
Figure 5.
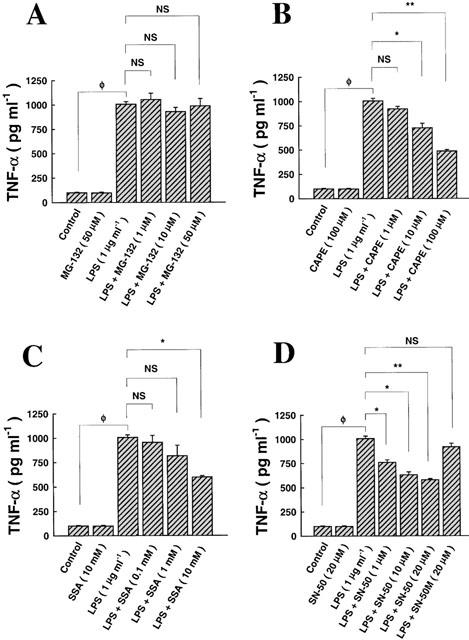
The role of the NF-κB signalling pathway in regulating LPS-mediated TNF-α biosynthesis. (A) Incubation of epithelial cells with medium alone (24 h) had no effect on TNF-α production, compared to pre-incubation with MG-132 (2 h), a proteasome inhibitor, followed by exposure to medium alone (24 h). Exposure to LPS (1 μg ml−1; 24 h) induced TNF-α secretion into the supernatant, an effect that was not affected or reversed by MG-132, at all doses tested. (B) The effect of CAPE on LPS-mediated TNF-α biosynthesis, where incubation with medium alone had no effect on TNF-α production, compared to pre-incubation with CAPE (2 h), an inhibitor of NF-κB, followed by exposure to medium alone. The effect of LPS was reversed by CAPE at doses ⩾10 μM. (C) The effect of SSA on LPS-mediated TNF-α biosynthesis, where pre-incubation with CAPE, followed by exposure to medium alone had no effect. The effect of LPS was reversed by SSA only at a dose of 10 mM. (D) The effect of SN-50 and its mutant, SN-50M, on LPS-mediated TNF-α biosynthesis showed that pre-incubation with SN-50, followed by exposure to medium alone had no effect on TNF-α. The effect of LPS was reversed by SN-50 in a dose-dependent manner. Mutation of the wild-type peptide (SN-50 M; 20 μM) reversed the inhibitory effect of SN-50 on LPS-dependent TNF-α secretion. ΦP<0.05, as compared with control; NS, non-specific, as compared with LPS alone. n=3, which represents the number of independent experiments run in duplicate.
Table 1.
Analysis of the 50% minimum effective inhibitory (IC50) concentrations of various NF-κB inhibitors on LPS-induced TNF-α biosynthesis in the alveolar epithelium
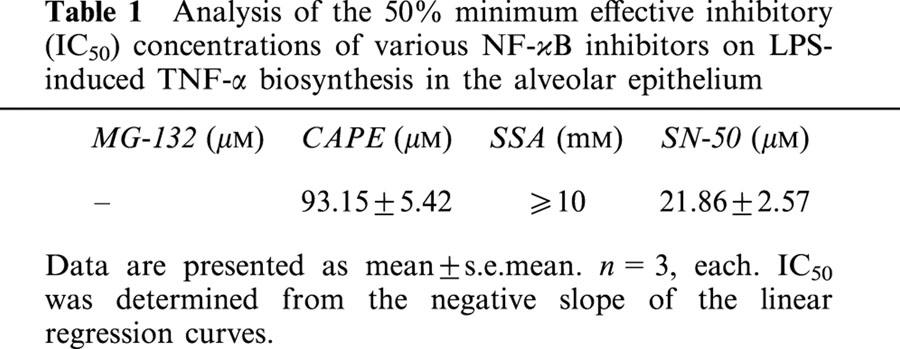
The effect of LPS on intracellular accumulation of ROS and the role of antioxidants and NAC
The underlying mechanism of the mode of action of LPS as a mediator of oxidative stress was investigated. LPS induced, in a time-dependent manner, the accumulation of H2O2 (Figure 6A), O2−. (Figure 6B) and ·OH (Figure 6C). In order to assess the likely source of ROS in response to LPS, cells were pre-treated with HMAP or NAC prior to exposure to LPS for 30 min. HMAP and NAC reduced H2O2 accumulation (Figure 6D), but to a lesser extent than O2−. (Figure 6E). The effect of HMAP on ·OH was milder than that of NAC (Figure 6F). This indicates that the membrane-bound NADPH oxidase and the mitochondrial complex are likely involved as potential resources for ROS in response to LPS, an effect which is redox-sensitive.
Figure 6.
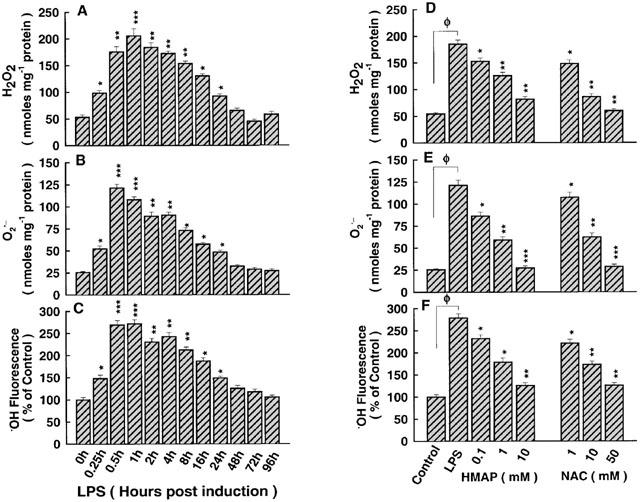
Analysis of ROS accumulation in response to LPS and the role of selective antioxidants. (A) H2O2 began to increase at 15 min and continued to be significantly higher than control until 24 h, thereafter declining. (B) O2−. appeared at 15 min and continued to be higher than control until 24 h, thereafter declining. (C) The concentration of ·OH (fluorescence) began to increase at 15 min and continued to be significantly higher than control until 24 h, thereafter declining. (D) HMAP and NAC pre-treatments reduced accumulation of H2O2 in response to LPS. (E) HMAP and NAC reduced accumulation of O2−. in response to LPS, in a dose-dependent manner. (F) HMAP and NAC mildly reduced accumulation of ·OH in response to LPS. ΦP<0.05, as compared with control; *P <0.05, **P<0.01 ***P<0.001, as compared with control in the absence or presence of LPS. n=4, which represents the number of independent experiments run in duplicate.
The effect of ROS-generating systems and antioxidants on LPS-mediated MAPKp38 phosphorylation
Exposure to O2−.-generating system (X/XO; 100 μM/2 mu ml−1) induced the phosphorylation of MAPKp38, as shown in Figure 7A. Similarly, H2O2 (100 μM), which is a major source of ·OH, up-regulated the phosphorylation of MAPKp38, as shown in Figure 7A. Pre-treatment with selective antioxidants, purported to reduce the intracellular accumulation of H2O2, O2−. and ·OH, abrogated LPS-mediated phosphorylation of MAPKp38, as shown in Figure 7B. The antioxidant and cell-permeant precursor of GSH, γ-GCE, reduced MAPKp38 phosphorylation (Figure 7B). The numbers shown below the upper panel of Figure 7B exhibit the per cent phosphorylation state of MAPKp38. The lower panels in Figure 7A,B show the steady state, phosphorylation-independent, MAPKp38.
Figure 7.
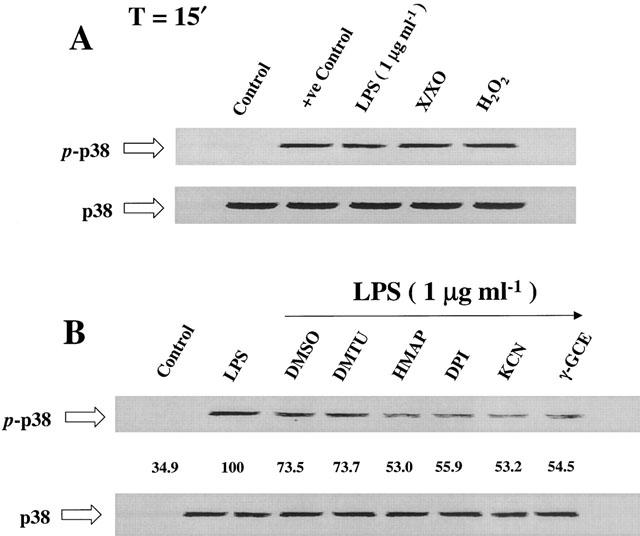
The effect of ROS-generating systems and selective antioxidants on MAPKp38 phosphorylation. (A) Xanthine (X)/xanthine oxidase (XO) system, which generates O2−., up-regulated the phosphorylation of MAPKp38, similar to the effect of exogenous H2O2, which is a major source for ·OH. (B) Selective antioxidants, including DMSO, DMTU, HMAP, DPI and KCN, attenuated LPS-dependent MAPKp38 phosphorylation. The antioxidant and cell-permeant precursor of GSH, γ-GCE, similarly attenuated the effect of LPS (p-p38 indicates the phosphorylated form and p38 that of the steady-state form). n=3, which represents the number of independent experiments.
The role of NAC in mediating glutathione (GSH) biosynthesis
The cysteine that is provided due to NAC administration is eventually fed into the biosynthetic pathway that brings about the formation of GSH by the action of γ-glutamylcysteine synthetase (γ-GCS), the rate-limiting enzyme in the biosynthesis of GSH (Meister, 1988; Hayes & McLellan, 1999; Haddad & Land, 2000a). Incubation of alveolar epithelial cells for 24 h with NAC induced, in a dose-dependent manner, the intracellular accumulation of GSH (Figure 8A). This effect was associated with a dose-dependent decrease in the intracellular level of the oxidized disulphide gultathione (GSSG), as reflected by an increasing redox ratio of GSH/GSSG (Figure 8A). The time-response curve for NAC incubation at 50 mM is shown in Figure 8B. Incubating cells with NAC for different time points induced the intracellular formation of GSH and reduced the levels of GSSG (Figure 8B). Within 2 h of NAC incubation there was significant accumulation of GSH, the concentration of which continued to elevate in the continuiing presence of NAC and maximized at around 16–24 h, thereafter declining (48 h) but persistently found to be still significantly different from control deproteinated monolayers (Figure 8B). Redox equilibrium assessment of glutathione ratio homeostasis reported in alveolar cells treated with NAC for 24 h at 37°C is given in Table 2.
Figure 8.
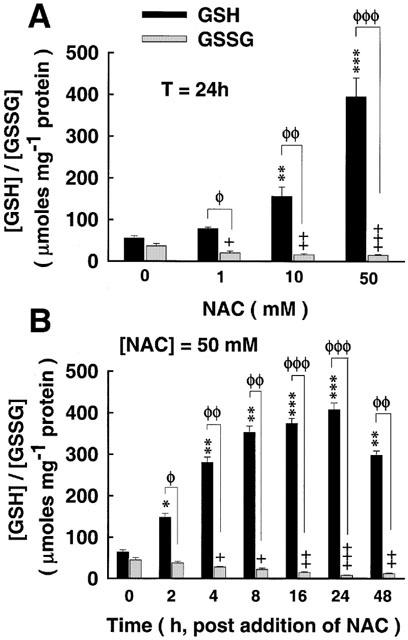
The effect of NAC on intracellular redox potential (GSH/GSSG). (A) Cells were exposed to NAC treatment for 24 h, thereby inducing the intracellular accumulation of the reduced form of glutathione (GSH) in a dose-dependent manner. Intracellular accumulation of GSH due to NAC was at the expense of the oxidized form of glutathione (GSSG), as evident from the reduced GSSG/GSH ratio. (B) Time-dependent analysis of the effect of NAC (50 mM) on GSH and GSSG accumulation. GSH concentration prevailed with NAC treatment, statistically significant as early as 2 h post incubation with NAC, thereafter ascending to a maxima at 24 h, declining, but still persistently significant, at 48 h. This elevation of intracellular GSH due to NAC is partly at the expense of GSSG, as shown from the reduced GSSG/GSH equilibrium ratio. ΦP<0.05, ΦΦP<0.01, ΦΦΦP<0.001, for (GSH) as compared with (GSSG); *P<0.05, **P<0.01, ***P<0.001, as compared with control (NAC at 24 h and NAC at 50 mM); +P<0.05, ++P<0.01, +++P<0.001, for (GSSG) as compared with (GSSG)control.
Table 2.
Redox equilibrium assessment of glutathione ratio homeostasis reported in alveolar cells treated with NAC for 24 h at 37°C

The role of NAC in regulating the MAPKp38 signalling pathway
The amount of threonine/tyrosine phosphorylation of p38/RK MAP kinase was reduced, in a dose-dependent manner, with NAC pre-treatment (2 h) prior to exposure to LPS (1 μg ml−1) for 15 min (Figure 9A). LPS-mediated phosphorylation of p38/RK MAP kinase was maximally blockaded with NAC at 50 mM. The steady, phosphorylation-independent state of MAPKp38 is shown in the lower panel of Figure 9A. The dose-response curve exhibiting the inhibitory effect of NAC on MAPKp38 phosphorylation state is given in a form of histogram analysis in Figure 9B.
Figure 9.
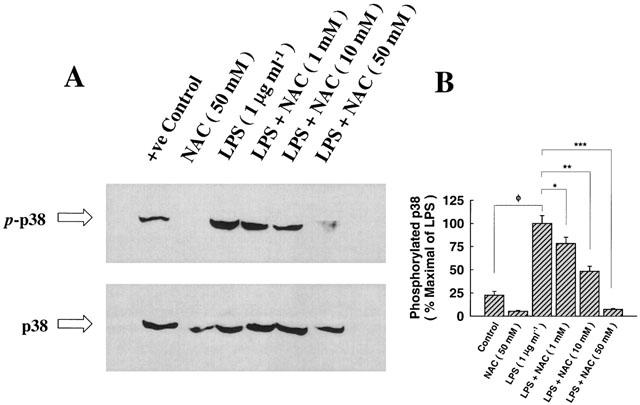
The attenuating effect of NAC on p38/RK phosphorylation. (A) Epithelial cells were cultured with medium alone (control), pre-treated with NAC (50 mM) for 2 h prior to exposure to medium alone for 15 min, exposed to LPS (1 μg ml−1) for 15 min, or pre-treated with NAC (1–50 mM) for 2 h followed by exposure to LPS (1 μg ml−1) for 15 min. Lysates were separated by a 7.5% SDS–PAGE and probed with specific antibodies directed against the phosphorylated threonine/tyrosine residues of p38/RK MAP kinase or the phosphorylation-independent state of p38. Pre-treatment with NAC (50 mM) followed by exposure to medium alone did not affect the phosphorylation of p38/RK, despite the observation it abolished the faint band appearing under the control lane. Pre-treatment with NAC attenuated the phosphorylation-mediated effect of LPS, in a dose-dependent manner, completely blocking its phosphorylation at 50 mM (IC50=20.62±3.08 mM). The steady state of p38 is largely not affected with any of the aforementioned treatments. (B) Histogram analysis of p38 phosphorylation for the inhibitory dose-response curve of NAC. ΦP<0.05, as compared with control; *P<0.05, **P<0.01, ***P<0.001, as compared with control (LPS=1 μg ml−1). n=3, which represents the number of independent experiments with separate cell preparations.
Discussion
TNF-α is a potent inflammatory cytokine, which exerts its pleiotropic activities through ligand-induced cross-linking of specific receptors, virtually present in almost all cell types (Vandenabeele et al., 1995). During inflammation, TNF-α released in the inflammatory environment transiently activates neutrophils and macrophages, thereby causing them to release O2−. as a consequence of the activation of the plasma membrane NADPH oxidase (Klebanoff et al., 1986). This oxidative burst features a rapid but transient release of ROS and is a crucial part of the defence mechanism against invading pathogens and tumour cell metastasis. Although TNF-α is primarily produced by macrophages, growing evidence suggested that other cell types, such as endothelial and epithelial cells, release TNF-α and other inflammatory mediators, thereby amplifying and boosting the inflammatory reaction by activating and recruiting inflammatory cells (Thompson et al., 1985; Hashimoto et al., 1999b; Haddad et al., 2001a, 2001c, 2001d).
There is increasing evidence implicating the alveolar epithelium, in particular, as a dynamic barrier that plays an important role in regulating the inflammatory and metabolic responses to oxidative stress and the accompanying inflammatory signal, sepsis, endotoxaemia, and other critical illnesses in the lung (Thompson et al., 1985; Pittet et al., 1997; Haddad et al., 2001a, 2001b, 2001c, 2001d). The respiratory epithelium is a primary target of an inflammatory/infectious condition at the epithelial-blood interface and is itself capable of amplifying an inflammatory signal by recruiting inflammatory cells and by producing inflammatory mediators. Many of the side effects of LPS, derived from the cell wall of gram-negative bacteria, are secondary to the overproduction of pro-inflammatory mediators. Inflammatory as well as autoimmune disease is often associated with deregulated expression and biosynthesis of pro-inflammatory cytokines, including TNF-α, which influence a plethora of cellular functions. Therefore, the down-regulation of an inflammatory signal is a major focus of the rational approach to the treatment of inflammatory diseases, such as chronic inflammation, sepsis and rheumatoid arthritis. For instance, a novel recent study by Haskó et al. (2000) reported a potential role for extracellular purines, including adenosine and ATP, and inosine, a degradation product of these purines, as potent endogenous immunomodulatory molecules that inhibit inflammatory cytokine biosynthesis and protect against endotoxin-induced shock. It has also been reported, in addition, that selective inhibition of phosphodiesterases, a family of enzymes involved in the degradation of cyclic AMP (Haskó et al., 1998; Doherty, 1999), steroids, such as glucocorticoids (Visser et al., 1998), pyrimidylpiperazine derivatives (Fukuda et al., 2000; Hanano et al., 2000; Hisadome et al., 2000; Haddad et al., 2001e), and ERK and p38/RK MAPK selective inhibitors (Su & Karin, 1996; Garrington & Johnson, 1999; Haddad, 2001) differentially regulate the transcription and biosynthesis of inflammatory cytokines.
The novel mammalian MAPKp38 was originally identified in murine pre-B lymphocytes transfected with the LPS-complex receptor CD14 and in murine macrophages where it was activated in response to LPS (Han et al., 1994). In parallel, MAPKp38 was also identified as a reactivating kinase (RK), which activates MAPKAP kinase-2 (MAPKAP-K2; MK-2), which in turn phosphorylates the small heat-shock protein, Hsp27, and regulates the stability of cytokine transcripts bearing the pentanucleotide sequence of AU-rich elements (ARE) (Rouse et al., 1994; Su & Karin, 1996; Rutault et al., 2001). Of note, MAPKp38, MAPKERK and the downstream pathways regulated are essentially crucial for LPS-induced cytokine gene expression and biosynthesis (Lee et al., 1994; Cano & Mahadevan, 1995; Su & Karin, 1996; Garrington & Johnson, 1999). For instance, knockout mice lacking MK-2 (MK-2−/−) exhibited a colossal reduction in the biosynthesis of TNF-α, a potent pro-inflammatory cytokine that is involved in many human diseases (Hudson et al., 1995; Pittet et al., 1997; Herlaar & Brown, 1999), and are remarkably resistant to shock induced by LPS (Kotlyarov et al., 1999). Recent evidence, furthermore, suggested that the MAPKp38-MAPK/MK-2 pathway is redox-sensitive and governs the transcription and biosynthesis of pro-inflammatory cytokines in a ROS and redox-dependent mechanism (de keulenaer et al., 2000; Kang et al., 2000; Chan et al., 2001; Hashimoto et al., 2001). However, redox/ROS regulation of the MAPKp38/ERK -MAPK/MK-2 pathway mediating TNF-α signalling in the foetal alveolar epithelium is not well characterized. It was subsequently hypothesized that MAPKp38/ERK-mediated regulation of LPS-dependent TNF-α biosynthesis is redox-sensitive and that intracellular glutathione manipulation modulates the capacity of the epithelium to produce this inflammatory cytokine.
The signal transduction cascade that regulates the biosynthesis and secretion of TNF-α in the foetal alveolar epithelium has not been well defined. Administration of LPS, derived from E. coli, up-regulated the extracellular accumulation of TNF-α and other cytokines (Haddad et al., 2001a,2001c,2001d). The effects of LPS are essentially mediated by the membrane-bound CD14 complex and, in part, through the circulating LPS-binding protein (LBP) (Wright et al., 1990; Rietschel et al., 1994). It was reported, furthermore, that LPS-mediated responses on TNF-α transcription and biosynthesis bifurcate at the level of Ras/Raf G-coupled proteins into two major signalling pathways: One that runs through NF-κB-inducing kinase (NIK) route, which regulates the phosphorylation of the inhibitory-κB (IκB) proteins, the cytosolic inhibitors of NF-κB, and another which is mediated through the extracellular signal-regulated kinase (MAPKERK) and MAPKp38 pathways (Su & Karin, 1996; Garrington & Johnson, 1999; Mercurio and Manning, 1999). The promoters of genes encoding cytokines contain multiple cis-acting motifs including those that bind such transcription factors as NF-κB. Furthermore, the release of free NF-κB upon extracellular stimulation due to IκB phosphorylation and degradation by the proteasome complex, leads to DNA binding to specific κB moieties in order to initiate transcription of related genes, including immunoreceptors, cytokines and, interestingly, its own inhibitor, IκB (Baldwin, 1996; Haddad et al., 2001d, 2001e). Two unique features of the NF-κB/IκB complex system are deduced from its feedback regulation. The transcriptional activation of NF-κB triggers the synthesis of IκB, and NF-κB activated transcription is maintained by continuous degradation of IκB, which is sustained by an extracellular stimulus. Thus, the expression of IκB parallels both NF-κB activity and the duration of the activating extracellular stimulation, suggesting that this temporal parallelism between IκB accumulation/degradation and an effective external stimulation is a mechanism allowing dual regulation of NF-κB within the alveolar space. The selective interference with the functioning of the proteasome complex in regulating the translocation and activation of NF-κB and the expression of its inhibitor IκB-α suggested that the IκB-α/NF-κB pathway is partially implicated in regulating LPS-mediated biosynthesis of TNF-α. This is rather unequivocally reinforced with the observation reported in this study that selective inhibition of the NF-κB pathway attenuated but did not abrogate the inductive effect of LPS on TNF-α production. Therefore, the IκB-α/NF-κB pathway could be partially dissociated from the presumably downstream pathway regulating TNF-α signalling, indicating the involvement of a possible cross-talk among several pathways, such as MAPKp38/ERK, working independently or in coherence to integrate signalling mechanisms governing the regulation of cytokines in the alveolar epithelium (Nemoto et al., 1998).
Exposure to LPS induced, in a time- and dose-dependent manner, the phosphorylation of MAPKp38, suggesting the involvement of an up-stream kinase, such as MAPK kinase (MKK), in LPS-mediated regulation of the MAPKp38 pathway (Widmann et al., 1999). Similar to other MAPK families, MAPKp38 is activated by dual phosphorylation of threonine (Thr) and tyrosine (Tyr) in the so-called Thr-Gly-Tyr activation loop or motif (Su & Karin, 1996; Garrington & Johnson, 1999; Widmann et al., 1999). Upon stimulation, MAPKp38 phosphorylates and activates the MK-2 pathway, which regulates the phosphorylation of Hsp27. The biological activity of MAPKp38 is selectively blocked by the pyridinyl-imidazole compound SmithKline Beecham ((SB-203580), which is postulated to possess anti-inflammatory activity (Widmann et al., 1999). In this respect, SB-203580 has been reportedly associated with the suppression and/or augmentation of the transcription and biosynthesis of a wide array of inflammatory and anti-inflammatory mediators, including IL-1β (Baldassare et al., 1999), IL-6, IL-8 (Griego et al., 2000; Hashimoto et al., 2001), IL-10 (Foey et al., 1998; de et al., 2000), IL-12 (Widmann et al., 1999), TNF-α (Yamakawa et al., 1999; Rutault et al., 2001) and prostaglandin-E2 (PGE2) (Scherle et al., 1998). Our results jibe with the proposal that selective blockade of the MAPKp38 signalling transduction pathway exerts an anti-inflammatory condition via suppression of the release of inflammatory mediators, including TNF-α. Despite the fact that selective blockade of the MAPKERK pathway partially attenuated LPS-dependent secretion of TNF-α, simultaneous inhibition of MAPKp38 and MAPKERK pathways synergistically inhibited this response, suggesting co-ordinate regulation. Although from the present data alone we could not infer whether the Hsp27 pathway, which is an immediate pathway regulated by the up-stream kinase MK-2 regulated by MAPKp38, is likely to exert a direct immunoregulatory effect on TNF-α biosynthesis, it is possible that Hsp27 might be implicated. This assumption is reinforced by recent evidence suggesting that Hsp27 can act as an endogenous protein circulating in the serum of breast cancer patients and a protein whose induction correlates with the onset of LPS shock (de et al., 2000). Moreover, it has been reported that Hsp27 mediates regulatory effects on the biosynthesis of TNF-α through an IL-10 dependent mechanism, a pathway known to exert an anti-inflammatory role in several systems (Widmann et al., 1999; de et al., 2000).
Redox and ROS signalling regulating MAPKp38-mediated induction of pro-inflammatory cytokines, and particularly TNF-α, has not been well characterized. It has been recently reported that ROS-initiated and redox-sensitive mechanisms converge on MAPKp38/ERK-mediated regulation of TNF-α signalling in a cascade circuit, emphasizing the highly organized interactive nature of intracellular signals emanating from cell membranes leading to gene regulation and induction (de keulenaer et al., 2000). Furthermore, a novel glutathione-sensitive antioxidant response element regulating the MAPKp38 cascade was reported in mediating the inducible expression of phase II enzymes, such as rGSTA2, known to be responsible for the protective adaptive responses to electrophiles and ROS (Kang et al., 2000). In a much more recent novel investigation, Hashimoto et al. (2001) reported a dual role for NAC in attenuating TNF-α-induced MAPKp38 activation and MAPKp38-mediating IL-8 biosynthesis in vitro. In the present study, LPS-mediated secretion of TNF-α was attenuated by selectively blockading the MAPKp38/ERK/MK-2 pathways and that MAPK-mediated regulation of TNF-α biosynthesis was shown to be ROS/redox-sensitive and modulated by the antioxidant and glutathione (GSH) precursor, NAC. NAC, a cysteine pro-drug (Aruoma et al., 1989; Bernard, 1991; Haddad et al., 2000), can suppress cytokine production (Jeannin et al., 1995; Matsumoto et al., 1998; Gosset et al., 1999; Haddad et al., 2001c) and protect against ROS-mediated lung injury (Bernard, 1991). The rate-limiting substrate for GSH biosynthesis is glutamate-cysteine (Km Glutamate=1.6−2 mM; Km Cysteine=0.3 mM) (Griffith & Meister, 1979; Meister, 1988; Aruoma et al., 1989; Bernard, 1991; Haddad et al., 2001c). Replenishing and sustaining intracellular GSH concentrations, therefore, is accomplished by administering compounds that increase the level of this amino acid (cysteine), or by promoting the activity of γ-glutamylcysteine synthetase (γ-GCS), the rate-limiting enzyme in the biosynthesis of GSH (Meister, 1988). The ability of NAC to induce intracellular accumulation of GSH suggested that its potential to suppress TNF-α secretion residues on two possible pathways: Firstly, the amino acid cysteine provided by the administration of NAC eventually feeds into the biosynthetic machinery regulated by γ-GCS. GSH that is formed during this conversion detoxifies accumulating ROS through the glutathione-peroxidase coupled reaction (Haddad & Land, 2000a). Therefore, the pathway implicated with cysteine is to complement the biosynthetic process, where GSH can directly scavenge ROS. Secondly, NAC has antioxidant properties in that it is capable of directly scavenging and dissimulating accumulating ROS (Aruoma et al., 1989). We have previously shown that NAC induced intracellular formation of GSH (Haddad & Land, 2000a; Haddad et al., 2000), consistent with the observation reported in this study and that NAC ability to detoxify intracellular ROS blockaded the regulated downstream pathway for cytokine signalling (Haddad et al., 20001a, 20001c). Taken together, these data argue for ROS as potential messengers in regulating TNF-α signalling and that the ability of NAC to blockade this pathway resides in its potential either to deliver GSH, a major antioxidant thiol, or to detoxify excess ROS accumulation (Aruoma et al., 1989; Bernard, 1991; Haddad & Land, 2000a).
This novel antioxidant potential of NAC was associated with the ability to suppress the phosphorylation and activation of the MAPKp38 pathway. Since ROS, derived from exposure to extracellular signals inducing stress, were reported to activate the MAPKp38 pathway (Su & Karin, 1996; Widmann et al., 1999), the likely possibility of NAC effectively acting as an antioxidant blockading this cascade is very probable, supported by the unequivocal observation that exogenous ROS up-regulated LPS-mediated MAPKp38 phosphorylation and selective antioxidants attenuated this effect. However, the possibility that NAC might be directly interacting with one or more of the components of the MAPK module and whether its blockading upstream kinases converging onto this pathway cannot be excluded. On the mechanism of action of NAC, as a thiol-modulating agent, in blockading the MAPKp38 pathway, we report the likely occurrence of several possible mechanisms. ROS, such as O2−., H2O2 and ·OH may function as second messengers in signal transduction. Although MAPKs may be directly activated by oxidants, the role of ROS in the activation of the MAPKs pathway by cytokines is largely inferred on the basis of the inhibition of MAPKs by NAC and other antioxidants (Chan et al., 2001). It was also reported that NAC acts as an inhibitor of the c-Jun NH2-terminal kinase (JNK) pathway regulated by TNF-α (Natoli et al., 1997) and that TNF-α stimulation of MAPKp42/44, MAPKp38 and MAPKJNK pathways was blockaded by NAC under serum-free conditions (Chan et al., 2001). The fact that the cell-permeable glutathione pro-drug, γ-GCE, was shown to be likewise potently effective in down-regulating LPS induced MAPKp38 phosphorylation, suggested that GSH is a major player in regulating this pathway and that NAC suppression of this response is GSH-sensitive. γ-GCE is rapidly de-esterified by intracellular esterase, thereby serving as an effective delivery agent for glutathione, which is a peptide incapable of crossing membranes in its native form. Although a distinction between the biological effects of γ-GCE and GSH is indiscriminate, intracellular conversion of γ-GCE suggests that its effects are mediated by GSH. Exogenous/endogenous glutathione, therefore, may feed into one of the well-characterized pathways of metabolism (Meister, 1988). For instance, GSH plays an important role in determining how readily pro-inflammatory genes can be regulated, and GSH/GSSG equilibrium is a major determinant of the activation of redox-sensitive transcription factors, including NF-κB (Dröge et al., 1994). Furthermore, apoptosis signal-regulating kinase-1 (ASK-1) was recently identified as a MAP kinase kinase that activates the MAPKp38 pathway (Saitoh et al., 1998). Additionally, it was shown that TNF-α-mediated regulation of ASK-1 is ROS- and redox-sensitive and that NAC and thioredoxin (TRX), a redox molecule, blockaded the dimerization and the activity of ASK-1 (Saitoh et al., 1998; Hashimoto et al., 2001). Subsequently, it was hypothesized that ASK-mediated regulation of the MAPK cascades depends on the interaction of ASK with the reduced form of a thiol modulating agent (NAC, GSH, TRX), thereby causing direct inhibition of the activity of the downstream pathway (Saitoh et al., 1998). Although the activity of ASK-1 has not been assessed in this investigation, it is very likely that MAPK-mediated regulation of TNF-α biosynthesis in the alveolar epithelium involves a ROS/redox-sensitive mechanism regulating the activity of ASK-1 and probably other components of the converging up-stream cascades, thereby attenuating the stimulatory effect of MAPK pathway in TNF-α signalling. Collectively, the MAPK pathway seems to be directly involved in cytokine signalling and that MAPK-mediated regulation of LPS-induced TNF-α biosynthesis is ROS/redox-sensitive.
In summary, the results of the present investigation could be highlighted as follows: (i) Exposure to E. coli-derived LPS induced a time- and dose-dependent activation of the MAPKp38 pathway; (ii) LPS mediated regulation of MAPKp38 cascade was associated with the activation of the MAPKAP-K2 (MK-2) pathway, thereby allowing phosphorylation of Hsp27; (iii) Selective blockade of either MAPKp38 and MAPKERK pathway attenuated TNF-α biosynthesis induced by LPS, an effect synergistically amplified by simultaneous inhibition; (iv) Selective inhibition of NF-κB attenuated, but did not abrogate, LPS-mediated secretion of TNF-α; (v) LPS induced intracellular accumulation of ROS, which up-regulated MAPKp38 phosphorylation, an effect attenuated by NAC and other antioxidants, including the GSH precursor, γ-GCE; (vi) NAC reduced LPS-induced TNF-α secretion, an effect associated with its ability to induce intracellular accumulation of GSH and lower than that of GSSG; and (vii) The antioxidant potential of NAC and its ability to feed cysteine onto the GSH biosynthetic machinery are likely to reside in its effectiveness to blockade the MAPKp38 pathway, thereby suppressing the downstream TNF-α signalling pathway. It is concluded that MAPK-mediated regulation of LPS induced TNF-α biosynthesis in the alveolar epithelium is ROS and redox-sensitive and requires the involvement of GSH-mediated signalling pathways.
Acknowledgments
This work was supported by grants from the Medical Research Council (MRC, U.K.), Anonymous Trust and Tenovus-Scotland (S.C. Land). Dr John Haddad is a recipient of the George John Livanos prize (London).
Abbreviations
- AEBSF
4-(2-aminoethyl)]-benzene sulphonyl fluoride-HCl
- ANOVA
analysis of variance
- ARE
AU-rich element
- BSA
bovine serum albumin
- DMEM
Dulbecco's modified Eagle medium
- DMSO
dimethyl sulphoxide
- DTT
dithiothreitol
- ELISA
enzyme-linked immuno-sorbent assay
- ERK
extracellular signal-regulated kinase
- fATII
foetal alveolar type II epithelial cells
- FCS
foetal calf serum
- Glutathione (GSH)
L-γ-glutamyl-L-cysteinyl-glycine
- glutathione (GSSG)
oxidized glutathione disulphide
- H2O2
hydrogen peroxide
- HBSS
Hanks' balanced salt solution
- Hsp27
heat-shock protein 27
- HRP
horseradish peroxidase
- IκB
inhibitory-κB
- IKK
IκB kinase
- IL
interleukin
- LPS
lipopolysaccharide
- LBP
lipopolysaccharide binding protein
- MKK
MAPK kinase
- MAPK
mitogen-activated protein kinase
- MAPKK
MAPK kinase
- NAC
N-acetyl-L-cysteine
- NF-κB
Nuclear factor-κB
- NIK
NF-κB inducing kinase
- O2−·
superoxide anion
- ·OH
hydroxyl radical
- PBS
phosphate buffered saline
- PGE2
prostaglandin-E2
- redox
reduction-oxidation
- RK
reactivating kinase
- ROS
reactive oxygen species
- SAPK
stress-activated protein kinase
- SDS–PAGE
sodium-dodecyl polyacrylamide gel electrophoresis
- Thr
threonine
- TMB
3,3,5,5-tetramethyl-benzidine dihydrochloride
- TNF-α
tumour necrosis factor-α
- TRX
thioredoxin
- Tyr
tyrosine
References
- ARUOMA O.I., HALLIWELL B., HOEY B.M., BEUTLER J. The antioxidant action of N-acetyl-L-cysteine: Its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochloric acid. Free Radic. Biol. Med. 1989;6:593–597. doi: 10.1016/0891-5849(89)90066-x. [DOI] [PubMed] [Google Scholar]
- BALDASSARE J.J., BI Y., BELLONE C.J. The role of p38 mitogen-activated protein kinase in IL-1β transcription. J. Immunol. 1999;162:5367–5373. [PubMed] [Google Scholar]
- BALDWIN A.S. The NF-κB and IκB proteins: New discoveries and insights. Annu. Rev. Immunol. 1996;14:649–681. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- BALLARD-CROFT C., WHITE D.J., MAASS D.L., HYBKI D.P., HORTON J.W. Role of p38 mitogen-activated kinase in cardiac myocyte secretion of the inflammatory cytokine TNF-α. Am. J. Physiol. Heart Circ. Physiol. 2001;280:H1970–H1981. doi: 10.1152/ajpheart.2001.280.5.H1970. [DOI] [PubMed] [Google Scholar]
- BARRETT E.G., JOHNSON C., OBERDÖRSTER G., FINKELSTEIN J.N. Antioxidant treatment attenuates cytokine and chemokine levels in murine macrophages following silica exposure. Toxicol. Appl. Pharmacol. 1999;158:211–220. doi: 10.1006/taap.1999.8716. [DOI] [PubMed] [Google Scholar]
- BERNARD G.R. N-acetyl-L-cysteine in experimental and clinical acute lung injury. Am. J. Med. 1991;91:54S–59S. doi: 10.1016/0002-9343(91)90284-5. [DOI] [PubMed] [Google Scholar]
- CANO E., MAHADEVAN L.C. Parallel signal processing among mammalian MAPKs. Trends Biochem. Sci. 1995;20:117–122. doi: 10.1016/s0968-0004(00)88978-1. [DOI] [PubMed] [Google Scholar]
- CHAN E.D., RICHES D.W.H., WHITE C.W. Redox paradox: Effect of N-acetylcysteine and serum on oxidation-reduction-sensitive mitogen-activated protein kinase signaling pathways. Am. J. Respir. Cell Mol. Biol. 2001;24:627–632. doi: 10.1165/ajrcmb.24.5.4280. [DOI] [PubMed] [Google Scholar]
- CHANDEL N.S., MALTEPE E., GOLDWASSER E., MATHIEU C.E., SIMON M.C., SCHUMACKER P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. U.S.A. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE A.K., KODYS K.M., YEH B.S., MILLER-GRAZIANO C. Exaggerated human monocyte IL-10 concomitant to minimal TNF-α induction by heat-shock protein 27 (Hsp27) suggests Hsp27 is primarily an anti-inflammatory stimulus. J. Immunol. 2000;165:3951–3958. doi: 10.4049/jimmunol.165.7.3951. [DOI] [PubMed] [Google Scholar]
- DE KEULENAER G.W., USHIO-FUKAI M., YIN Q.Q., CHUNG A.B., LYONS P.R., ISHIZAKA N., RENGARAJAN K., TAYLOR W.R., ALEXANDER R.W., GRIENDLING K.K. Convergence of redox-sensitive and mitogen-activated protein kinase signaling pathways in tumor necrosis factor-α-mediated monocyte chemoattractant protein-1 induction in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2000;20:385–391. doi: 10.1161/01.atv.20.2.385. [DOI] [PubMed] [Google Scholar]
- DOHERTY A.M. Phosphodiesterase 4 inhibitors as novel anti-inflammatory agents. Curr. Opin. Chem. Biol. 1999;3:466–473. doi: 10.1016/S1367-5931(99)80068-4. [DOI] [PubMed] [Google Scholar]
- DRÖGE W., SCHULZE-OSTHOFF K., MIHM S., GALTER D., SCHENK H., ECK H.P., ROTH S., GMUNDER H. Functions of glutathione and glutathione disulfide in immunology and immunopathology. FASEB J. 1994;8:1131–1138. [PubMed] [Google Scholar]
- FOEY A.D., PARRY S.L., WILLIAMS L.M., FELDMANN M., FOXWELL B.M.J., BRENNAN F.M. Regulation of monocyte IL-10 synthesis by endogenous IL-1 and TNF-α: Role of the p38 and p42/44 mitogen-activated protein kinases. J. Immunol. 1998;160:920–928. [PubMed] [Google Scholar]
- FOX R.B. Prevention of granulocyte-mediated oxidant lung injury in rats by a hydroxyl radical scavenger, dimethylthiourea. J. Clin. Invest. 1984;74:1456–1464. doi: 10.1172/JCI111558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FUKUDA T., SUMICHIKA H., MURATA M., HANANO T., ADACHI K., HISADOME M. A novel dual regulator of tumour necrosis factor-α and interleukin-10 protects mice from endotoxin-induced shock. Eur. J. Pharmacol. 2000;391:317–320. doi: 10.1016/s0014-2999(00)00096-0. [DOI] [PubMed] [Google Scholar]
- GARRINGTON T.P., JOHNSON G.L. Organisation and regulation of mitogen-activated protein kinase signalling pathways. Curr. Opin. Cell Biol. 1999;11:211–218. doi: 10.1016/s0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- GOSSET P., WALLAERT B., TONNEL A.B., FOURNEAU C. Thiol regulation of the production of TNF-α, IL-6 and IL-8 by human alveolar macrophages. Eur. Respir. J. 1999;14:98–105. doi: 10.1034/j.1399-3003.1999.14a17.x. [DOI] [PubMed] [Google Scholar]
- GRIEGO S.D., WESTON C.B., ADAMS J.L., TAL-SINGER R., DILLON S.B. Role of p38 mitogen-activated protein kinase in rhinovirus-induced cytokine production by bronchial epithelial cells. J. Immunol. 2000;165:5211–5220. doi: 10.4049/jimmunol.165.9.5211. [DOI] [PubMed] [Google Scholar]
- GRIFFITH O.W., MEISTER A. Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine) J. Biol. Chem. 1979;254:7558–7560. [PubMed] [Google Scholar]
- HADDAD J.J. VX-745 Vertex Pharmaceuticals: A novel MAPKp38 inhibitor with anti-inflammatory actions. Curr. Opin. Invest. Drugs. 2001;2:1070–1076. [PubMed] [Google Scholar]
- HADDAD J.J., LAND S.C. O2-evoked regulation of HIF-1α and NF-κB in perinatal lung epithelium requires glutathione biosynthesis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000a;278:L492–L503. doi: 10.1152/ajplung.2000.278.3.L492. [DOI] [PubMed] [Google Scholar]
- HADDAD J.J., LAND S.C. The differential expression of apoptosis factors in the alveolar epithelium is redox sensitive and requires NF-κB (RelA)-selective targeting. Biochem. Biophys. Res. Commun. 2000b;271:257–267. doi: 10.1006/bbrc.2000.2607. [DOI] [PubMed] [Google Scholar]
- HADDAD J.J., OLVER R.E., LAND S.C. Antioxidant/pro-oxidant equilibrium regulates HIF-1α and NF-κB redox sensitivity: Evidence for inhibition by glutathione oxidation in alveolar epithelial cells. J. Biol. Chem. 2000;275:21130–21139. doi: 10.1074/jbc.M000737200. [DOI] [PubMed] [Google Scholar]
- HADDAD J.J., SAFIEH-GARABEDIAN B., SAADÉ N.E., KANAAN S.A., LAND S.C. Chemioxyexcitation (ΔpO2/ROS)-dependent release of IL-1β, IL-6 and TNF-α: Evidence of cytokines as oxygen sensitive mediators in the alveolar epithelium. Cytokine. 2001a;13:138–147. doi: 10.1006/cyto.2000.0789. [DOI] [PubMed] [Google Scholar]
- HADDAD J.J., CHOUDHARY K.K., LAND S.C. The ex vivo differential expression of apoptosis signalling cofactors in the developing lung: Essential role of oxygenation during the transition from placental to pulmonary-based respiration. Biochem. Biophys. Res. Commun. 2001b;281:311–316. doi: 10.1006/bbrc.2001.4350. [DOI] [PubMed] [Google Scholar]
- HADDAD J.J., SAFIEH-GARABEDIAN B., SAADÉ N.E., LAND S.C. Thiol regulation of pro-inflammatory cytokines reveals a novel immunopharmacological potential of glutathione in the alveolar epithelium. J. Pharmacol. Exp. Therap. 2001c;296:996–1005. [PubMed] [Google Scholar]
- HADDAD J.J., LAUTERBACH R., SAADÉ N.E., SAFIEH-GARABEDIAN B., LAND S.C. α-Melanocyte-related tripeptide, Lys-D-Pro-Val, ameliorates endotoxin-induced nuclear factor κB translocation and activation: Evidence for involvement of an interleukin-1β193–195 receptor antagonism in the alveolar epithelium. Biochem. J. 2001d;355:29–38. doi: 10.1042/0264-6021:3550029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HADDAD J.J., SAFIEH-GARABEDIAN B., SAADÉ N.E., LAND S.C. The biphasic immunoregulation of pyrimidylpiperazine (Y-40138) is IL-10 sensitive and requires NF-κB targeting in the alveolar epithelium. Br. J. Pharmacol. 2001e;133:49–60. doi: 10.1038/sj.bjp.0704041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAN J., LEE J.D., BIBBS L., ULEVITCH R.J. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. 1994;265:808–811. doi: 10.1126/science.7914033. [DOI] [PubMed] [Google Scholar]
- HANANO T., ADACHI K., AOKI Y., MORIMOTO H., NAKA Y., HISADOME M., FUKUDA T., SUMICHIKA H. Novel DMARDs on the basis of a new concept of dual cytokine regulation, TNF-α suppression and IL-10 augmentation. Bioorg. Med. Chem. Lett. 2000;10:881–884. doi: 10.1016/s0960-894x(00)00129-3. [DOI] [PubMed] [Google Scholar]
- HASHIMOTO S., MATSUMOTO K., GON Y., FURUICHI S., MARUOKA S., TAKESHITA I., HIROTA K., YODOI J., HORIE T. Thioredoxin negatively regulates p38 MAP kinase activation and IL-6 production by tumour necrosis factor-α. Biochem. Biophys. Res. Commun. 1999a;258:443–447. doi: 10.1006/bbrc.1999.0658. [DOI] [PubMed] [Google Scholar]
- HASHIMOTO S., MATSUMOTO K., GON Y., MARUOKA S., TAKESHITA I., HAYASHI S., KOURA KI., KUJIME K., HORIE T. p38 mitogen-activated protein kinase regulates IL-8 expression in human pulmonary vascular endothelial cells. Eur. Respir. J. 1999b;13:1357–1364. [PubMed] [Google Scholar]
- HASHIMOTO S., GON Y., MATSUMOTO K., TAKESHITA I., HORIE T. N-Acetylcysteine attenuates TNF-α-induced p38 MAP kinase activation and p38 MAP kinase-mediated IL-8 production by human pulmonary vascular endothelial cells. Br. J. Pharmacol. 2001;132:270–276. doi: 10.1038/sj.bjp.0703787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HASKÓ G., KUHEL D.G., NÉMETH Z.H., MABLEY J.G., STACHLEWITZ R.F., VIRÁG L., LOHINAI Z., SOUTHAN G.J., SALZMAN A.L., SZABÓ C. Inosine inhibits inflammatory cytokine production by a post-transcriptional mechanism and protects against endotoxin-induced shock. J. Immunol. 2000;164:1013–1019. doi: 10.4049/jimmunol.164.2.1013. [DOI] [PubMed] [Google Scholar]
- HASKÓ G., SZABÓ C., NÉMETH Z.H., SALZMAN A.L., SYLVESTER VIZI E. Suppression of IL-12 production by phosphordiesterase inhibition in murine endotoxemia is IL-10 independent. Eur. J. Immunol. 1998;28:468–472. doi: 10.1002/(SICI)1521-4141(199802)28:02<468::AID-IMMU468>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- HAYES J.D., MCLELLAN L.I. Glutathione and glutathione-dependent enzymes represent a coordinately regulated defense against oxidative stress. Free Radic. Res. 1999;31:273–300. doi: 10.1080/10715769900300851. [DOI] [PubMed] [Google Scholar]
- HERLAAR E., BROWN Z. p38 MAPK signalling cascades in inflammatory disease. Mol. Med. Today. 1999;5:439–447. doi: 10.1016/s1357-4310(99)01544-0. [DOI] [PubMed] [Google Scholar]
- HISADOME M., FUKUDA T., SUMICHIKA H., HANANO T., ADACHI K. A novel anti-rheumatic drug suppresses tumour necrosis factor-α and augments interleukin-10 in adjuvant arthritic rats. Eur. J. Pharmacol. 2000;409:331–335. doi: 10.1016/s0014-2999(00)00866-9. [DOI] [PubMed] [Google Scholar]
- HUDSON L.D., MILBERG J.A., ANARDI D., MAUNDER R.J. Clinical risks for the development of the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1995;151:293–301. doi: 10.1164/ajrccm.151.2.7842182. [DOI] [PubMed] [Google Scholar]
- JEANNIN P., DELNESTE Y., LECOANET-HENCHOZ S., GAUCHAT J.F., LIFE P., HOLMES D, BONNEFOY J.Y. Thiols decrease human interleukin (IL)-4 production and IL-4-induced immunoglobulin synthesis. J. Exp. Med. 1995;182:1785–1792. doi: 10.1084/jem.182.6.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KANG K.W., RYU J.H., KIM S.G. The essential role of phosphatidylinositol 3-kinase and of p38 mitogen-activated protein kinase activiation in the antioxidant response element-mediated rGSTA2 induction by decreased glutathione in H4IIE cells. Mol. Pharmacol. 2000;58:1017–1025. doi: 10.1124/mol.58.5.1017. [DOI] [PubMed] [Google Scholar]
- KLEBANOFF S.J., VADAS M.A., HARLAN J.M., SPARKS L.H., GAMBLE J.R., AGOSTI J.M., WALTERSDORPH A.M. Stimulation of neutrophils by tumor necrosis factor. J. Immunol. 1986;136:4220–4225. [PubMed] [Google Scholar]
- KOTLYAROV A., NEININGER A., SCHUBERT C., ECKERT R., BIRCHMEIER C., VOLK H.D., GAESTEL M. MAPKAP kinase 2 is essential for LPS-induced TNF-α biosynthesis. Nature Cell. Biol. 1999;1:94–97. doi: 10.1038/10061. [DOI] [PubMed] [Google Scholar]
- LAPPERRE T.S., JIMENEZ L.A., ANTONICELLI F., DROST E.M., HIEMSTRA P.S., STOLK J., MACNEE W., RAHMAN I. Apocynin increases glutathione synthesis and activates AP-1 in alveolar epithelial cells. FEBS Lett. 1999;443:235–239. doi: 10.1016/s0014-5793(98)01723-2. [DOI] [PubMed] [Google Scholar]
- LEE J.C., LAYDON J.T., MCDONNELL P.C., GALLAGHER T.F., KUMAR S., GREEN D., MCNULTY D., BLUMENTHAL M.J., HEYS J.R., LANDVATTER S.W., STRICKLER J.E., MCLAUGHLIN M.M., SIEMENS I.R., FISHER S.M., LIVI G.P., WHITE J.R., ADAMS J.L., YOUNG P.R. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- MARSHALL C.J. Specificity of receptor tyrosine kinase signalling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- MATSUMOTO K., HASHIMOTO S., GON Y., NAKAYAMA T., HORIE T. N-Acetylcysteine inhibits IL-1α-induced IL-8 secretion by human bronchial epithelial cells. Respir. Med. 1998;92:512–515. doi: 10.1016/s0954-6111(98)90300-6. [DOI] [PubMed] [Google Scholar]
- MEISTER A. Glutathione metabolism and its selective modification. J. Biol. Chem. 1988;263:17205–17208. [PubMed] [Google Scholar]
- MERCURIO F., MANNING A.M. Multiple signals converging on NF-κB. Curr. Opin. Cell Biol. 1999;11:226–232. doi: 10.1016/s0955-0674(99)80030-1. [DOI] [PubMed] [Google Scholar]
- MORIGUCHI T., TOYOSHIMA F., MASUYAMA N., HANAFUSA H., GOTOH Y., NISHIDA E. A novel SAPK/JNK kinase, MKK7, stimulated by TNF-α and cellular stresses. EMBO J. 1997;16:7045–7053. doi: 10.1093/emboj/16.23.7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NATOLI G., COSTANZO A., IANNI A., TEMPLETON D.J., WOODGETT J.R., BALSANO C., LEVRERO M. Activation of SAPK/JNK by TNF receptor I through a non-cytotoxic TRAF2-dependent pathway. Science. 1997;275:200–203. doi: 10.1126/science.275.5297.200. [DOI] [PubMed] [Google Scholar]
- NEMOTO S., DIDONATO J.A., LIN A. Co-ordinate regulation of IκB kinases by mitogen-activated protein kinase kinase kinase 1 and NF-κB-inducing kinase. Mol. Cell. Biol. 1998;18:7336–7343. doi: 10.1128/mcb.18.12.7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEUSCHWANDER-TETRI B.A., BELLEZZO J.M., BRITTONM R.S., BACON B.R., FOX E.S. Thiol regulation of endotoxin-induced release of tumor necrosis factor α from isolated rat Kupffer cells. Biochem. J. 1996;320:1005–1010. doi: 10.1042/bj3201005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NICK J.A., AVDI N.J., YOUNG S.K., LEHMAN L.A., MCDONALD P.P., FRASCH S.C., BILLSTROM M.A., HENSON P.M., JOHNSON G.L., WORTHEN G.S. Selective activation and functional significance of p38α mitogen-activated protein kinase in lipopolysaccharide-stimulated neutrophils. J. Clin. Invest. 1999;103:851–858. doi: 10.1172/JCI5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARKER N.B., BERGER E.M., CURTIS W.E., MULDROW M.E., LINAS S.L., REPINE J.E. Hydrogen peroxide causes dimethylthiourea consumption while hydroxyl radical causes dimethyl sulfoxide consumption in vitro. J. Free Radic. Biol. Med. 1985;1:415–419. doi: 10.1016/0748-5514(85)90155-2. [DOI] [PubMed] [Google Scholar]
- PARTRICK D.A., MOORE E.E., OFFNER P.J., MELDRUM D.R., TAMURA D.Y., JOHNSON J.L., SILLIMAN C.C. Maximal human neutrophil priming for superoxide production and elastase release requires p38 mitogen-activated protein kinase activation. Arch. Surg. 2000;135:219–225. doi: 10.1001/archsurg.135.2.219. [DOI] [PubMed] [Google Scholar]
- PENA L.R., HILL B.B., MCCLAIN C.J. Treatment with glutathione precursor decreases cytokine activity. J. Parenter. Enteral. Nutr. 1999;23:1–6. doi: 10.1177/014860719902300101. [DOI] [PubMed] [Google Scholar]
- PITTET J.F., MACKERSIE R.C., MARTIN T.R., MATTHAY M.A. Biological markers of acute lung injury: prognostic and pathogenetic significance. Am. J. Respir. Crit. Care Med. 1997;155:1187–1205. doi: 10.1164/ajrccm.155.4.9105054. [DOI] [PubMed] [Google Scholar]
- RAHMAN I. Inflammation and the regulation of glutathione level in lung epithelial cells. Antiox. Redox Signal. 1999;1:425–447. doi: 10.1089/ars.1999.1.4-425. [DOI] [PubMed] [Google Scholar]
- RAHMAN I. Regulation of nuclear factor-κB, activator protein-1, and glutathione levels by tumour necrosis factor-α and dexamethasone in alveolar epithelial cells. Biochem. Pharmacol. 2000;60:1041–1049. doi: 10.1016/s0006-2952(00)00392-0. [DOI] [PubMed] [Google Scholar]
- RAINGEAUD J., GUPTA S., ROGERS J.S., DICKENS M., HAN J., ULEVITCH R.J., DAVIS R.J. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J. Biol. Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- REIMUND J.-M., ALLISON A.C., MULLER C.D., DUMONT S., KENNEY J.S., BAUMANN R., DUCLOS B., POINDRON P. Antioxidants inhibit the in vitro production of inflammatory cytokines in Crohn's disease and ulcerative colitis. Eur. J. Clin. Invest. 1998;28:145–150. doi: 10.1046/j.1365-2362.1998.00257.x. [DOI] [PubMed] [Google Scholar]
- RIDLEY S.H., SARSFIELD S.J., LEE J.C., BIGG H.F., CAWSTON T.E., TAYLORM D.J., DEWITT D.L., SAKLATVALA J. Actions of IL-1 are selectively controlled by p38 mitogen-activated protein kinase. Regulation of prostaglandin H synthase-2, metalloproteinases, and IL-6 at different levels. J. Immunol. 1997;158:3165–3173. [PubMed] [Google Scholar]
- RIETSCHEL E.T., KIRIKAE T., SCHADE U., MAMAT U., SCHMIDT G., LOPPNOW H., ULMER A., ZAHRINGER U., SEYDEL U., PADOVA F., SCHRIER M., BRADE H. Bacterial endotoxin: Molecular relationships of structure to activity and function. FASEB J. 1994;8:217–225. doi: 10.1096/fasebj.8.2.8119492. [DOI] [PubMed] [Google Scholar]
- ROUSE J., COHEN P., TRIGON S., MORANGE M., ALONSO-LLAMAZARES A., ZAMANILLO D., HUNT T., NEBREDA A.R. A novel kinase cascade triggered by stress and heat shock stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell. 1994;78:1027–1037. doi: 10.1016/0092-8674(94)90277-1. [DOI] [PubMed] [Google Scholar]
- ROVIN B.H., DICKERSON J.A., TAN L.C., FASSLER J. Modulation of IL-1-induced chemokine expression in human mesangial cells through alterations in redox status. Cytokine. 1997;9:178–185. doi: 10.1006/cyto.1996.0152. [DOI] [PubMed] [Google Scholar]
- RUTAULT K., HAZZALIN C.A., MAHADEVAN L.C. Combinations of ERK and p38 MAPK inhibitors ablate tumour necrosis factor-α (TNF-α) mRNA induction: Evidence for selective destabilisation of TNF-α transcripts. J. Biol. Chem. 2001;276:6666–6674. doi: 10.1074/jbc.M005486200. [DOI] [PubMed] [Google Scholar]
- SAFIEH-GARABEDIAN B., KANAAN S.A., HADDAD J.J., ABOU JAOUDE P., JABBUR S.J., SAADÉ N.E. Involvement of interleukin-1β, nerve growth factor and prostaglandin E2 in endotoxin-induced localised inflammatory hyperalgesia. Br. J. Pharmacol. 1997;121:1619–1626. doi: 10.1038/sj.bjp.0701313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAITOH M., NISHITOH H., FUJI M., TAKEDA K., TOBIUM K., SAWADA Y., KAWABATA M., MIYAZONO K., ICHIJO H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal regulating kinase (ASK)-1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHERLE P.A., JONES E.A., FAVATA M.F., DAULERIO A.J., COVINGTON M.B., NURNBERG S.A., MAGOLDA R.L., TRZASKOS J.M. Inhibition of MAP kinase kinase prevents cytokine and prostaglandin E2 production in lipopolysaccharide-stimulated monocytes. J. Immunol. 1998;161:5681–5686. [PubMed] [Google Scholar]
- STEIN B., YANG M.X., YOUNG D.B., JANKNECHT R., HUNTER T., MURRAY B.W., BARBOSA M.S. p38-2, a novel mitogen-activated protein kinase with distinct properties. J. Biol. Chem. 1997;272:19509–19517. doi: 10.1074/jbc.272.31.19509. [DOI] [PubMed] [Google Scholar]
- SU B., KARIN M. Mitogen-activated protein kinase cascades and regulation of gene expression. Curr. Biol. 1996;8:402–411. doi: 10.1016/s0952-7915(96)80131-2. [DOI] [PubMed] [Google Scholar]
- THOMPSON A.B., ROBBINS R.A., ROMBERGER D.J. Immunological functions of the pulmonary epithelium. Eur. Respir. J. 1985;8:127–149. doi: 10.1183/09031936.95.08010127. [DOI] [PubMed] [Google Scholar]
- TSUJI F., MIYAKE Y., AONO H., KAWASHIMA Y., MITA S. Effects of bucillamine and N-acetyl-L-cysteine on cytokine production and collagen-induced arthritis (CIA) Clin. Exp. Immunol. 1999;115:26–31. doi: 10.1046/j.1365-2249.1999.00749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VANDENABEELE P., DECLERCQ W., BEYAERT R., FIERS W. Two tumour necrosis factor receptors: Structure and function. Trends Cell Biol. 1995;5:392–399. doi: 10.1016/s0962-8924(00)89088-1. [DOI] [PubMed] [Google Scholar]
- VISSER J., VAN BOXEL-DEZAIRE A., METHORST D., BRUNT T., RONALD DE KLOET E., NAGELKERKEN L. Differential regulation of interleukin-10 (IL-10) and IL-12 by glucocorticoids in vitro. Blood. 1998;91:4255–4264. [PubMed] [Google Scholar]
- WANG X.S., DIENER K., MANTHEY C.L., WANG S., ROSENZWEIG B., BRAY J., DELANEY J., COLE C.N., CHAN-HUI P.Y., MANTLO N., LICHENSTEIN H.S., ZUKOWSKI M., YAO Z. Molecular cloning and characterisation of a novel p38 mitogen-activated protein kinase. J. Biol. Chem. 1997;272:23668–23674. doi: 10.1074/jbc.272.38.23668. [DOI] [PubMed] [Google Scholar]
- WASIL M., HALLIWELL B., GROOTVELD M., MOORHOUSE C.P., HUTCHISON D.C.S., BAUM H. The specificity of thiourea, dimethylthiorea and dimethyl sulphoxide as scavengers of hydroxyl radicals: Their protection of α 1-antiproteinase against inactivation by hypochlorous acid. Biochem. J. 1987;243:867–870. doi: 10.1042/bj2430867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WIDMANN C., GIBSON S., JARPE M.B., JOHNSON G.L. Mitogen-activated protein kinase: Conservation of a three-kinase module from yeast to human. Physiol. Rev. 1999;79:143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- WRIGHT S.D., RAMOS R.A., TOBIAS P.S., ULEVITCH R.J., MATHISON J.C. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science. 1990;249:1431–1433. doi: 10.1126/science.1698311. [DOI] [PubMed] [Google Scholar]
- YAMAKAWA T., EGUCHI S., MATSUMOTO T., YAMAKAWA Y., NUMAGUCHI K., MIYATA I., REYNOLDS C.M., MOTLEY E.D., INAGAMI T. Intracellular signalling in rat cultured vascular smooth muscle cells: Roles of nuclear factor-κB and p38 mitogen-activated protein kinase on tumour necrosis factor-α production. Endocrinol. 1999;140:3562–3573. doi: 10.1210/endo.140.8.6914. [DOI] [PubMed] [Google Scholar]
- ZU Y.L., QI J., GILCHRIST A., FERNANDEZ G.A., VAZQUEZ-ABAD D., KREUTZER D.L., HUANG C.K., SHA'AFI R.I. p38 mitogen-activated protein kinase activation is required for human neutrophil function triggered by TNF-α or FLMP stimulation. J. Immunol. 1998;160:1982–1989. [PubMed] [Google Scholar]