

Abstract
We have examined the effects of 12 glucocorticoids as inhibitors of A549 cell growth.
Other than cortisone and prednisone, all the glucocorticoids inhibited cell growth and this was strongly correlated (r=0.91) with inhibition of prostaglandin (PG)E2 formation.
The molecular mechanism by which the active steroids prevented PGE2 synthesis was examined and three groups were identified. Group A drugs did not inhibit arachidonic acid release but inhibited the induction of COX2. Group B drugs were not able to inhibit the induction of COX2 but inhibited arachidonic acid release through suppression of cPLA2 activation. Group C drugs were apparently able to bring about both effects.
The inhibitory actions of all steroids was dependent upon glucocorticoid receptor occupation since RU486 reversed their effects. However, group A acted through the NF-κB pathway to inhibit COX2 as the response was blocked by the inhibitor geldanamycin which prevents dissociation of GR and the effect was blocked by APDC, the NF-κB inhibitor. On the other hand, the group B drugs were not inhibited by NF-κB inhibitors or geldanamycin but their effect was abolished by the src inhibitor PP2. Group C drugs depended on both pathways.
In terms of PGE2 generation, there is clear evidence of two entirely separate mechanisms of glucocorticoid action, one of which correlates with NF-κB mediated genomic actions whilst the other, depends upon rapid effects on a cell signalling system which does not require dissociation of GR. The implications for these findings are discussed.
Keywords: Glucocorticoids, non-genomic, cell signalling, genome-independent
Introduction
Since 1949, glucocorticoids (GCs) have been widely used in the treatment of inflammatory diseases (Hench et al., 1949) and despite undesirable side effects they are still regarded as one of the most potent and, in some cases, life saving therapies. Since their introduction, our insight into their molecular mechanism of action has grown considerably. Many investigators are convinced that a nuclear or ‘genomic' mechanism is responsible for most, if not all, of the effects of GCs (Beato et al., 1995; de waal, 1994). According to this idea the expression of key genes is modulated by a glucocorticoid receptor (GR)-dependent process. Following passive diffusion through the cell membrane, GCs bind with GRs which are believed to be located predominantly in the cytoplasm (Gustafsson et al., 1987) as multi-protein complexes including heat shock proteins (hsp), immunophillins and several kinases (Pratt et al., 1999). Following the binding of ligand the receptor undergoes key steric changes where many of the hsps and other factors are dissociated and the molecule acquires a dimer conformation (Beato et al., 1996). This multi-step process also brings about a recruitment/activation of several transcription factors and, as a result, the GR complex acquires a higher affinity for binding to specific DNA sequences – glucocorticoid response elements (GREs) in the nucleus (Sanchez et al., 1990). Due to the nature of these interactions the expression of target genes controlling the inflammatory process may be either up or down-regulated.
The relative importance of the contributions of transcription factor/response element binding versus GR/GRE binding of the GR complex in mediating GC effects has been widely debated. Some investigators believe that most of the anti-inflammatory actions of GCs can be accounted for by transcription factor interaction alone (Barnes, 1999). Indeed, the A458T mutated GR (GRdim), which is unable to dimerize and bind to classical nuclear GREs, is nevertheless capable of functionally interacting with transcription factors AP1 and NFκB (Reichardt et al., 1998) and therefore gives credence to this. These are profound observations which appear to support the notion that interaction with GREs and transcription factors are functionally separate molecular events which follow ligand binding.
However, it is increasingly clear that due to the pluripotent nature of GCs upon gene expression (approximately 1% of the genome is believed to be affected; Merkulova et al., 1997) systemic side effects including reduced bone mass, hypertension, diabetes, skin bruising and weight gain severely compromise their therapeutic effectiveness (Kimberly, 1991). In order to diminish the contribution of unwanted side effects to GC therapy several strategies have been employed. These include altering the route of administration. For example, a topical application or inhalation of an aerosol may target GCs to their site of action whilst reducing the amount in systemic circulation (Barnes et al., 1998). Similarly, using GCs with short half-lives (either through hepatic clearance or localized metabolism) may have the same effect. The creation of a new generation of GCs including mometasone, fluticasone and budesonide has gone some way in achieving this (Boobis, 1998; Johnson, 1998; Nathan et al., 2001). These GCs have significantly reduced side effects whilst retaining their potent anti-inflammatory properties and in some cases this has been attributed to a more rapid systemic clearance. Alternatively, a novel class of NO-derivatized GCs has also been shown to exhibit enhanced anti-inflammatory properties (Paul-Clark et al., 2000). The concept of dissociating the transcription factor-mediated responses from GRE-mediated effects by using highly specific synthetic GCs has also been advanced (Heck et al., 1994). A useful outcome of this remains to be proven – but, nevertheless, a differential selection of the beneficial effects of GCs still remains an important pharmacological target.
Many aspects of inflammation involve the over-production of eicosanoid metabolites such as the prostaglandins (PGs) (Vane, 1978). These are formed following the action of PLA2 enzymes on the phospholipid bilayer membrane and the subsequent conversion of liberated arachidonic acid by the cyclo-oxygenase (COX) enzymes. PLA2 activity and COX expression are therefore not only control points for regulating inflammation, but also important targets for pharmacological intervention. In studies using the A549 human lung adenocarcinoma cell line, we have shown that the synthetic GC dexamethasone, may, following binding to GR, inhibit the induction of COX2 (Newman et al., 1994). Furthermore, we have also shown that dexamethasone can modulate cytokine activation of arachidonic acid release (Croxtall et al., 1995) by inhibiting key components in the signal transduction pathway leading to the control of MAPK and specifically cPLA2 activity (Croxtall et al., 1996a,1996b; 2000). More importantly, we have shown that this effect upon arachidonic acid release precedes the down regulation of COX2 and is reversed by neither inhibitors of protein synthesis nor inhibitors of nuclear translocation such as geldanamycin (Croxtall et al., 2000). In this study we have investigated two major points. Firstly, whether the inhibition of cPLA2 activity and the down regulation of COX2 expression by dexamethasone are functionally separate events. Secondly, we have sought to compare and contrast the efficacy of a range of conventional GCs together with the new generation GCs on these two processes.
Methods
Cell culture
A549 cells (Flow) were maintained in continuous log phase growth in Dulbecco's modified Eagle medium/F-12 (DMEM/F-12) containing phenol red, 10% foetal calf serum (FCS) and 1% penicillin/streptomycin (PS) in T-150 flasks (Greiner) at 37°C, 5% CO2. The cells were not allowed to reach confluence at any time as this diminishes their response to growth factors, stimulators and GCs.
Cell proliferation experiments
Sub-confluent A549 cells were seeded into 12-place multi-well plates (Falcon) at a density of 5×104 cells ml−1 well−1 in DMEM/F-12, 10% FCS and 1% PS. Following incubation overnight, the cells were washed in 2 ml of sterile PBS and 1 ml of fresh DMEM/F-12 containing 1% PS (without phenol red) plus various GCs (10−12–10−7 M) or vehicle control added (either ethanol or DMSO). On day 2, cells were replenished with fresh experimental media containing test GCs or vehicle control. On day 3, media was removed from each well and 1 ml of PBS containing 0.05% trypsin and 0.02% EDTA was added to each well. The dispersed cells then counted with a Coulter Multisizer II counter. The percentage inhibition of cell proliferation for each GC treated culture was calculated compared to control well. We found this experimental template best revealed the growth inhibitory effects of GC treatment. Trypan Blue was used to determine cell viability. The data reported are due to inhibition of cell proliferation and not cell death due to toxicity of the GCs tested.
Measurements of IL-1β-stimulated PGE2 release
Sub-confluent A549 cells were seeded into 12-place multi-well plates at a density of 5×104 cells ml−1 well−1 in DMEM/F-12, 10% FCS and 1% PS. Following incubation overnight, the cells were washed in 2 ml of sterile PBS and 1 ml of fresh DMEM/F-12 containing 1% PS (without phenol red) plus 1 ng ml−1 IL-1β together with the various GCs (10−10–10−5 M) or vehicle control (either ethanol or DMSO) were added for 3 h. After which time 0.5 ml of experimental media was removed and PGE2 was measured in the samples using an enzyme-immunoassay (EIA) kit.
Measurement of arachidonic acid release
Sub-confluent A549 cells were seeded into 12-place multi-well plates (Falcon) at a density of 3×105 cells ml−1 well−1 in DMEM/F-12, 10% FCS, 1% PS and incubated overnight. [3H]-arachidonic acid [3H]-AA in ethanol was evaporated to dryness under N2 and resuspended in an appropriate volume of DMEM/F-12 (without phenol red) and after vortex mixing left at 37°C for 1 h. After the cells had been washed with PBS, 9.25 KBq of [3H]-AA in 0.5 ml DMEM/F-12 (without phenol red or FCS) was added to each well and incubated overnight. The media containing free [3H]-AA was then removed and the cells washed three times with 1 ml DMEM/F-12 containing 1 mg ml−1 BSA. The cells thus labelled with [3H]-AA were then treated for 3 h with various GCs (10−10–10−5 M) or vehicle control (either ethanol or DMSO). Then 10 nM EGF and 50 nM thapsigargin was added for 30 min. After incubation, 0.4 ml of medium was removed from each well for scintillation counting.
Determination of the activation of cPLA2 and expression of COX2
The activation of cPLA2 was determined by measuring the phosphorylation status of serine residues of the immunoprecipitated protein by Western blotting (Croxtall et al., 2000) and the same cell lysates were also Western blotted for COX 2 protein induction. A549 cells were incubated with 1 ng ml−1 IL-1β together with various GCs (10−10–10−5 M) or vehicle control (either ethanol or DMSO) for 3 h (Figure 1). In inhibitor studies the various GCs (1 μM) were incubated together with either 1 μM PP2, 10 μM APDC or 10 μM geldanamycin for 3 h. The medium from T-75 flasks was aspirated and the A549 cell monolayer washed with PBS, 1 mM EDTA to remove adherent surface-bound proteins. The monolayer was dispersed with 0.05% trypsin in PBS, 10 mM EDTA. Cell pellets were snap-frozen in 3 ml of PBS, 10 mM EDTA containing 1 mg ml−1 soyabean trypsin inhibitor, 0.01% leupeptin, 1 mM PMSF and 1 mM sodium orthovanadate. Once thawed, cell lysates were clarified by centrifugation at 13,000×g for 20 min. Protein concentrations were measured by Bradford assay and identical concentrations were used in each immunoprecipitation. One millilitre of cell lysate was incubated with 5 μg of precipitating monoclonal antibody to cPLA2 (Santa Cruz) for 16 h with continuous rocking. Then 20 mg Protein A-Sepharose was added for a further 2 h. The Protein A-Sepharose bound immunocomplexes were washed three times in PBS, 10 mM EDTA and then incubated with 250 μl sample buffer for 5 min at 90°C prior to SDS–PAGE analysis by Western blotting with anti-phospho serine monoclonal antibody (10 μg ml−1) and detection by DAB. The same cell lysate was also Western blotted for COX2 (Santa Cruz) expression. Western blots were scanned using an Agfa Snapscan 1236S and the image composite transferred into Power Point (Microsoft, WA, U.S.A.) running on an Apple Macintosh. Densitometric analysis was performed with NIH Image 1.54 and relative band intensities reported as per cent changes within each blot. The calculated values are semiquantitative and are only meant to give some numerical guide to the ratio of band intensities. The blots are presented graphically to enable easier comparisons between treatments and are typical examples of at least three such experiments. Although overall band intensities varied between experiments, the ratio of band intensities remained the same.
Figure 1.
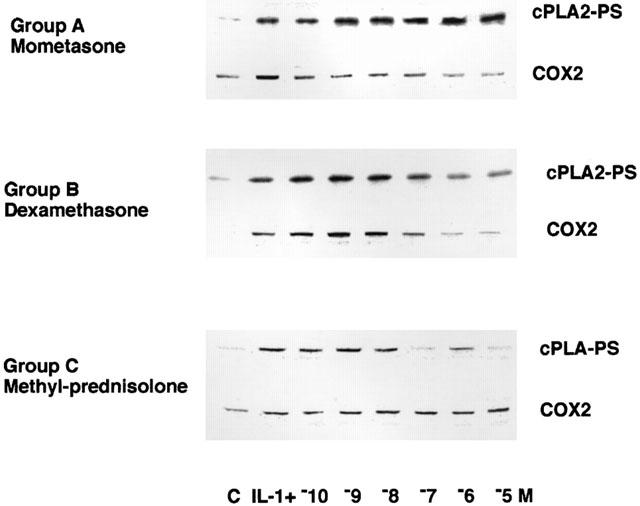
Representative examples of the effect of GCs from each group upon IL-1β activation of cPLA2 and induction of COX2 expression. A549 cells were treated with 1 ng ml−1 for 3 h in the presence of a range of concentrations of each GC (10−10–10−5 M). Immunoprecipitates of cPLA2 were examined for activation status by Western blotting with antibodies to phosphoserine. COX2 expression was assessed by Western blotting the total cell lysate with COX2 antibody.
Materials
EGF, thapsigargin, geldanamycin, Protein A sepharose, anti-phospho serine monoclonal antibodies, GCs (except below) and all other general purpose, cell culture or blotting reagents were from Sigma (Poole, U.K.). PP2 was from Calbiochem-Novabiochem (U.K.). APDC was from Tocris Cookson. RU486 was a gift from Roussel-Uclaf (Romainville, France). [5,6,8,9,11,12,14,15-3H]-(N)-arachidonic acid was from NEN Du Pont (Belgium). Immunoprecipitation of activated cPLA2 and Western blotting for COX2 was performed using monoclonal antibodies from Santa Cruz Biotechnology. PGE2 EIA kits were from Amersham. Budesonide was from AstraZeneca, Lund, Sweden. We are very grateful to Dr William Kreutner, Schering-Plough, New Jersey for mometasone and GlaxoSmithKline, The Netherlands for fluticasone proprionate.
Statistical analysis
Each experiment was performed in triplicate (n=3) and each experiment is a typical example of at least three such experiments. Results were calculated as the mean±s.e.mean and are presented as the per cent inhibition±s.e.mean. Statistical differences were calculated on raw data using the ANOVA test with post analysis Bonferroni correction. A threshold value of P<0.05 was taken as significant.
Results
Effects of glucocorticoids on the proliferation of A549 cells
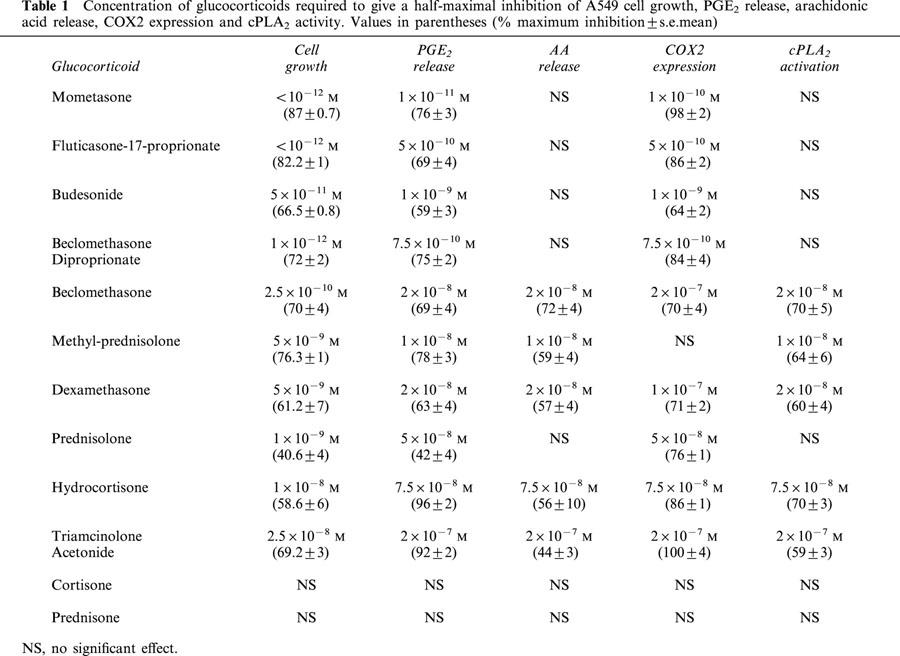
We have previously described the growth inhibitory response of A549 cells to dexamethasone (Croxtall & Flower, 1992). The data presented in Table 1 (column 1) show that this property is shared by many of the various GCs tested, however, there are marked differences of efficacy between these compounds. Following a 3 day incubation period, dexamethasone (our reference GC) significantly (P<0.05) inhibited proliferation (within the concentration range 1–100 nM) with a half-maximal inhibitory effect at 5 nM and maximal inhibition of 61.2% at 100 nM. Whereas hydrocortisone and triamcinolone acetonide were less active, with half-maximal inhibitory effects at 10 and 25 nM respectively, albeit with similar maximal potencies to dexamethasone. On the other hand, prednisolone and methyl-prednisolone had similar half-maximal inhibitory concentations to dexamethasone (1 and 5 nM respectively) but with different degrees of maximal inhibition of proliferation (40.6% at 100 nM for prednisolone and 76.3% at 100 nM for methyl-prednisolone). Both beclomethasone and beclomethasone diproprionate were more potent than dexamethasone with maximal inhibition of 70 and 72% at 100 nM and half-maximal inhibitory effects at 0.25 nM and 1 pM respectively. Likewise, budesonide was also more active than dexamethasone with a half-maximal inhibitory effect at 50 pM but a similar maximal potency of 66.5% at 100 nM. Mometasone and fluticasone differed markedly from the rest of this group of GCs in their degree of efficacy with maximal inhibitions at 100 nM of 87 and 82.2% respectively and half-maximal inhibitory concentrations that were assessed to be <10−12 M. Due to the lipophillic nature of mometasone and fluticasone there is a residue of GC bound to the cell culture plasticware over the 3-day culture period making an accurate measurement unreliable at high dilution. Therefore, specific values for half-maximal inhibitory concentrations have not been given. Ranking this group of GCs in order of efficacy for growth inhibition of A549 cells would be as follows:- Mometasone=fluticasone>beclomethasone diproprionate>budesonide>beclomethasone>prednisolone>methyl prednisolone = dexamethasone>hydrocortisone>triancinolone acetonide. Cortisone and prednisone are inactive GCs that lack the 11-hydoxyl substituent and are not converted to other active metabolites under these experimental conditions.
Table 1.
Concentration of glucocorticoids required to give a half-maximal inhibition of A549 cell growth, PGE2 release, arachidonic acid release, COX2 expression and cPLA2 activity. Values in parentheses (% maximum inhibition±s.e.mean)
Effects of glucocorticoids on the release of PGE2 from A549 cells
The dexamethasone inhibitory response of A549 cell growth is mediated, at least in part, by suppression of the release of growth stimulating PGE2 (Croxtall & Flower, 1992). In the experiments presented in Table 1 (column 2) we have measured the inhibitory effect of the various GCs co-incubated with IL-1β for 3 h on the stimulation of PGE2 release. Again we have found that there is a significant correlation (r=0.91, data not shown) between the degree of inhibition of PGE2 release and cell growth. Il-1β typically stimulated PGE2 release from a basal concentration of 30 ng ml−1 to approximately 200 ng ml−1. Our reference GC dexamethasone significantly (P<0.05) inhibited the release of PGE2 (within the concentration range 10 nM–10 μM) with a half-maximal inhibitory effect at 20 nM. Lower concentrations of GCs are required for inhibition of cell growth compared to inhibition of PGE2 release due to a residue of bound GC in the cell culture plasticware. Hydrocortisone and triamcinolone acetonide were less active with half-maximal inhibitory effects at 75 and 200 nM respectively. On the other hand, prednisolone and methyl-prednisolone had half-maximal inhibitory concentrations (50 and 10 nM respectively) to dexamethasone but again with differing degrees of maximal inhibition which matched their growth inhibitory profiles. Beclomethasone was of comparable activity to dexamethasone with a half-maximal inhibitory effect at 20 nM, however, beclomethasone diproprionate was more active with a half-maximal inhibitory effect at 0.75 nM. Similarly, budesonide was also more potent than the reference compound with a half-maximal inhibitory effect at 1 nM. Again mometasone and fluticasone differed significantly from the rest of this group of GCs showing high activity at low concentrations with half-maximal inhibitory effects at 10−11 M and 0.5 nM respectively. Ranking the group of GCs in order of efficacy for inhibition of PGE2 release produces a similar profile as for growth inhibition. Mometasone>fluticasone>beclomethasone diproprionate>budesonide>methyl prednisolone>dexamethasone= beclomethasone>prednisolone>hydrocortisone>triancinolone acetonide.
Effects of glucocorticoids on the release of arachidonic acid from A549 cells
Arachidonic acid is released from cellular phospholipids by PLA2 enzymes in response to a variety of extracellular stimuli and is therefore an important control point in eicosanoid biosynthesis. We have previously shown that dexamethasone suppresses the release of arachidonic acid from A549 cells by inhibiting the activation of cPLA2 (Croxtall et al., 1998). In the experiments presented in Table 1 (column 3) we have measured the inhibitory effect (following pretreatment for 3 h) of the various GCs on the release of arachidonic acid stimulated by 10 nM EGF and 50 nM thapsigargin. In these experiments EGF and thapsigargin typically stimulated arachidonic acid release by approximately 500%. Dexamethasone significantly (P<0.05) inhibited arachidonic acid release (within the concentration range 10 nM–10 μM) with half-maximal inhibitory effect at 20 nM. This is a similar profile to that reported above for inhibition of PGE2 release at similar contact times. Both hydrocortisone and triamcinolone acetonide were less active with half-maximal inhibitory effects at 75 and 200 nM respectively. Prednisolone, which, in contrast to the growth inhibition and PGE2 release assays reported above, was not significantly active within the range of concentrations tested up to 10 μM. Conversely, methyl-prednisolone appears to be more active than dexamethasone with a half-maximal inhibitory effect at 10 nM. A similar finding was also seen with beclomethasone diproprionate which failed to significantly inhibit arachidonic acid release within the concentration range tested. Whereas, beclomethasone was found to be similarly active to dexamethasone with a half-maximal inhibitory effect at 20 nM. Mometasone, fluticasone and budesonide failed to inhibit the release of arachidonic acid at any concentration tested. Ranking the GCs in this assay produces a clearly different profile of activity:- methyl-prednisolone>beclomethasone=dexamethasone>hydrocortisone>triamcinolone acetonide with the remainder inactive. These results are contrary to what might be expected but imply that different GCs have profoundly different activities in regulating arachidonic acid generation and PGE2 release.
Effects of glucocorticoids on activation of cPLA2 and expression of COX2
The agonist-activated release of arachidonic acid from A549 cells is largely mediated by the activation of cPLA2 (Croxtall et al., 1995; Choudhury et al., 2000). Furthermore, we have described the co-ordinate activation of cPLA2 and up-regulation of COX2 expression in these cells following treatment with IL-1β (Croxtall et al., 1996a). In A549 cells this activation of cPLA2 and induction of COX2 expression is inhibited by pretreatment with dexamethasone (Croxtall et al., 1996b; Newman et al., 1994). The experiments presented in Table 1 (columns 4 and 5) show the comparative effects of co-incubation of the various GCs with IL-1β for 3 h on activation of cPLA2 and induction of COX2. IL-1β typically stimulated cPLA2 activation by approximately 100%, whereas COX2 expression was undetectable in untreated cells. Dexamethasone inhibits both cPLA2 activation and induction of COX2 expression, albeit, with differing half-maximal inhibitory concentrations (20 and 100 nM respectively). Hydrocortisone and triamcinolone acetonide also inhibited both pathways in a similar manner but, with less activity. However, prednisolone did not significantly inhibit cPLA2 activation, whereas COX2 expression was inhibited with a half-maximal inhibitory effect at 50 nM. On the other hand, methyl-prednisolone inhibited COX2 expression very weakly (P<0.05, 1 and 10 μM), whereas cPLA2 activity was inhibited with a half-maximal inhibitory effect at 10 nM. This differential regulation was also seen with beclomethasone diproprionate where no inhibition of cPLA2 activity was seen in the concentration range tested. However, COX2 induction was inhibited with a half-maximal inhibitory effect at 0.75 nM. On the other hand beclomethasone inhibited cPLA2 activity with a half-maximal inhibitory effect at 20 nM, whereas, COX2 induction was only significantly (P<0.05) inhibited at concentrations of 1 and 10 μM (half-maximal inhibitory effect at 200 nM). Again, mometasone, fluticasone and budesonide were markedly different from the other GCs. All of these compounds exhibited high inhibitory activities against COX2 induction without appearing to significantly effect the activation of cPLA2. These differential activities on the regulation of cPLA2 activity and COX2 expression imply that they are functionally separable events that may be mediated by different mechanisms of action.
Which signalling pathways controlling cPLA2 activity and COX2 expression are regulated by glucocorticoids?
Inhibition of arachidonic acid release by dexamethasone, which, although dependent upon occupation of GR does not require translocation of the receptor to the nucleus (Croxtall et al., 2000). Rather, it appears that a perturbation of src-mediated pathways is involved instead. In the experiments described below we have investigated the contribution of src-mediated pathways to the GC inhibition of cPLA2 activity and COX2 expression by pre-treating the cells with the src inhibitor PP2. We have then compared this to cells pre-treated with either geldanamycin, which prevent translocation of the GR to the nucleus, or with APDC, which prevents activation of NFκB-dependent regulation of gene transcription.
In agreement with our previously published data, dexamethasone (1 μM, 3 h) inhibited both cPLA2 activity and COX2 expression. However, the inhibition of cPLA2 activity was significantly (P<0.05) reversed by pre-treatment with 1 μM PP2, whereas the inhibition of COX2 expression remained unaffected. On the other hand, pre-treatment with either 10 μM geldanamycin or 10 μM APDC did not affect inhibition of cPLA2 activity but significantly (P<0.05) reversed the inhibition of COX2 expression. Both inhibition of cPLA2 activity and COX2 expression were reversed in the presence of the GR antagonist RU486 (10 μM) confirming the requirement for GR occupation in both processes. These observations clearly show that the inhibition of cPLA2 activity and COX2 expression by dexamethasone are functionally separable events. The inhibition of cPLA2 activity does not require translocation of the GR complex to the nucleus nor the activation of NF-κB-dependent processes but nevertheless, the activation of src-mediated pathways is required. Conversely, the inhibition of COX2 expression is dependent upon nuclear translocation of GR and the participation of NF-κB-dependent mechanisms.
Both hydrocortisone and triamcinolone acetonide (1 μM, 3 h) also appeared to regulate these pathways in a similar manner. In both cases the inhibition of cPLA2 activity was significantly (P<0.05) reversed by 1 μM PP2 and unaffected by 10 μM geldanamycin or 10 μM APDC. Whereas the inhibition of COX2 expression was significantly (P<0.05) reversed by geldanamycin and APDC but unaffected by PP2. Prednisolone (1 μM, 3 h) did not significantly affect cPLA2 activity, nevertheless, the inhibition of COX2 expression was significantly (P<0.05) reversed by both geldanamycin and APDC but unaffected by PP2. On the other hand methyl-prednisolone (1 μM, 3 h) significantly (P<0.05) inhibited cPLA2 activity but only weakly inhibited COX2 expression. The inhibition of cPLA2 activity was again significantly (P<0.05) reversed by PP2 but unaffected by geldanamycin or APDC. At a concentration of 1 μM beclomethasone behaved as dexamethasone. However, beclomethasone diproprionate inhibited only COX2 expression and this was reversed by geldanamycin and APDC, whereas PP2 was without effect. Of the remaining group of GCs mometasone, fluticasone and budesonide, all of these inhibited only COX2 expression and this was significantly (P<0.05) reversed by geldanamycin and APDC, whereas, PP2 was without effect.
Discussion
The anti-inflammatory and anti-proliferative effects of physiological and synthetic GCs are thought to rely upon their ability to influence gene expression, and, a key parameter in determining efficacy is thought to be their binding affinity to GR. The data we have presented in Table 1 shows clearly the close correlation between inhibition of PGE2 release and the arrest of cell proliferation (r=0.91) for all GCs tested. However, the activities of these GCs upon PGE2 release and cell proliferation we have described do not always strictly correlate to their reported binding affinities for GR. The new generation of highly active GCs mometasone, fluticasone and budesonide do indeed exhibit much higher binding affinities than the other members of the group. However, there are several significant anomalies such as triamcinolone acetonide which has a binding affinity of 3.6 fold higher than dexamethasone yet is the least active GC of the group. Conversely, beclomethasone diproprionate which has a binding affinity of less than half that for dexamethasone yet is one of the most active members of the group (Johnson, 1998; Boobis, 1998; Wurthwein et al., 1992). The problem of GR binding affinity as a measure of efficacy is further highlighted by the data presented in Table 1 where it is clear that several GCs have significantly different half-maximal inhibitory concentrations for cPLA2 activity and COX2 expression. This issue, is perhaps, also reflected by the fact that many reported binding affinities appear to be ‘assay-dependent' resulting in significant differences in the values described. For example the IC50's of a panel of GCs in assays including inhibition of T-cell IL-5 release, T-cell proliferation, basophil histamine release and eosinophil apoptosis not only vary considerably between assay but in some cases result in a different ranking of GC potency (Johnson, 1998). Whilst hepatic clearance, serum half-life, lipophilicity and sequestration by binding globulins etc may be important factors that may account for variations in GC efficacy in vivo, it is clearly difficult to accommodate binding affinity alone as a key parameter in determining GC efficacy in the serum-free in vitro assays used in this study.
Despite our knowledge of the molecular detail of GR function it is increasingly apparent that many familiar actions of GCs cannot be adequately explained by the classical genomic mechanism outlined in the Introduction. In particular, it seems that some GC actions are more rapid than could be explained by a protein synthesis-dependent mechanism. For example, the rapid response of patients with acute adrenal insufficiency (Merry et al., 1994) and the rapid inhibition of ACTH or prolactin release (Buckingham, 1996) are both evident within minutes following GC administration. Similarly, increases in inositol tri-phosphate production (Steiner et al., 1988) and prolonged growth arrest of cells in culture (Kawai et al., 1998) have been shown following only a brief exposure to GCs. Therefore this has lead investigators to postulate that GCs can influence cellular events directly – without invoking transcriptional and/or translational processes. Buttgereit et al. (1998) has extended this concept further and proposed a ‘modular' mechanism to GC action which is both concentration- and time-dependent. In this hypothesis genomic processes are activated by low GC concentrations (>10−12 M) and require at least 30 min to work. Whereas, non-genomic receptor-mediated processes require concentrations of GC >10−9 M and occur within minutes (Buttgereit et al., 1998). So far, the relative importance of these processes in determining GC efficacy has not been determined. However, a ‘two-concentration' model would be congruent with our findings. Intriguingly, low doses of mometasone have been shown to bind to monomeric forms of GR and ellicit selective effects upon histone acetylation whereas higher doses bind to dimeric forms of the receptor and ellicit gene expression events (Barnes, 2001).
Early indications to provide direct evidence for the concept of non-genomic, GR-mediated actions of GCs first came from in vitro cell culture studies. Brief treatment (mins) of cultured human endometrial cells with GCs resulted in a rapid polymerization and stabilization of the actin cytoskeleton (Koukouritaki et al., 1997). This process did not require de novo protein synthesis, but rather, a rapid activation of PTK's was involved instead (Koukouritaki et al., 1999). More specifically, it has also been shown that rapid inactivation of JNK signalling pathways by GCs (Caelles et al., 1997) may account for antagonism of AP-1 function by activated steroid receptors. How this might happen remains an open question, however, the demonstration of an interaction between GR and 14-3-3 and Raf-1 may provide a mechanistic solution to this (Widen et al., 2000).
Exposure to dexamethasone significantly arrests A549 human lung epithelial cell growth by inhibiting the release of PGE2, an autocrine regulator of proliferation in these cells (Croxtall & Flower, 1992). There are two significant observations connected to this. Firstly, the continued presence of dexamethasone in the culture media is not required to inhibit cell growth since a brief exposure (and subsequent withdrawal) is almost as effective. Secondly, it is also apparent that dexamethasone rapidly modulates cytokine activation of arachidonic acid (and hence PGE2) release by inhibiting key components in the signal transduction pathway leading to the control of MAPK and cPLA2 activity (Croxtall et al., 2000). This process occurs within minutes and is reversed by the GR antagonist RU486 but not by inhibitors of protein synthesis (Croxtall et al., 2000). Dexamethasone causes an immediate increase in the phosphorylation of annexin 1 (ANXA1) which shares homologous domains to the signal recruitment factor Grb2 (Croxtall et al., 1998; 2000). In its phosphorylated form it appears that ANXA1 can displace Grb2 from growth factor receptor signalling complexes and thereby block the transducing signal leading to activation of JNK1, MAPK's and cPLA2 (Croxtall et al., 2000). In A549 cells pre-treated with either the benzoquinone ansamycin geldanamycin, which selectively binds hsp90 and prevents nuclear translocation of GR, the dexamethasone-induced inhibition of cPLA2 is completely unaffected (Croxtall et al., 2000). However, this rapid signalling effect of dexamethasone is reversed by pre-treatment with the src inhibitor PP2. We have now shown in this report that rapid effects of dexamethasone upon arachidonic acid release and cPLA2 activity are also unaffected by pre-treatment with the NF-κB inhibitor APDC. Conversely, what might be regarded as ‘classical genomic' effects of dexamethasone, the suppression of COX2 expression, are inhibited by geldanamycin and APDC but not PP2.
These observations perhaps become more significant when the differential effects of other GCs upon cPLA2 activity and COX2 expression are also taken into consideration. The data presented in Table 1 shows clearly the distinct activities of the GCs tested and we have arbitrarily assigned these into three groups. Firstly, the members of group A comprise the new generation of GCs, mometasone, fluticasone and budesonide and also beclomethasone diproprionate and prednisolone, and have no significant effect on arachidonic acid release and cPLA2 activity. However, they retain a profound inhibitory activity upon COX2 expression (mometasone>fluticasone>beclomethasone diproprionate>budesonide>prednisolone) which presumably accounts for their growth inhibitory effects. Secondly, group B, comprises methyl-prednisolone which, in complete contrast to group A, inhibits only cPLA2 activity without significantly inhibiting COX2 expression. Finally, in group C there is beclomethasone, dexamethasone, hydrocortisone and triamcinolone acetonide which inhibit both cPLA2 activity and COX2 expression. However, even within this group beclomethasone and dexamethasone express a differential effect upon cPLA2 activity and COX2 expression that appears to be concentration-dependent.
These differences in activity for each group are reflected in apparent differences in mechanism of action as reported in the results section. The effects of GCs from group A, which only inhibit COX2 expression, are reversed in the presence of geldanamycin and NF-κB but unaffected by PP2. Whereas for group B, the cPLA2-inhibitory action is reversed by PP2 but unaffected by geldanamycin and NF-κB. Even in group C where both cPLA2 activity and COX2 expression are inhibited, these pathways are still mechanistically distinct. These observations would appear to support the notion that the genomic versus non-genomic actions of GCs are not merely concentration-dependent, but that they are also mediated via distinct cellular pathways following GR activation. This perhaps gives some credence to the proposed modular mechanism of action (Buttgereit et al., 1998) where it is possible that, for some GCs, sub-threshold concentrations of hormone would activate only genomic pathways leaving signalling pathways unactivated or vice versa.
The ability of various GCs to differentially influence transactivation, NF-κB transrepression and apoptosis has been described in a study by Hofmann et al. (1998) where significant differences were found. Prednisolone and hydrocortisone were significantly less effective than dexamethasone in activating MMTV-luciferase reporter constructs and inducing apoptosis in CEM C7 cells. However, no significant differences were observed in the ability of these GCs to regulate NF-κB transrepression, rather, it appears that IκB-α degradation was instead affected. The authors commented on the structural similarities of modifications at C16 and C17 of the active GCs. In our study mometasone, fluticasone, budesonide and beclomethasone diproprionate were not only the most active GCs, but also formed a distinct group (A) in terms of their mechanism of action. It may not be coincidental, but these GCs all have significant modifications at their C16 and C17 positions. The implication of these findings is, that, changes in key structural determinants of GCs may elicit differential functional interactions of GR leading to either classical genomic or rapid non-genomic effects, or both. Whether GCs are able to differentially influence GR structure in a manner analagous to that which has already been established for oestrogen antagonists upon ER (Brzozowski et al., 1997) remains to be determined.
The data presented in this study appears to support the concept that the inhibitory action of GCs upon genomic versus non-genomic mechanisms as evidenced by changes in cPLA2 activity and COX2 expression are indeed functionally separate events. Furthermore, it is also apparent that many GCs have widely differing activities in these two processes. These differences between GCs may have important clinical implications. For example, in asthma therapy although inhaled GCs reduce clinical symptoms, there are, in some cases, further beneficial effects when used in combination with either long-acting β2-sympathicomimetics (Bateman et al., 1998) or leukotriene receptor antagonists (LTRA) (Nayak et al., 1998). It may be postulated that the clinical benefits of such drug combinations may depend on the choice of inhaled GC. Adding a LTRA to fluticasone, mometasone or budesonide is theoretically justified as none of these GCs inhibit AA release. However, it might be expected that adding a LTRA to methyl-prednisolone would be of less value since as this GC suppresses AA release, leukotriene generation would in any case be reduced.
For the new generation of GCs mometasone, fluticasone and budesonide the therapeutic benefits are more widely acknowledged. This has been attributed to higher binding affinities for GR and reduced systemic effects (Barnes et al., 1998). However, the lack of any detectable effect upon arachidonic acid release reported here may also be another important factor. On the other hand, the therapeutic benefits of methyl-prednisolone, which we and others (Buttgereit et al., 1997) have shown, operates through a signalling-dependent mechanism, remain to be fully evaluated, although, its neuroprotective effects have been known for some time. This was originally attributed to free-radical scavenging activity of high doses of this steroid (Hall, 1992). However, such an effect could not explain our observations since low doses of methyl-prednisolone elicit a response and they are reversed by RU486. Clearly, a more detailed insight into the molecular mechanisms of action of GCs may enable more selective therapeutic strategies to be employed.
Acknowledgments
J.D. Croxtall, Q. Choudhury and R.J. Flower are supported by The Wellcome Trust, P.Th.W. van Hal is supported by The Niels Stensen Foundation and Stichting Astmabestrijding.
Abbreviations
- AA
arachidonic acid
- APDC
pyrrolidinedithiocarbamate
- COX
cyclo-oxygenase
- EGF
epidermal growth factor
- GA
geldanamycin
- GC
glucocorticoid
- GR
glucocorticoid receptor
- GRE
glucocorticoid response element
- hsp
heat shock protein
- PG
prostaglandin
- PP2
4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine
- RU 486
glucocorticoid receptor antagonist
References
- BARNES P.J. Therapeutic strategies for allergic diseases. Nature. 1999;402 Suppl.:B31–B38. doi: 10.1038/35037026. [DOI] [PubMed] [Google Scholar]
- BARNES P.J. Optimizing the anti-inflammatory effects of corticosteroids. Eur. Respir. Rev. 2001;11:7815–7822. [Google Scholar]
- BARNES P., PEDERSEN S., BUSSE W.W. Efficacy and safety of inhaled corticosteroids: an update. Am. J. Respir. Crit. Care Med. 1998;157:S1–S53. doi: 10.1164/ajrccm.157.3.157315. [DOI] [PubMed] [Google Scholar]
- BATEMAN E.D., BRITTON M., CARRILLO J., ALMEIDA J., WIXON C. Salmeterol/fluticasone combination inhaler. A new, effective and well-tolerated treatment for asthma. Clin. Drug Invest. 1998;16:193–201. doi: 10.2165/00044011-199816030-00003. [DOI] [PubMed] [Google Scholar]
- BEATO M., HERRLICH P., SCHUTZ G. Steroid hormones receptors: many actors in search of a plot. Cell. 1995;83:851–857. doi: 10.1016/0092-8674(95)90201-5. [DOI] [PubMed] [Google Scholar]
- BEATO M., TRUSS M., CHAVEZ S. Control of transcription by steroid hormones. Ann N.Y. Acad. Sci. 1996;784:93–123. doi: 10.1111/j.1749-6632.1996.tb16231.x. [DOI] [PubMed] [Google Scholar]
- BRZOZOWSKI A.M., PIKE A.C.W., DAUTER Z., HUBBARD R.E., BONN T., ENGSTROM O., OHMAN L., GREENE G.L., GUSTAFSSON J.-A., CARLQUIST M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- BOOBIS A.R. Comparitive physiochemical and pharmokinetic profiles of inhaled beclomethasone diproprionate and budesonide. Respir. Med. 1998;92 Suppl. B:2–6. doi: 10.1016/s0954-6111(98)90434-6. [DOI] [PubMed] [Google Scholar]
- BUCKINGHAM J.C. Stress and the neuroendocrine-immune axis: the pivotal role of glucocorticoids and lipocortin 1. Br. J. Pharmacol. 1996;118:1–19. doi: 10.1111/j.1476-5381.1996.tb15360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUTTGEREIT F., KRAUSS S., BRAND M.D. Methylprednisolone inhibits uptake of Ca2+ and Na+ into concanavalin A-stimulated thymocytes. Biochem. J. 1997;326:329–332. doi: 10.1042/bj3260329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUTTGEREIT F., WEHLING M., BURMESTER G.-R. A new hypothesis of modular glucocorticoid actions. Arthritis Rheum. 1998;41:761–767. doi: 10.1002/1529-0131(199805)41:5<761::AID-ART2>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- CAELLES C., GONZALEZ-SANCHO J.M., MONOZ A. Nuclear hormone receptor antagonism with AP-1 by inhibition of the JNK pathway. Gene Dev. 1997;11:3351–3364. doi: 10.1101/gad.11.24.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOUDHURY Q.G., MCKAY D.T., FLOWER R.J., CROXTALL J.D. Investigation into the involvement of phospholipases A2 and MAP kinases in modulation of AA release and cell growth in A549 cells. Br. J. Pharmacol. 2000;131:255–265. doi: 10.1038/sj.bjp.0703573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CROXTALL J.D., CHOUDHURY Q., FLOWER R.J. Inhibitory effect of peptides derived from the N-terminus of lipocortin 1 on arachidonic acid release and proliferation in the A549 cell line: identification of E-Q-E-Y-V as a crucial component. Br. J. Pharmacol. 1998;123:975–983. doi: 10.1038/sj.bjp.0701679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CROXTALL J.D., CHOUDHURY Q., FLOWER R.J. Glucocorticoids act within minutes to inhibit recruitment of signalling factors to activated EGF receptors through a receptor-dependent, transcription-independent mechanism. Br. J. Pharmacol. 2000;130:289–298. doi: 10.1038/sj.bjp.0703272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CROXTALL J.D., FLOWER R.J. Lipocortin 1 mediates dexamethasone-induced inhibition of A549 cell growth and eicosanoid release. P.N.A.S. (USA) 1992;89:3571–3575. doi: 10.1073/pnas.89.8.3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CROXTALL J.D., NEWMAN S.P., CHOUDHURY Q., FLOWER R.J. The concerted regulation of cPLA2, COX2, and Lipocortin 1 expression by IL-1β in A549 cells. Biochem. Biophys. Res. Comm. 1996a;220:491–495. doi: 10.1006/bbrc.1996.0432. [DOI] [PubMed] [Google Scholar]
- CROXTALL J.D., CHOUDHURY Q., NEWMAN S., FLOWER R.J. Lipocortin 1 and the control of cPLA2 activity in A549 cells. Biochem. Pharmacol. 1996b;52:351–356. doi: 10.1016/0006-2952(95)02442-5. [DOI] [PubMed] [Google Scholar]
- CROXTALL J.D., CHOUDHURY Q., TOKUMOTO H., FLOWER R.J. Lipocortin-1 and the control of arachidonic acid release in cell signalling. Biochem. Pharmacol. 1995;50:465–474. doi: 10.1016/0006-2952(95)00156-t. [DOI] [PubMed] [Google Scholar]
- DE WAAL R.M.W. The anti-inflammatory activity of glucocorticoids. Mol. Biol. Rep. 1994;19:81–88. doi: 10.1007/BF00997151. [DOI] [PubMed] [Google Scholar]
- GUSTAFSSON J.-A., CARLSTEDT-DUKE J., POELLINGER L., OKRET S., WIKSTROM A.-C., BRONNEGARD M., GILLNER M., DONG Y., FUXE K., CINTRA A., HARFSTRAND A., AGNATI L. Biochemistry, molecular biology, and physiology of the glucocorticoid receptor. Endocrine Rev. 1987;8:185–234. doi: 10.1210/edrv-8-2-185. [DOI] [PubMed] [Google Scholar]
- HALL E.D. The neuroprotective pharmacology of methylprednisolone. Ann. Neurol. 1992;1:201–203. doi: 10.3171/jns.1992.76.1.0013. [DOI] [PubMed] [Google Scholar]
- HECK S., KULLMANN M., GAST A., PONTA H., RAHMSDORF H.J., HERRLICH P., CATO A.C.B. A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. EMBO J. 1994;13:4087–4095. doi: 10.1002/j.1460-2075.1994.tb06726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HENCH P., KENDALL E.C., SLOCUMB C.H., POLLEY H.F. The effects of a hormone of the adrenal cortex and of pituitary adrenocorticotropic hormone on rheumatoid arthritis. Proc. Mayo. Clin. 1949;24:181–197. [PubMed] [Google Scholar]
- HOFMANN T.G., HEHNER S.P., BACHER S., DROGE W., SCHMITZ M.L. Various glucocorticoids differ in their ability to induce gene expression, apoptosis and to repress NF-κB-dependent transcription. FEBS Lett. 1998;441:441–446. doi: 10.1016/s0014-5793(98)01609-3. [DOI] [PubMed] [Google Scholar]
- JOHNSON M. Development of fluticasone proprionate and comparison with other inhaled corticosteroids. J. Allergy Clin. Immunol. 1998;101:S434–S439. doi: 10.1016/s0091-6749(98)70155-1. [DOI] [PubMed] [Google Scholar]
- KAWAI Y., HAYASHI T., EGUCHI K., ASAZUMA K., MASAMURA K., IWAMURO A., TAKANO Y., TADA H., MATSUKAWA S., MIYAMORI I. Effects of brief exposure on growth of vascular smooth muscle cell in culture. Biochem. Biophys. Res. Commun. 1998;245:493–496. doi: 10.1006/bbrc.1998.8462. [DOI] [PubMed] [Google Scholar]
- KIMBERLY R.P. Mechanism of action, dosage schedules, and side effects of steroid therapy. Curr. Opin. Rheumatol. 1991;3:373–379. doi: 10.1097/00002281-199106000-00008. [DOI] [PubMed] [Google Scholar]
- KOUKOURITAKI S.B., GRAVANIS A., STOURNARAS C. Tyrosine phosphorylation of focal adhesion kinase and paxillin regulates the signaling mechanism of the rapid nongenomic mechanism of dexamethasone on actin cytoskeleton. Mol. Med. 1999;5:731–742. [PMC free article] [PubMed] [Google Scholar]
- KOUKOURITAKI S.B., MARGIORIS A.N., GRAVANIS A., HARTIG R., STOURNARAS C. Dexamethasone induces rapid actin assembly in human endometrial cells without affecting its synthesis. J. Cell. Biochem. 1997;65:492–500. doi: 10.1002/(sici)1097-4644(19970615)65:4<492::aid-jcb5>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- MERKULOVA T.I., MERKULOV V.M., MITINA R.L. Glucocorticoid regulation mechanisms and glucocorticoid-controlled gene regulatory regions: Description in the TRRD database. Mol. Biol. 1997;31:605–615. [PubMed] [Google Scholar]
- MERRY W.H., CAPLAN R.H., WICKUS G.G., REYNERTSON R.H., KISKEN W.A., COGBILL T.H., LANDCASPER J. Post operative acute adrenal failure caused by transient corticotropin deficiency. Surgery. 1994;116:1095–1100. [PubMed] [Google Scholar]
- NATHAN R.A., NAYAK A.S., GRAFT D.F., LAWRENCE M., PICONE F.J., AHMED T., WOLFE J., VANDERWALKER M.L., NOLOP K.B, HARRISON J.E. Mometasone furoate: efficacy and safety in moderate asthma compared with beclomethasone diproprionate. Ann. Allergy Asthma Immunol. 2001;86:203–210. doi: 10.1016/S1081-1206(10)62692-0. [DOI] [PubMed] [Google Scholar]
- NAYAK A.S., ANDERSON P., CHAROUS B.L., WILLIAMS K., SIMONSON S. Equivalence of adding zafirlukast versus double-dose inhaled corticosteroids in asthmatic patients symptomatic on low-dose inhaled corticosteroids. J. Allergy Clin. Immunol. 1998;101:S233. [Google Scholar]
- NEWMAN S.P., FLOWER R.J., CROXTALL J.D. Dexamethasone suppression of IL-1β induced cyclooxygenase 2 expression is not mediated by lipocortin-1 in A549 cells. Biochem. Biophys. Res. Commun. 1994;202:931–939. doi: 10.1006/bbrc.1994.2019. [DOI] [PubMed] [Google Scholar]
- PAUL-CLARK M., DEL SOLDARTO P., FIORUCCI S., FLOWER R.J., PERRETTI M. 21-NO-prednisolone is a novel nitric oxide-releasing derivative of prednisolone with enhanced anti-inflammatory properties. Br. J. Pharmacol. 2000;131:1345–1354. doi: 10.1038/sj.bjp.0703704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRATT W.B., SILVERSTEIN A.M., GALIGNIANA M.D. A model for the cytoplasmic trafficking of signalling proteins involving the hsp90-binding immunophilins and p50cdc37. Cell. Signal. 1999;11:839–851. doi: 10.1016/s0898-6568(99)00064-9. [DOI] [PubMed] [Google Scholar]
- REICHARDT H.M., KAESTNER K.H., TUCKERMANN J., KRETZ O., GASS P., SCHMID W., HERRLICH P., ANGEL P., SCHUTZ G. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. 1998;93:531–541. doi: 10.1016/s0092-8674(00)81183-6. [DOI] [PubMed] [Google Scholar]
- SANCHEZ E.R., HIRST M., SCHERRER L.C., TANG H.-Y., WELSH M.J., HARMON J.M., SIMONS S.S., JR, RINGOLD G.M., PRATT W.B. Hormone-free mouse glucocorticoid receptors overexpressed in chinese hamster ovary cells are localized to the nucleus and are associated with both hsp70 and hsp90. J. Biol. Chem. 1990;265:20123–20130. [PubMed] [Google Scholar]
- STEINER A., VOGT E., LOCHER R., VETTER W. Stimulation of phosphoinositide signalling system as a possible mechanism for glucocorticoid action in blood pressure control. J. Hypertens. Suppl. 1988;6:S366–S368. doi: 10.1097/00004872-198812040-00114. [DOI] [PubMed] [Google Scholar]
- VANE J.R. The mode of action of asprin-like drugs. Agents Actions. 1978;8:430–431. doi: 10.1007/BF01968671. [DOI] [PubMed] [Google Scholar]
- WIDEN C., ZILLIACUS J., GUSTAFSSON J.-A., WIKSTROM A.-C. Glucocorticoid receptor interaction with 14-3-3 and Raf-1, a proposed mechanism for cross-talk of two signal transduction pathways. J. Biol. Chem. 2000;275:39296–39301. doi: 10.1074/jbc.M006943200. [DOI] [PubMed] [Google Scholar]
- WURTHWEIN G., REHDER S., ROHDEWALD P. Lipophilicity and receptor affinity of glucocorticoids. Pharm. Ztg. Wiss. 1992;5:161–167. [Google Scholar]