

Abstract
In this study we have determined the different signalling pathways involved in adenosine A1-receptor (A1-receptor)-dependent inhibition of contractility in rat isolated atria.
N-cyclopentyladenosine (CPA) stimulation of A1-receptor exerts: negative inotropic response, inositol phosphates accumulation, stimulation of nitric oxide synthase (NOS), increased production of nitric oxide (NO) and cyclic GMP.
Inhibitors of phospholipase C (PLC), protein kinase C (PKC), calcium/calmodulin, NOS and guanylate cyclase shifted the dose-response curve of CPA on contractility to the right. Those inhibitors also attenuated the A1-receptor-dependent increase in cyclic GMP and activation of NOS.
These results suggest that CPA activation of A1-receptors exerts a negative inotropic effect associated with increased production of nitric oxide and cyclic GMP. The mechanism appears to occur secondarily to stimulation of phosphoinositide turnover via PLC activation. This, in turn, triggers cascade reactions involving calcium/calmodulin and PKC, leading to activation of NOS and soluble guanylate cyclase.
Keywords: Heart contractility, A1 receptors, nitric oxide synthase, cyclic GMP, phosphoinositides turnover, binding assay
Introduction
Adenosine is recognized as an important regulator of myocardial function providing myocardial protection during time of stress. Studies on paced rat left atria have demonstrated that adenosine compounds caused a dual inotropic effect: first a rapid decrease in contractility and second an increase in contractile tension. The negative inotropism was mediated by direct stimulation on A1-adenosine receptors (A1) and the positive inotropism by activation on A2-adenosine receptors (A2) (Froldi et al., 1994). Adenosine effects on myocardium are mediate predominantly by the type A1 receptor and the effect were demonstrated in different mammalian preparations including human isolated cardiac preparations (de jong et al., 2000). An indirect anti ß-adrenergic effect of A1 receptor activation on guinea-pig ventricular myocytes was also showed (Zhang et al., 2001).
Studies on signalling events coupled to adenosine receptor subtypes has revealed differences in the abilities to modulate many different signal transduction pathways. The signal transduction process subsequent to agonist binding to A1 receptors include the pertussis-toxin sensitive G-protein (Endoh et al., 1993) that mediate the decrement of cyclic AMP production (Stein et al., 1994) while A2 receptors increase intracellular levels of cyclic AMP via a pertussis-toxin insensitive Gs protein (Kitakaze et al., 1993; Stein et al., 1993; 1994). Neither the cyclic AMP elevating A2 receptors (Stein et al., 1993; 1994) nor the cyclic AMP reducing A1 receptors (Endoh et al., 1993) appears to be associated with changes in the direct inotropic effect of the specific receptor agonists. However, the anti ß-adrenergic effect of A1 receptor stimulation is linked to a pertussis-toxin sensitive Gi protein (Zhang et al., 2001).
Thus, the signal transduction mechanism that mediate the action of A1 receptor agonists on heart contractility have not been clearly defined. It is possible that A1 receptor was coupled to more than one G protein regulating signal transduction pathways.
It is demonstrated that A1 receptor activation decrease the basal influx of calcium acting on the L-type calcium channel current, but simultaneously elevating intracellular calcium concentration (Eckert et al., 1993). These responses were not sensitive to pertussis-toxin, but were reduced by guanosine-diphosphate- beta sulphate and by intracellular calcium release blockers; whereas intracellularly applied inositol-1,4,5-tris-phosphate (IP3) mimicked the A1 receptor activation (Eckert et al., 1993). These findings suggest a non-sensitive pertussis toxin-A1 receptor dependent activation of cyclic GMP synthesis with increase in intracellular calcium.
Among the mechanisms involved in the activation of receptor coupled to G proteins that activate cyclic GMP synthesis, an increase intracellular calcium are those mediated by phospholipase C (PLC) activation followed by generation of IP3, which releases calcium from intracellular calcium stores, that in turn triggers cascade reactions involving calcium/calmodulin-dependent nitric oxide synthase (NOS) that leads to activation of soluble guanylate cyclase (Sterin-Borda et al., 1995).
It has been reported that myocytes produced endogenous nitric oxide (NO) and some of the physiological effect of NO appear to be mediated by activation of guanylate cyclase (Balligand et al., 1993; Sterin-Borda et al., 1995). Moreover, A1 receptor activation and NO release from preconditioned heart have been implicated in protecting the myocardium from subsequent ischemic events (Liu et al., 1991; Vegh et al., 1992).
The aim of the present study was to determine the different signalling events involved in the A1 receptor dependent inhibition of contractility in rat isolated atria. Therefore, we investigated whether (i) CPA-stimulation of A1 receptor excerted negative inotropic response, stimulation of phosphoinositide (PI) turnover, activation of NOS and increase production of NO and cyclic GMP; (ii) if there is an association between PLC, PKC and calcium/calmodulin system with NOS activation, and (iii) if the negative inotropic effect of A1 receptor activation is related with NOS activation and cyclic GMP accumulation at low concentration of CPA.
Methods
Animals
Adult male Wistar strain rats (250–300 g) were used. The animals housed in standard environmental conditions were fed with a commercial pelleted diet and water ad libitum. Experimental protocol were performed following the Guide to The Care and Use of Experimental Animals (DHEW Publication, NIH 80-23).
Radioligand binding assay
Membranes were prepared as described previously (Goin et al., 1994). In brief, atria were homogenized in an Ultraturrax at 4°C in 6 vol of potassium phosphate buffer, 1 mM MgCl2, 0.25 M sucrose (buffer A) pH 7.5 supplemented with 0.1 mM phenylmethylsulphonylfluoride (PMSF), 1 mM EDTA, 5 μg ml−1 leupeptin, 1 μM bacitracin and 1 μM pepstatin A. The homogenate was centrifuged for 10 min at 3 000×g, then at 10,000×g and 40,000×g at 4°C for 15 and 90 min, respectively. The resulting pellets were resuspended in 50 mM phosphate buffer with the same protease inhibitors pH 7.5 (buffer B). Receptor ligand binding was performed as described previously (Bacman et al., 1990). Aliquots of the membrane suspension (30–50 μg protein) were incubated with different concentrations of [3H]-cyclopentyl 1,3-dipropilxantine ([3H]-DPCPX) (Tocris, Sp. Act: 98.2 Ci mmol) for 60 min at 25°C in a total volume of 150 μl of buffer B. Binding was stopped by adding 2 ml ice-cold buffer followed by rapid filtration (Whatman GF/c). Filters were rinsed with 12 ml of ice-cold buffer, transferred into vials containing 10 ml of scintillation cocktail and counted in a liquid scintillation spectrometer. Nonspecific binding was determined in the presence of 5×10−6 M DPCPX and never exceeded 10% of total binding. Radioactivity bound was lower than 10% of total counts.
Atrial preparation for contractility
Male Wistar rats were killed by decapitation. The left atria were carefully dissected from the ventricles, attached to a glass holder and immersed in a tissue bath containing Krebs–Ringer Bicarbonate (KRB) solution gassed with 5% CO2 in oxygen and maintained at pH 7.4 and 30°C. KRB solution was composed as described previously (Sterin-Borda et al., 1986). A preload tension of 750 mg was applied to the atria and tissues were allowed to equilibrate for 1 h. The initial control values for contractile tension of the isolated atria were recorded by use of a force transducer coupled to an ink writing oscillograph (Borda et al., 1984). The preparations were paced with a bipolar electrode and an SK4 Grass Stimulator. The stimuli had a duration of 2 ms and the voltage was 10% above threshold. Inotropic effects (dF/dt) were assessed by recording the maximum rate of isometric force development during electrical stimulation at a fixed frequency of 150 beats min−1. Control values (=100%) refer to the dF/dt before the addition of drugs. The absolute value for dF/dt at the end of the equilibration period (60 min) was 7.8±0.5 g s−1. Cumulative dose-response curves were obtained according to the method of van rossum (1963). A maximal effect was achieved within 5 min after each dose. Dose-response curves of CPA were done on untreated atria and those from chemically sympathectomized rats injected intravenously 24 h prior to sacrifice with 6-hydroxydopamine (6-OHDA) (16.5 mg Kg−1). In order to assess an adequate denervation, the in vitro influence of tyramine (10−6 M) and norepinephrine (NE) (10−8 M) were assayed. As expected 6-OHDA-treated atria showed supersensitive to NE and refractory to tyramine (Sterin-Borda et al., 1996a).
Measurement of total labelled inositol phosphates (IPs)
Rat atria were incubated for 120 min in 0.5 ml of KRB gassed with 5% CO2 in O2 with 1 μCi [myo-3H]-inositol ([3H]-MI) (Sp. Act. 15 Ci mmol−1) from Dupont/New England Nuclear. LiCl (10 mM) was added for inositol monophosphate accumulation, according to the technique of Berridge et al. (1982). CPA was added 30 min before the end of the incubation period and the blockers 30 min before the addition of CPA. Water-soluble IPs were extracted after 120 min incubation as previously described (Sterin-Borda et al., 1995). Determination of nitric oxide synthase activity
Nitric oxide synthase (NOS) activity was measured in atria by production of [U−14C]-citrulline from [U−14C]-arginine according to the procedure described by Bredt & Snyder (1989) for brain slices and by Sterin-Borda et al. (1995) for rat atria. Briefly, after 20 min preincubation in KRB solution, atria were transferred to 500 μl of prewarmed KRB equilibrated with 5% CO2 in O2 in the presence of [U-14C]-arginine (0.5 μCi). Appropriate concentrations of drugs were added and the atria were incubated for 20 min under 5% CO2 in O2 at 37°C. Measurement of basal NOS activity in whole atria by the above mentioned procedure was inhibited 95% in the presence of 0.5 mM NG-monomethyl-L-arginine (L-NMMA). The results (pmol g−1 tissue wet wt) obtained for whole atria were expressed as the difference between values in the absence (252±12; n=9) and in the presence (12±2, n=9) of L-NMMA.
Cyclic GMP assay
Tissues were incubated in 1 ml KRB for 30 min and CPA was added in the last 5 min. When blockers were used, they were added 25 min before the addition of CPA. After incubation, atria were homogenized in 2 ml of absolute ethanol and centrifuged at 6000×g for 15 min at 4°C. Pellets were then rehomogenized in ethanol-water (2 : 1) and supernatants collected and evaporated to dryness as indicated above. Cyclic GMP in the residue was dissolved in 400 μl of 0.05 M sodium acetate buffer pH 6.2. Aliquots of 100 μl were taken for the nucleotide determination using RIA procedure with a cyclic GMP 125I-RIA KIT from Dupont/New England Nuclear.
Drugs
N-cyclopentyladenosine (CPA), 8-cyclopentyl-1,3-dipropylxantine (DPCPX), 5′-(N-cyclopropyl)-carboxamidoadenosine (CPCA), and 3,4-dimethyl-1-propargylxanthine (DMPX) were purchased from RBI; Atropine, L-arginine, NG-monomethyl-L-arginine (L-NMMA), trifluoperazine (TFP) and staurosporine from Sigma Chemical Company; 1-6-17ß-3-methoxgestra-1,3,5 (10)-trien-17yl-aminohexyl-1-H-pirrole-2,5-dione (U-73122) from ICN Pharmaceuticals Inc; 1H-[1,2,4]-oxadiazola-[4,3-2]-quinoxaline-1-one (ODQ) from Tocris Cookson Inc. Stock solutions were freshly prepared in the corresponding buffers. The drugs were diluted in the bath to achieve the final concentration stated in the text.
Statistical analysis
Student's t-test for unpaired values was used to determine the levels of significance. When multiple comparisons were necessary, after analysis of variance, the Student-Newman-Keuls test was applied. Differences between means were considered significant if P<0.05.
Results
To assess the adenosine receptor subtypes involved in the inotropic negative effect of adenosine in rat atria, the concentration response curves of both A1 (CPA) and A2 (CPCA) agonists were performed. Table 1 shows that the potency (Kd) of CPA was greater than CPCA, while the efficacy (Emax) was similar (100% of inhibition). Moreover, a selective A1 antagonist DPCPX could antagonize the negative inotropism of CPA, but the A2 antagonist DMPX and atropine were without effects. It is note that the effect of CPA was not modified when experiments were carried out on atria from rats that had been chemically sympathectomized with 6-OHDA (normal: Kd 8.5×10−9 M; 6-OHDA 8.7×10−9 M; n=5). Results show that the negative inotropism is a direct effect triggered by adenosine receptor activation mediated preferentially by A1 receptor subtype. The binding characteristics of [3H] DPCPX in rat atria calculated by scatchard plots were Kd: 1.35±0.08 nmol l−1 and Bmax: 24.2±2.6 fmol mg−1 protein (n=9). [3H] DPCPX binding to atria membranes was a saturable process to a single binding site and reversible manner with high affinity.
Table 1.
Action of adenosine receptor antagonist on the inotropic negative effect of adenosine receptor agonists

To determine if an endogenous NO signalling system participates in the contractile action of CPA, rat isolated atria were incubated with different inhibitors of the enzymatic pathways involved in the activation of NO and cyclic GMP synthesis. As can be seen in Figure 1 upper panel, the inhibition of NOS activity by L-NMMA (2×10−6 M) (Sterin-Borda et al., 1995) or the selective inhibition of NO-sensitive guanylyl cyclase by ODQ (5×10−5 M) (Garthwaite et al., 1995) shifted to the right the dose response curve of CPA. The inhibitory action of L-NMMA on the A1 receptor agonist effect was reversed by L-arginine (Figure 1 lower panel).
Figure 1.
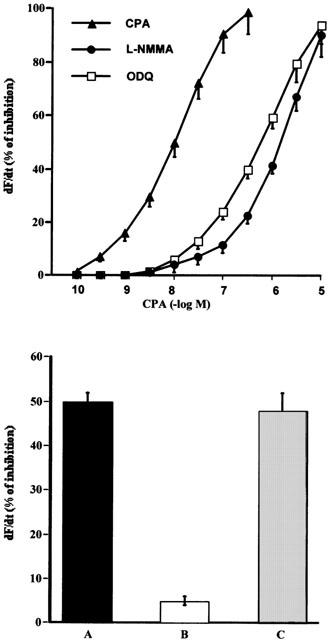
Decrease in contractility (dF/dt) of rat isolated atria by increasing concentrations of CPA (upper panel). The inhibitory action of L-NMMA (2×10−6 M) and ODQ (5×10−5 M) is also shown. Inhibition of negative inotropic effect of 1×10−8 M CPA (A) by treatment of atria with 2×10−6 M L-NMMA (B) and reversal of inhibition by L-arginine 1×10−5 M (C) (lower panel). Values represent the mean±s.e.mean of six different determinations in each group. Tissues were incubated for 30 min in presence or absence of different enzymatic inhibition and then CPA was added. Values are expressed as percentage changes calculated by comparison with the absolute values prior to the addition of CPA. No inotropic effects were observed with either inhibitors or L-arginine at the concentrations used.
To determine the participation of PLC in the negative inotropic effect of CPA, the action of U-73122 (5×10−6 M) (Smith et al., 1996) was explored. As can be seen in Figure 2, the inhibition of PLC shifted to the right the dose-response curve of the A1 receptor agonist. To elucidate which pathways gated by PI turnover could be involved in this effect, atria were incubated in the presence of an inhibitor of protein-kinase C (PKC), staurosporine (1×10−9 M) (Sterin-Borda et al., 1996b) and an inhibitor of calcium-calmodulin (TFP, 5×10−6 M) (Sterin-Borda et al., 1995). Figure 2 shows that in the presence of either staurosporine or TFP the dose-response curve of CPA was also shifted to the right.
Figure 2.
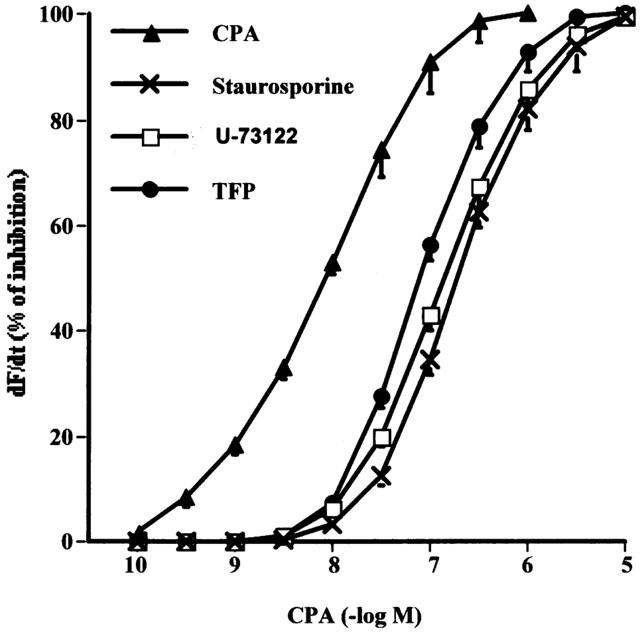
Effect of 5×10−6 M of U-73122, 5×10−6 M TFP and 1×10−9 M staurosporine on the dose-response curve of CPA upon atria dF/dt. Tissues were incubated for 30 min in presence or absence of different inhibitors and then the dose-response curves to CPA were obtained. Values represent the mean±s.e.mean seven experiments in each group. For more details see Figure 1.
To assess if A1 receptor is coupled to PI turnover in rat atria, the effect of CPA in the presence and absence of A1 receptor antagonist was investigated. As can be seen in Table 2, 1×10−8 M of CPA (at the EC50, which induced the inotropic negative effect) increased PI turnover. The specific A1 antagonist DPCPX but not DMPX (A2 antagonist), significantly inhibited the stimulatory action of CPA. As a control, U-73122 abrogated this effect, indicating the PLC activation would be implicated in A1 receptor activation of CPA upon PI hydrolysis.
Table 2.
Effect of CPA upon phosphoinositide (PI) turnover

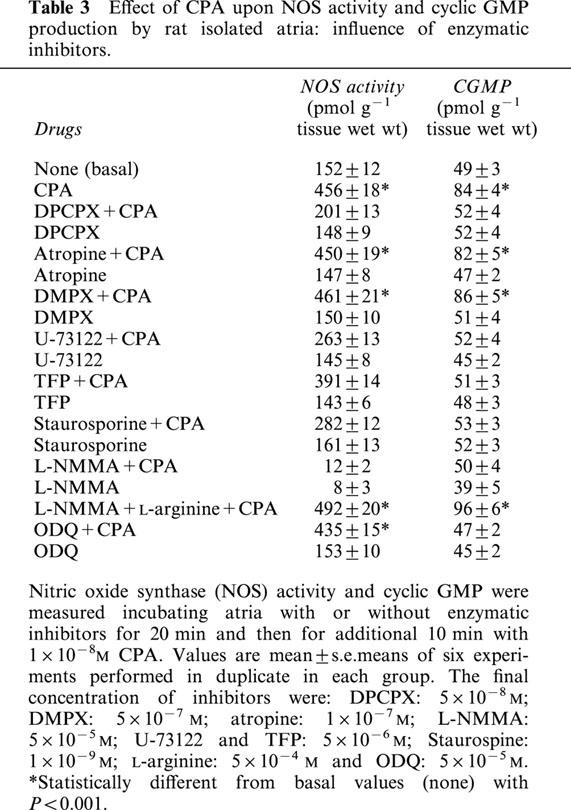
The A1 receptor agonist also increased in a concentration dependent manner the NOS activity and cyclic GMP production in rat isolated atria (Figure 3). But, while NOS activity showed a bell-shaped curve, cyclic GMP did not follow this scheme. Determination of nitrate/nitrite by Griess reaction in the incubation media followed similar profile (basal: 5.2±0.3; 10−8 M CPA: 11.2±0.4; 10−6 M CPA: 4.3±0.2 μM/100 μl, n=5). So the increment on cyclic GMP accumulation triggered by high CPA concentrations did not reflect parallel change in NOS activity and in cardiac NO release. The inhibition of NOS activity by L-NMMA (2×10−6 M) or the selective inhibition of NO-sensitive guanylyl cyclase by ODQ (5×10−5 M); blocked the action of CPA upon the enzymes (Figure 3). To verify the nature of the mechanism by which the activation of A1 receptor increased cyclic GMP synthesis and NOS activity, rat atria were incubated with several inhibitors. Table 3 shows that DPCPX, but not atropine or DMPX inhibited the stimulatory action of CPA on both NOS activity and cyclic GMP production. Also the inhibition of PLC (by U-73122), calcium-calmodulin (by TFP), and PKC (by staurosporine) attenuated the A1 receptor-dependent activation of cyclic GMP production and NOS activity. As expected, L-NMMA (5×10−5 M) inhibited 95% basal NOS activity and was reversed by L-arginine (5×10−4 M) (see Methods). On the other hand, ODQ (5×10−5 M) blunted cyclic GMP-CPA stimulation. Atropine (1×10−7 M) was without any effect. All inhibitors at the concentration used, had not effect per se on basal values of PI hydrolysis (Table 2), NOS activity and cyclic GMP levels (Table 3) or cardiac contractility (data not shown). It is important to note that on contractile experiments L-NMMA was used at 2×10−6 M, concentration that inhibited by 57% basal NOS activity (basal: 153±11; basal+L-NMMA 2×10−6 M: 86.6±0.5, n=6) but did not modify basal dF/dt values.
Figure 3.
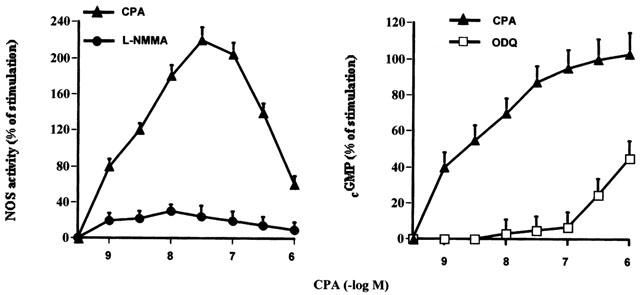
Increasing in NOS activity and cyclic GMP accumulation by increasing concentration of CPA. Inhibition of CPA effects by treatment atria with L-NMMA (2×10−6 M) or ODQ 5×10−5 M. Values are expressed as percentage of changes calculated by comparison with the absolute values prior to the addition of CPA. Values are mean±s.e.mean of eight different determination in each group. For other details see legend of Table 3.
Table 3.
Effect of CPA upon NOS activity and cyclic GMP production by rat isolated atria: influence of enzymatic inhibitors.
Discussion
The current studies give a new insight into the pathway by which NO and cyclic GMP are involved in the A1 receptor-mediated negative inotropic effect of CPA in rat isolated atria. The results point to a role for PKC and calcium mobilization in the rapid activation of NOS and the accumulation of cyclic GMP. The presence of A1 receptors on rat atria membranes which activation triggered negative inotropism on rat atria was demonstrated (de jong et al., 2000); being CPA the most potent functional agonist (Gurden et al., 1993).
Several results support these conclusions: the concentration response curve of the CPA acting on A1 receptor were shifted to the right when either NOS or soluble guanylate cyclase activity were inhibited. The mechanism seems to involve an increase of PI hydrolysis, whose intermediates would turn on a calcium/calmodulin constitutive NOS; since agents known to interfere with PLC and with calcium mobilization also cause a right-ward shift of the negative inotropic effect of CPA. The inhibition of PKC activity exerted the same effect.
In support of this, CPA activation of A1 receptor led to increased inositol phosphate, cyclic GMP accumulation and NOS activation. The CPA-induced contractile response NOS activity and cyclic GMP production were blunted by the stereospecific NOS inhibitors and by the selective inhibition of NO-sensitive guanylate cyclase at low concentrations of the A1 receptor agonist. The nature of this inhibition suggest that NO-cyclic GMP-mediate pathway predominate at low doses of CPA, while more than one signaling cascade accounts for the maximal negative inotropic effect of the A1 receptor agonist in rat atria (Cinel & Gur, 2000; Zhang et al., 2001). Martynyuk et al. (1997) have shown that the inhibitory effect of adenosine could be related to a mechanism involving the interaction with superoxide anion in addition to the NO and cyclic GMP pathway. The lack of the correlation between myocardial A1 receptor stimulation promonted increase in NOS activation and NO release with cyclic GMP accumulation could indicate that cardiac cyclic GMP content does not necesarilly reflect changes in cardiac NO (Csont et al., 1998). The physiological effect of NO are believed to be mediated by cyclic GMP, however recent studies suggested that the glyceryl trinitrate derived NO induced direct cardioprotective effect involves a cyclic GMP-independent activation of KATP in the isolated rat heart (Csont et al., 1999).
It is known that myocardial contractility is regulated by changes in the free intracellular calcium concentrations which is determined by concerted interaction of calcium influx through voltage dependent calcium channel, release of calcium from intracellular pools and calcium extrusion systems (Xu & Narayanan, 2000). Adenosine through A1 receptors decreases the L-type calcium channel but simultaneously elevate intracellular calcium concentration (Eckert et al., 1993). According to our results, the increment of intracellular calcium concentration by A1-receptor activation could be related to activation of PLC followed by the generation of IPs, which releases calcium from intracellular calcium stores. This, in turn, activate constitutive atrial NOS with cyclic GMP accumulation. Both messengers mediate the CPA-induced negative inotropic effect.
Our results showed that A1 receptor stimulation activating PKC, increased the NOS-cyclic GMP pathway. In support of this, adenosine receptor activation modulating rat myocardial PKC activity was demonstrated (Lasley et al., 1994). Also, PKC translocation had been previously proposed as a transduction mechanism of ischemic preconditioning mediated by A1 receptor (Borst et al., 1999). Moreover, preconditioning through the activation of phosphatidylinositol-3-kinase, upstream PKC and NO has been reported (Tong et al., 2000).
Adenosine has been demonstrated that interacts with adrenergic neurons at both pre and post junctional sites through altering catecholamine release and through direct alteration of catecholamine effect at the receptor level (Pelleg, 1987). Adenosine modulates ß-adrenoceptor contractile response via activation of both A1 and A2 myocardial receptors (Sawmiller et al., 1996). A1 receptor activation coupled to Gi protein attenuates ß-adrenoceptor mediated increase in myocardial contractility accounts to indirect adenosine-inhibited adenylate cyclase system (Kitakaze et al., 1993; Mudumbi et al., 1995). The fact that in 6-hydroxydopaminized rat atria persisted the negative inotropic effect of CPA observed in this paper, demonstrated a direct action on atria A1 receptor independent of endogenous adrenergic mechanism.
On the other hand, adenosine has several muscarinic cholinergic interactions. Vagal stimulation will have enhanced negative chronotropic effect (Pelleg et al., 1985). Cardiac muscarinic receptor activation mediates inotropic negative response via NOS-cyclic GMP pathway in rat isolated atria (Sterin-Borda et al., 1995). However, in this work, atropine did not modify the CPA-induced contractile inhibition, indicating that muscarinic receptor are not involved in A1 receptor induced contractile effect. A dissociation between adenosine induced bradichardia and M2 receptor mediated muscarinic response, was described in M2 receptor knockout mice (Stengel et al., 2000).
So far, adenosine A1 receptor (Liu et al., 1991), NO (Vegh et al., 1992) and cGMP (Szilvassy et al., 1994) have been identified as potential mediators of preconditioning Here we give a new insight into the pathway by which A1 receptor activation induced rapid stimulation of NOS with subsequent accumulation of cyclic GMP that may be the final mediators of protection by ischemia preconditioning.
These results suggest that CPA activation of A1-receptors exerts a negative inotropic effect associated with increased production of nitric oxide and cyclic GMP. The mechanism appears to occur secondarily to stimulation of phosphoinositide turnover via PLC activation. This, in turn, triggers cascade reactions involving calcium / calmodulin and PKC, leading to activation of NOS and soluble guanylate cyclase.
Acknowledgments
This work has been supported by Grants UBACYT from Buenos Aires University and PIP from CONICET, Argentina. We thank Mrs Elvita Vannucchi for her outstanding technical assistance.
Abbreviations
- A1-receptor
adenosine A1 receptor
- A2-receptor
adenosine A2 receptor
- NO
nitric oxide
- NOS; nitric oxide synthase; PLC
phospholipase C
- PKC
protein kinase C
- 6-0HDA
6-hydroxydopamine
References
- BACMAN S.R., STERIN-BORDA L., LUSTIG L., DENDUCHIS B., BORDA E. Antilaminin IgG binds and interacts with cardiac cholinergic receptors. Can. J. Physiol. Pharmacol. 1990;68:539–544. doi: 10.1139/y90-078. [DOI] [PubMed] [Google Scholar]
- BALLIGAND J.L., KELLY R.A., MARSDEN P.A., SMITH T.W., MICHEL T. Control of cardiac muscle cell functions by an endogenous nitric oxide signaling system. Proc. Natl. Acad. Sci. U.S.A. 1993;90:347–351. doi: 10.1073/pnas.90.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERRIDGE M.J., DOWNES C.P., HAULEY M.R. Lithium amplifies agonist-dependent phosphatidylinositol responses in brain and salivery glands. Biochem. J. 1982;206:587–595. doi: 10.1042/bj2060587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BORDA E.S., PASCUAL J., COSSIO P.R., DE LA VEGA M., ARANA R.P., STERIN-BORDA L. A circulating IgG in Chagas' disease which binds to β adrenoreceptors of myocardium and modulates their activity. Clin. Exp. Immunol. 1984;57:679–686. [PMC free article] [PubMed] [Google Scholar]
- BORST M.M., SIMONIS G., ROTHELE J., GERLACH E., MARQUETANT R., STRASSER R.H. Blockade of A1 adenosine receptors prevents the ischaemia-induced sensitisation of adenylyl cyclase: evidence for a protein kinase C-mediated pathway. Basic Res. Cardiol. 1999;94:472–480. doi: 10.1007/s003950050163. [DOI] [PubMed] [Google Scholar]
- BREDT D.S., SNYDER S.H. Nitric oxide mediates glutamate-linked enhancement of cyclic GMP levels in the cerebellum. Proc. Natl. Acad. Sci. U.S.A. 1989;86:9030–9033. doi: 10.1073/pnas.86.22.9030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CINEL I., GUR S. Direct inotropic effects of propofol and adenosine on rat atrial muscle: possible mechanisms. Pharmacol. Res. 2000;42:123–128. doi: 10.1006/phrs.2000.0666. [DOI] [PubMed] [Google Scholar]
- CSONT T., SZILVASSY Z., FÜLÖ P., NEDEIANU S., PALI T., TOSAKI A., DUX L., FERDINANDY P. Direct myocardial anti-hischaemic effect of GTN in both nitrate-tolerant and non-tolerant rats: a cyclic GMP-independent activation of KATP. Br. J. Pharmacol. 1999;128:1427–1434. doi: 10.1038/sj.bjp.0702929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CSONT T., PALI T., SZILVASSY Z., FERDINANDY P. Lack of correlation between myocardial nitric oxide and cyclic guanosine monophosphate content in both nitrate-tolerant and non-tolerant rat. Biochem. Pharmacol. 1998;56:1139–1144. doi: 10.1016/s0006-2952(98)00167-1. [DOI] [PubMed] [Google Scholar]
- DE JONG J.W., DE JONGE R., KEIJZER E., BRADAMONTE S. The role of adenosine in preconditioning. Pharmacol. Ther. 2000;87:141–149. doi: 10.1016/s0163-7258(00)00044-9. [DOI] [PubMed] [Google Scholar]
- ECKERT R., UTZ J., TRAUTWEIN W., MENTZER R.M., , JR Involvement of intracellular Ca2+ release mechanism in adenosine-induced cardiac Ca2+ current inhibition. Surgery. 1993;114:334–342. [PubMed] [Google Scholar]
- ENDOH M., TAKANASHI M., NOROTA I., KAWABATA Y., ASANO T. Pronounced direct inhibitory action mediated by adenosine A1 receptor and pertussis toxin-sensitive G protein on the ferret ventricular contraction. Naunyn Schmiedebergs Arch. Pharmacol. 1993;348:282–289. doi: 10.1007/BF00169157. [DOI] [PubMed] [Google Scholar]
- FROLDI G., PANDOLFO L., CHINELLATO A., RAGAZZI E., CAPARROTTA L., FASSINA G. Dual effect of ATP and UTP on rat atria: which types of receptors are involved. Naunyn Schmiedebergs Arch. Pharmacol. 1994;349:381–386. doi: 10.1007/BF00170884. [DOI] [PubMed] [Google Scholar]
- GARTHWAITE J., SOUTHAM E., BOULTON C.L., NIELSEN E.B., SCHIMIDT K., MAYER B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4] oxadiozolo [4,3-a] quinoxalin-1-one (ODQ) Mol. Pharmacol. 1995;48:184–188. [PubMed] [Google Scholar]
- GOIN J.C., BORDA E.S., PEREZ LEIROS C., STORINO R., STERIN-BORDA L. Identification of antibodies with muscarinic cholinergic activity in human Chagas disease: pathological implications. J. Auton. Nerv. Syst. 1994;47:45–52. doi: 10.1016/0165-1838(94)90064-7. [DOI] [PubMed] [Google Scholar]
- GURDEN M.F., COATES J., ELLIS F., EVANS B., FOSTER M., HORNBY E., KENNEDY I., MARTIN D.P., STRONG P., VARDEY C.J. Functional characterization of three adenosine receptor types. Br. J. Pharmacol. 1993;109:693–698. doi: 10.1111/j.1476-5381.1993.tb13629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KITAKAZE M., HORI M., KAMADA T. Role of adenosine and its interaction with alpha adrenoceptor activity in ischaemic and reperfusion injury of the myocardium. Cardiovasc. Res. 1993;27:18–27. doi: 10.1093/cvr/27.1.18. [DOI] [PubMed] [Google Scholar]
- LASLEY R.D., NOBLE M.A., PAULSEN K.L., MENTZER R.M., , JR Adenosine attenuates phorbol ester-induced negative inotropic and vasoconstrictive effects in rat hearts. Am. J. Physiol. 1994;266:H2159–H2166. doi: 10.1152/ajpheart.1994.266.6.H2159. [DOI] [PubMed] [Google Scholar]
- LIU G.S., THORNTON J., VAN WINKLE D.M., STANLEY A.W., OLSSON R.A., DOWNEY J.M. Protection against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit heart. Circulation. 1991;84:350–356. doi: 10.1161/01.cir.84.1.350. [DOI] [PubMed] [Google Scholar]
- MARTYNYUK A.E., KANE K.A., COBBE S.M., RANKIN A.C. Role of nitric oxide, cyclic GMP and superoxide in inhibition by adenosine of calcium current in rabbit atrioventricular nodal cells. Cardiov. Res. 1997;34:360–367. doi: 10.1016/s0008-6363(97)00043-6. [DOI] [PubMed] [Google Scholar]
- MUDUMBI R.V., OLSON R.V., HUBLER B.E., MONTAMAT S.C., VESTAL R.E. Age-related effects in rabbit hearts of N6-R-phenyisopropyladenosine, an adenosine A1 receptor agonist. J. Gerontol. A. Blol. Sci. Med. Sci. 1995;50:B351–357. doi: 10.1093/gerona/50a.6.b351. [DOI] [PubMed] [Google Scholar]
- PELLEG A. Cardiac electrophysiology and pharmacology of adenosine and ATP: modulation by the autonomic nervous system. J. Clin. Pharmacol. 1987;27:366–372. doi: 10.1002/j.1552-4604.1987.tb03032.x. [DOI] [PubMed] [Google Scholar]
- PELLEG A., BELHASSEN B., ILIA R., LANIADO S. Comparative electrophysiologic effects of adenosine triphosphate and adenosine in the canine heart: influence of atropine, propranolol, vagotomy, dipyridamole and aminophylline. Am. J. Cardiol. 1985;55:571–576. doi: 10.1016/0002-9149(85)90249-8. [DOI] [PubMed] [Google Scholar]
- SAWMILLER D.R., FENTON R.A., DOBSON J.G., JR Myocardial adenosine A1 and A2 receptor activities during juvenile and adult stages of development. Am. J. Physiol. 1996;271:H235–H243. doi: 10.1152/ajpheart.1996.271.1.H235. [DOI] [PubMed] [Google Scholar]
- SMITH R.J., JUSTEN J.M., MCNAB A.R., ROSENBLOOM C.L., STEELE A.N., DETMERS P.A., ANDERSON D.C., MANNING A.M. U-73122: a potent inhibitor of human polymorphonuclear neutrophil adhesion on biological surfaces and adhesion-related effector functions. J. Pharmaco. Exp. Ther. 1996;278:320–329. [PubMed] [Google Scholar]
- STEIN B., MENDE U., NEUMANN J., SCHMITZ W., SCHOLZ H. Pertussis toxin unmasks stimulatory myocardial A2-adenosine receptors on ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 1993;25:655–659. doi: 10.1006/jmcc.1993.1078. [DOI] [PubMed] [Google Scholar]
- STEIN B., SCHMITZ W., SCHOLZ H., SEELAND C. Pharmacological characterization of A2-adenosine receptors in guinea-pig ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 1994;26:403–414. doi: 10.1006/jmcc.1994.1049. [DOI] [PubMed] [Google Scholar]
- STENGEL P.W., GOMEZA J., WESS J., COHEN M.L. M(2) and M(4) receptor knockout mice: muscarinic receptor function in cardiac and smooth muscle in vitro. J. Pharmacol. Exp. Ther. 2000;292:877–885. [PubMed] [Google Scholar]
- STERIN-BORDA L., CANTORE M., PASCUAL J., BORDA E.S., COSSIO P., ARANA R., PASSERON S. Chagasic IgG binds and interacts with cardiac beta adrenoreceptor-coupled adenylate cyclase system. Int. I. Immunopharmacol. 1986;8:581–588. doi: 10.1016/0192-0561(86)90029-9. [DOI] [PubMed] [Google Scholar]
- STERIN-BORDA L., PEREZ LEIROS C., BORDA E.S., DE BRACCO M.M. Effects of IL-2 on the myocardium. Participation of the sympathetic system. J. Mol. Cell. Cardiol. 1996a;28:2457–2465. doi: 10.1006/jmcc.1996.0238. [DOI] [PubMed] [Google Scholar]
- STERIN-BORDA L., CREMASCHI G., GENARO A., VILA ECHAGUE A., GOIN J.C., BORDA E. Involvement of NO and PKC activation on chagasic antibody activity upon myocardial contractility. Mol. Cell Biochem. 1996b;160/161:75–82. doi: 10.1007/BF00240034. [DOI] [PubMed] [Google Scholar]
- STERIN-BORDA L., VILA ECHAGUE A., PEREZ LEIROS C., GENARO A., BORDA E. Endogenous nitric oxide signalling system and the cardiac muscarinic acetylcholine receptor-inotropic response. Br. J. Pharmacol. 1995;115:1525–1531. doi: 10.1111/j.1476-5381.1995.tb16646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SZILVASSY Z., FERDINANDY P., BOR P., JAKAB I., LONOVICS J., KOLTAI M. Ventricular overdrive pacing-induced anti-ischemic effect: a conscious rabbit model of preconditioning. Am. J. Physiol. 1994;266:H2033–H2041. doi: 10.1152/ajpheart.1994.266.5.H2033. [DOI] [PubMed] [Google Scholar]
- TONG H., CHEN W., STEENBERGEN C., MURPHY E. Ischemic preconditioning activates phosphatidylinositol-3-kinase upstream of protein kinase C. Circ. Res. 2000;87:309–315. doi: 10.1161/01.res.87.4.309. [DOI] [PubMed] [Google Scholar]
- VAN ROSSUM J.M. Cumulative dose-response curves. Arch. Int. Pharmacodyn. Ther. 1963;143:299–330. [PubMed] [Google Scholar]
- VEGH A., SZEKERES L., PARRATT J. Preconditioning of the ischaemic myocardium involvement of the L-arginine nitric oxide pathway. Br. J. Pharmacol. 1992;107:648–652. doi: 10.1111/j.1476-5381.1992.tb14501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XU A., NARAYANAN N. Reversible inhibition of the calcium-pumping ATPase in native cardiac sarcoplasmic reticulum by a calmodulin-binding peptide. Evidence for calmodulin-dependent regulation of the V(max) of calcium transport. J. Biol. Chem. 2000;275:4407–4416. doi: 10.1074/jbc.275.6.4407. [DOI] [PubMed] [Google Scholar]
- ZHANG Y.H., HINDE A.K., HANCOX J.C. Anti-adrenergic effect of adenosine on Na(+)-Ca(2+)exchange current recorded from guinea-pigventricular myocytes. Cell. Calcium. 2001;29:347–358. doi: 10.1054/ceca.2001.0197. [DOI] [PubMed] [Google Scholar]