

Abstract
Inhibition of cardiomyocyte-specific ATP-sensitive potassium (KATP) channels prolongs the action potential during intense ischaemia with attendant antiarrhythmic effects. However, this is accompanied by contractile depression in some models. These changes may be particularly troublesome in dilated cardiomyopathic hearts that display basal systolic dysfunction, limited energy reserve, and prolonged repolarization favouring arrhythmia. Mechanical effects of selective myocyte KATP channel blockade on basal, β-adrenergic stimulated, and ischemic responses were therefore tested in dogs with cardiac failure induced by tachypacing.
Cardiovascular function was assessed by pressure – dimension relationships in 10 conscious, chronically instrumented dogs (sonomicrometry/micromanometry), with or without cardiac failure. Cardiomyocyte KATP channels were inhibited by HMR 1098, and data obtained under basal conditions, during epinephrine infusion to raise metabolic demand, during regional ischaemia, and with combined ischaemia+epinephrine.
HMR 1098 had no effect on baseline cardiac function nor did it induce arrhythmia in normal or failing hearts. Epinephrine raised cardiac work 65% and oxygen consumption 55%, yet HMR 1098 had no functional effect in either heart condition. Regional ischaemia with or without epinephrine co-stimulation depressed regional and global function, yet both were also unaffected by HMR 1098. There was minimal arrhythmia without HMR 1098, and drug infusion did not alter this.
Thus, myocyte-KATP channels play a negligible role modulating intact in vivo cardiac contraction or arrhythmia in normal and failing heart with and without increased metabolic demand and/or regional ischaemia. This supports the feasibility of administering such agents to depressed hearts, despite underlying contractile and electrophysiologic abnormalities.
Keywords: KATP channels, HMR 1098, heart failure, ischaemia, ventricular function, contractility
Introduction
ATP-sensitive potassium channels (KATP) are widely expressed in many cell types including β-islet pancreatic, cardiac and vascular smooth muscle cells (Yokoshiki et al., 1998). In myocytes, these channels are normally closed but open in response to a marked decline in intracellular ATP (Grover & Garlid, 2000) such as with anoxia or metabolic inhibition, and this in turn can result in interstitial K+ accumulation in ischaemic myocardium (Hill & Gettes, 1980). Cardiac myocyte KATP channel opening shortens the action potential (Wilde et al., 1990) and combined with increased interstitial K+, this effect can be arrhythmogenic, leading to sudden death (Janse & Wit, 1989). KATP channel activation exacerbates ischaemia-induced ventricular fibrillation (VF), and its blockade attenuates reperfusion-triggered VF in working rat hearts (Ferdinandy et al., 1995; Wirth et al., 1999a). Blockade of these channels by gliburide reduced fibrillation, while a more recently developed myocyte-specific KATP-channel antagonist (the sulfonylthiourea HMR 1883, or its sodium salt HMR 1098) prevented fibrillation induced by 2-min coronary occlusion superimposed on exercise in conscious dogs (Billman et al., 1993; 1998). Moreover, ST segment elevation was attenuated by HMR 1883 in a porcine ischaemia model (Wirth et al., 1999b), while transgenic mice lacking the myocyte-specific KATP-channel displayed no ST-segment elevation during coronary occlusion, suggesting its key role in mediating the injury current (Li et al., 2000).
The failing heart reflects various abnormal basal substrates compared to the normal ventricle or even normal heart made acutely ischemic. In particular, the failure state is characterized by basal prolongation of the action potential related in part to a decline in key repolarizing K+ current proteins and respective currents, and to Ca2+ cycling abnormalities (Kääb et al., 1996; O'rourke et al., 1999; Tomaselli & Marban, 1999), and energetic inefficiency with limited high energy phosphate reserve (Ekelund et al., 1999; Neubauer et al., 1995). Both changes likely contribute to the increased incidence of malignant arrhythmias and sudden cardiac death with heart failure. Given these changes, one might speculate that modulation of KATP channels could play a more prominent role in failure, and that their blockade might actually be pro-arrhythmic associated with APD lengthening. Furthermore, recent studies in isolated rat heart subjected to low-flow ischaemia raised the concern of a decline in contractile performance associated with HMR 1098 treatment (Gögelein et al., 2001). Such decrement would be poorly tolerated in the failing heart. Accordingly, we performed studies in a canine tachycardia-pacing model of failure to establish the influence of selective myocyte KATP channel blockade on basal function, stimulated function (epinephrine), ischaemia response, and combined ischaemia and epinephrine. The rapid-pacing model of cardiac failure has been shown to recapitulate many of the relevant mechanical, biochemical, molecular and electrophysiologic changes observed in human heart failure, and thus served as a useful platform to test these interactions.
Methods
The protocol was approved by the Animal Care and Use Committee of the Johns Hopkins University. Ten adult mongrel dogs weighing 25 – 30 kg were used. Dilated cardiomyopathy was induced after 4 weeks of pacing (210 beats min−1×3 weeks, 250 beats min−1×1 – 2 weeks). The evolution of cardiac failure was monitored and accepted when the maximal rate of pressure rise (dP/dtmax) declined by >50% of the pre-pacing baseline, and LVEDP >20 mm Hg.
Animals were premedicated with acepromazine (0.1 mg kg−1 IM) and diphenhydramine (25 mg IM), anesthesia induced with thiopentathal (15 mg kg−1 i.v.) and maintained following endotracheal intubation with inhaled oxygen (3 L min−1) and isofluorane (1 – 2%), maintaining arterial pO2, pCO2, and pH in the physiological range. The chest was exposed by left lateral thoracotomy and the animal instrumented with a perivascular cuft occluder placed around the inferior vena cava (IVC); sonomicrometer crystals to measure left ventricular antero-posterior diameter and lateral wall shortening, an ultrasonic flow probe (Transonics, NY, U.S.A.) and balloon occluder placed around the mid circumflex artery, a coronary sinus, aortic, and right atrial catheter for blood sampling, a micromanometer (Konigsberg, CA, U.S.A.) placed in the LV cavity via an apical stab to measure LV pressure, a screw-in epicardial pacing lead at the LV apex attached to a programmable pacemaker, and atrial pacing wires. Dogs were provided 10 days for full surgical recovery prior to baseline study.
Experimental protocol
Studies were performed in conscious animals standing quietly in a sling apparatus, with LV pacing suspended at least 30 min prior to study. Data were collected under five different conditions: (1) baseline, (2) epinephrine infusion (1 – 3 μg kg−1 min−1 i.v) (3) re-baseline, 30 min after discontinuing epinephrine, (4) HMR 1098 (3 mg kg−1 i.v bolus injection followed by continuous infusion of 17 μg kg−1 min−1 i.v), and (5) co-infusion of HMR 1098 and epinephrine (same doses used). This dose of HMR 1098 yielded plasma levels of its free-acid (HMR 1883) of 6.8±1.9 μg ml−1. This is 4 – 5 times the level previously reported to inhibit ventricular fibrillation in a canine-post-infarction ischaemia model (Billman et al., 1998). We also measured and confirmed normal blood glucose with this dose of HMR 1098 (mean 5.2±0.73 mmol l−1), concordant with prior results (Billman et al., 1998).
Each experimental condition was studied in five control and five failing hearts. For each, resting haemodynamics and surface ECG were recorded. Blood withdrawn from the coronary sinus and aorta was used to assess arterial – venous oxygen difference (AVO2), and myocardial oxygen consumption (MVO2) was indexed by the product of AVO2 and circumflex coronary flow. Cardiac efficiency was indexed by the ratio of oxygen consumption (MVO2) to stroke work (SW/MVO2). Data obtained during transient IVC balloon inflation were used to generate pressure – dimension relations (P-D). For the ischaemia protocols, circumflex artery blood flow was monitored and a graded occlusion obtained to lower flow by 75% of baseline, and regional function indexed by segment length in the later wall region.
Data analysis and statistics
Haemodynamic and electrophysiologic data were digitized at 200 Hz, and stored to disk. Pressure – dimension (P-D) loops were analysed both at steady-state (average of 5 – 10 sequential cycles), and during transient load-reduction to derive P-D relationships. The latter yielded load-independent measures of cardiac contractile function. Algorithms used for this analysis have been previously reported (Senzaki et al., 2000). Electrocardiograms were obtained from two leads. Analysis of the QT interval was performed using previously reported custom designed software (Berger et al., 1997).
Data were analysed using repeated measures ANOVA, with two drug condition factors (epinephrine and HMR 1098), and a dummy variable coding for each animal. A similar analysis was performed on the change in response to epinephrine or to ischaemia, with HMR 1098 status as the primary categorical variable. Statistical significance was accepted at P<0.05.
Results
Table 1 provides results for the effect of HMR 1098 on basal cardiac function in both normal and failing hearts. Cardiac failure was easily discerned by a marked decline in systolic function, prolonged relaxation, and elevated diastolic pressures. None of these haemodynamic function variables were altered by the administration of HMR 1098. Table 2 provides summary results for the influence of HMR 1098 on epinephrine-stimulated cardiac function in both normal and failing hearts. Epinephrine increased cardiac stroke work by 65% in controls and 51% in failing hearts associated with an increase in contractile function and rise in LVEDP. HMR 1098 did not influence the responses in any of these variables, indicating that despite the higher level of energy demand, myocyte KATP channel activation was not observed. There were no significant changes in mean coronary flow or myocardial oxygen consumption due to HMR 1098, consistent with myocyte selectivity of channel blockade.
Table 1.
HMR 1098 effect on control CHF
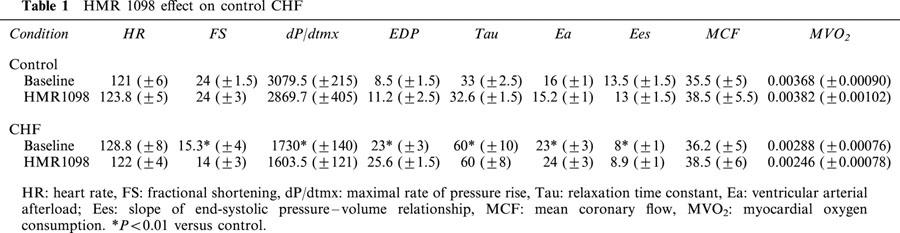
Table 2.
Delta epinephrine pre and post HMR 1098
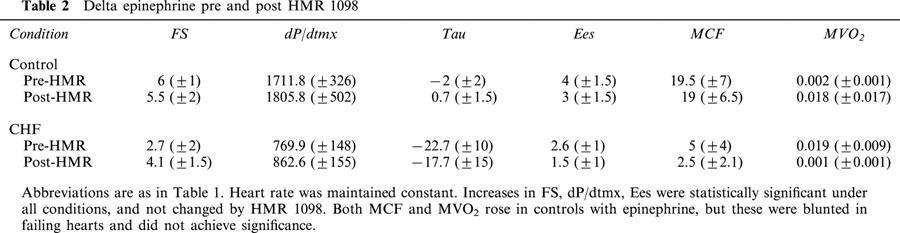
Figure 1 displays example pressure – dimension data for basal and epinephrine stimulated conditions in normal and failing hearts. These figures demonstrate hallmark features of the pacing-failure model, including chamber dilation, contractile depression, and increased steepness of the diastolic pressure – dimension relation. As demonstrated, none of these changes were further influenced by HMR 1098. Enhanced contractile response to epinephrine in both control and failing heart was demonstrated by an upward-left shift of the end-systolic pressure – dimension relation. This change was virtually identical with or without HMR 1098 treatment.
Figure 1.
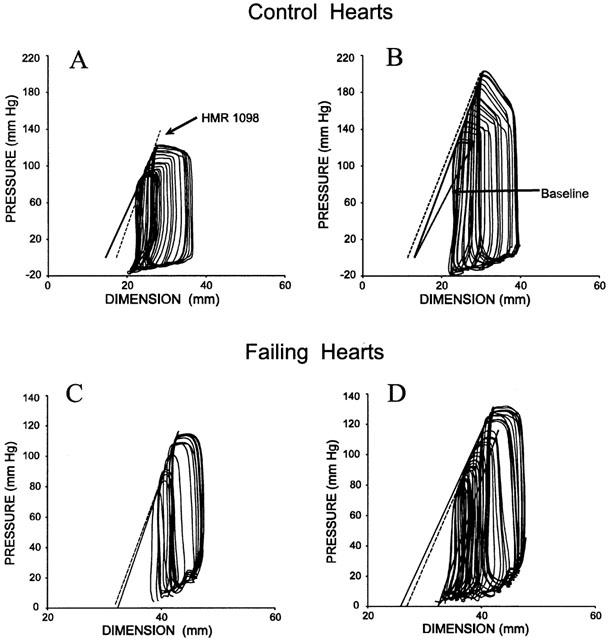
Pressure – volume loops from an example control and heart failure dog demonstrating minimal influence of HMR 1098 on either basal (A,C) or epinephrine-stimulated (B,D) cardiac function. The baseline end-systolic pressure – dimension relation(s) is shown at the upper left of each plot, and indexes chamber contractile function. The solid line relates to the loop which is in the presence of HMR 1098. Dashed lines depict the relevant comparison data (either with or without epinephrine). Lastly, in the right panels, the pre-epinephrine relations are shown (baseline) to assist in comparison.
Table 3 provides data before and after regional ischaemia. Coronary flow was reduced 50 – 75% resulting in a substantial decline in regional fractional shortening. The decline in flow was greater in failing hearts and led to regional dyskinesis. As shown in Figure 2, in failing hearts the dysfunction was quite marked, with a mean decline of fractional shortening of 80 – 100%. However, neither regional or global ischaemia responses were altered by the addition of HMR 1098.
Table 3.
Data for ischaemia under epinephrine and HMR 1098

Figure 2.
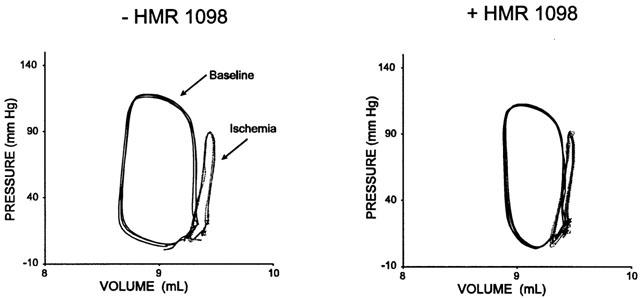
Regional myocardial function depicted as pressure-segment length loops before and after ischaemia in a failing heart. Data are shown before and after treatment with HMR 1098. The effect of ischaemia was to substantially lower fractional shortening, and this effect was unaltered by myocyte KATP channel blockade.
Under basal conditions, HMR 1098 did not alter the QT interval, a reflection of action potential duration as well as inhomogeneous repolarization. However, during ischaemia in control hearts either under basal or epinephrine stimulated conditions, HMR 1098 significantly prolonged the QT interval (235±32.4 to 257±31.1 ms, n=6; P<0.05 by Wilcoxon). In failing hearts, however, there was no analogous change in QT interval due to HMR 1098 (225±20 vs 233±15 ms, P=0.29 and Figure 3.). In the absence of KATP blockade, neither ventricular ectopy nor malignant arrhythmia (tachycardia or fibrillation) were observed in control and heart failure dogs. This was particularly striking in the failure dogs given their severe basal depression, severity of ischaemia in a dominant coronary distribution (i.e. circumflex territory), and superimposition of epinephrine-induced increase in metabolic demand. Again, KATP blockade did not elicit arrhythmia or alter surface ECG morphology with this manoeuvre in either control or failing hearts.
Figure 3.
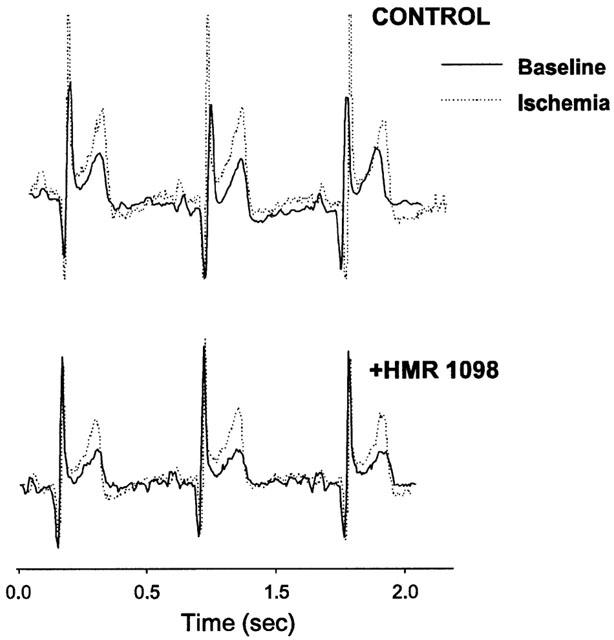
Examples of electrocardiographic recording in failing heart before and during regional ischaemia, in the presence or absence of HMR 1098. Slight ST elevation and peaked T-waves are observed with ischaemia in both conditions, but there is minimal change in the QT interval.
Discussion
Failing myocardium, including that induced by the rapid-pacing model, displays abnormal repolarization with prolonged action potential duration, reduced repolarizing potassium currents, abnormal calcium currents and sarcoplasmic reticular buffering (O'rourke et al., 1999; Kääb et al., 1996). In the whole heart, we and others have shown that this results in an arrhythmogenic heart (Pak et al., 1997), with electrophysiologic behaviour that shares features with the long-QT syndromes (Tomaselli et al., 1994; Tomaselli & Marban, 1999). Additionally, studies have shown that high energy phosphate metabolism is abnormal, particularly in the endocardium, and many have hypothesized that local supply/demand imbalance further contributes to regional electrical and mechanical dysfunction, inhomogeneity, and arrhythmia. Given these findings, it is reasonable to anticipate that KATP channels could play more of a role in the failing ventricle – particularly under conditions of increased demand.
The present study is the first to test a role of these channels in the intact failing heart. The finding that their blockade by HMR 1098 had negligible effects on contraction/relaxation presumably reflects their lack of activation and/or minimal contribution to regional or global myocardial function. This was true for basal function, but more importantly, it was also observed in hearts operating at high workload with or without superimposed regional ischaemia. While our experimental conditions were physiologically relevant and reflected substantial basal cardiodepression in failing ventricles, they nonetheless did not trigger marked ST-segment elevation or ventricular fibrillation as produced by global intense ischaemia models (Wirth et al., 1999a, 1999b). This may partially explain the lack of HMR 1098 effects. Recently, HMR 1098 was shown to depress systolic function in isolated rat heart following an ischaemia and reperfusion protocol (Gögelein et al., 2001). A similar trend was observed in intact hearts, although this was less pronounced. The current data suggests that the conscious animal with a fully intact heart is not susceptible to a similar effect – even in the presence of marked basal cardiodepression. Given concerns regarding a depressed energy state of the failing heart, these results are important and provide substantial insight regarding the level of ATP depletion required to unmask functional effects from myocyte KATP channels in this disease.
One potential cause for a limited effect of KATP channel blockade in failing hearts would be downregulation of these channels with cardiac failure. While no prior study specifically has examined myocyte KATP channel function in the pacing-induced model of heart failure, vascular KATP channels have been assessed and appear enhanced (Yamamoto et al., 2000). Furthermore, while short-term pacing (minutes) can induce a delayed pre-conditioning effect (Kis et al., 1999), there is no evidence that this leads to KATP channel downregulation, or that sustained pacing with the development of heart failure confers a similar effect. Furthermore, the present study examined changes shortly after suspending pacing (not after 24 – 48 h), and found regional dysfunction during ischaemia in CHF hearts was if anything worse than in controls. Lastly, several studies have examined KATP channel activity in human myocytes and found them to be present or even enhanced. For example, Koumi et al. (1997) reported similar KATP channel properties in normal and failing human atrial myocytes, but more rapid and larger action potential shortening associated with metabolic inhibition in failing cells. Other investigators (Kääb et al., 1999) have reported HMR 1883 inhibits KATP currents in human ventricular myocytes obtained from explanted hearts with varying levels of function. Taken together, these data do not support the notion that KATP channel downregulation likely explained the present results.
Several study limitations should be noted. Given the complexity of the study protocol – we did not assess multiple doses of HMR 1098, but rather employed a dose that had been previously reported to inhibit ventricular fibrillation and ischaemic action-potential shortening in various animal models – including those with post-infarction dysfunction (Billman et al., 1998). Still, it remains possible that cardiac failure may alter the dose-response. We also did not examine electrophysiologic behaviour in detail, as the principal focus was on global and regional mechanical function and gross chamber arrhythmogenicity.
The lack of mechanical effects by HMR 1098 under a myriad of experimental conditions, using sensitive and cardiospecific parameters, is promising in view of the potential therapeutic use of this agent in ischemic cardiomyopathy patients who are under sustained risk of ischaemia-induced arrhythmia. KATP channel activation shortens the action potential, and blockade of this channel prolongs it. DCM hearts display basal prolonged APD, and further APD lengthening, as induced by 4-AP in isolated myocytes (Kääb et al., 1996), or cesium chloride in whole heart (Pak et al., 1997), is associated with early after-depolarizations and increased arrhythmia. The lack of global arrhythmogenicity of HRM 1098 in intact failing myocardium suggests that this is unlikely to be a limitation in this setting.
Acknowledgments
We thank the technical assistance of Richard S. Tunin. This study was supported by a grant from Adventis Pharma Deutschland GmBH, and by National Institute of Health Grant P50-52307.
Abbreviations
- 4-AP
4-aminopyridine
- APD
action potential duration
- AVO2
cardiac arterial-venous oxygen difference
- CHF
congestive heart failure
- dP/dtmx
maximal rate of pressure rise
- Ea
ventricular arterial afterload
- Ees
slope of end-systolic pressure-volume relationship
- FS
fractional shortening
- HR
heart rate
- IVC
inferior vena cava
- KATP
adenosine triphosphate sensitive potassium channel
- LV
left ventricle
- LVEDP
left ventricular end-diastolic pressure
- MCF
mean coronary flow
- MVO2
myocardial oxygen consumption
- P-D
pressure-dimension
- SW
stroke work
- Tau
relaxation time constant
References
- BERGER R.D., KASPER E.K., BAUGHMAN K.L., MARBAN E., CALKINS H., TOMASELLI G.F. Beat-to-beat QT interval variability: novel evidence for repolarization lability in ischemic and nonischemic dilated cardiomyopathy. Circulation. 1997;96:1557–1565. doi: 10.1161/01.cir.96.5.1557. [DOI] [PubMed] [Google Scholar]
- BILLMAN G.E., AVENDANO C.E., HALLIWILL J.R., BURROUGHS J.M. The effects of the ATP-dependent potassium channel antagonist, glyburide, on coronary blood flow and susceptibility to ventricular fibrillation in unanesthetized dogs. J. Cardiovasc. Pharmacol. 1993;21:197–204. doi: 10.1097/00005344-199302000-00003. [DOI] [PubMed] [Google Scholar]
- BILLMAN G.E., ENGLERT H.C., SCHOLKENS B.A. HMR 1883, a novel cardioselective inhibitor of the ATP-sensitive potassium channel. Part II: effects on susceptibility to ventricular fibrillation induced by myocardial ischemia in conscious dogs. J. Pharmacol. Exp. Ther. 1998;286:1465–1473. [PubMed] [Google Scholar]
- EKELUND U.E., HARRISON R.W., SHOKEK O., THAKKAR R.N., TUNIN R.S., SENZAKI H., KASS D.A., MARBAN E., HARE J.M. Intravenous allopurinol decreases myocardial oxygen consumption and increases mechanical efficiency in dogs with pacing-induced heart failure. Circ. Res. 1999;85:437–445. doi: 10.1161/01.res.85.5.437. [DOI] [PubMed] [Google Scholar]
- FERDINANDY P., SZILVASSY Z., DROY-LEFAIX M.T., TARRADE T., KOLTAI M. KATP channel modulation in working rat hearts with coronary occlusion: effects of cromakalim, cicletanine, and glibenclamide. Cardiovasc. Res. 1995;30:781–787. [PubMed] [Google Scholar]
- GÖGELEIN H., RUETTEN H., ENGLERT H.C., BUSCH A.E. Effects of the cardioselective KATP channel blocker HMR 1098 on cardiac function in isolated perfused working rat hearts and anesthetized rats during ischemia and reperfusion. Naunyn Schmiedebergs Arch. Pharmacol. 2001;362:480–488. doi: 10.1007/s002100000391. [DOI] [PubMed] [Google Scholar]
- GROVER G.J., GARLID K.D. ATP-Sensitive potassium channels: a review of their cardioprotective pharmacology. J. Mol. Cell. Cardiol. 2000;32:677–695. doi: 10.1006/jmcc.2000.1111. [DOI] [PubMed] [Google Scholar]
- HILL J.L., GETTES L.S. Effect of acute coronary artery occlusion on local myocardial extracellular K+ activity in swine. Circulation. 1980;61:768–778. doi: 10.1161/01.cir.61.4.768. [DOI] [PubMed] [Google Scholar]
- JANSE M.J., WIT A.L. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol. Rev. 1989;69:1049–1169. doi: 10.1152/physrev.1989.69.4.1049. [DOI] [PubMed] [Google Scholar]
- KÄÄB S., NUSS B., CHIAMVIMONVAT N., O'ROURKE B., PAK P.H., KASS D.A., MARBAN E., TOMASELLI G.F. Ionic mechanism of action potential prolongation in ventricular myocytes from dogs with pacing-induced heart failure. Circ. Res. 1996;78:262–273. doi: 10.1161/01.res.78.2.262. [DOI] [PubMed] [Google Scholar]
- KÄÄB S., ZWERMANN A., BARTH M., HINTERSEER H.C., ENGLERT H.C., GÖGELEIN H., NABAUR M. Inhibition of the ATP-dependent potassium current IK(ATP) by HMR 1883 in human cardiomyocytes. Eur. Heart J. 1999;20:574. [Google Scholar]
- KIS A., VEGH A., PAPP J.G., PARRATT J.F. Repeated cardiac pacing extends the time during which canine hearts are protected against ischaemia-induced arrhythmias: role of nitric oxide. J. Mol. Cell. Cardiol. 1999;31:1229–1241. doi: 10.1006/jmcc.1999.0955. [DOI] [PubMed] [Google Scholar]
- KOUMI S.I., MARTIN R.L., SATO R. Alterations in ATP-sensitive potassium channel sensitivity to ATP in failing human hearts. Am. J. Physiol. 1997;272:H1656–H1665. doi: 10.1152/ajpheart.1997.272.4.H1656. [DOI] [PubMed] [Google Scholar]
- LI R.A., LEPPO M., MIKI T., SEINO S., MARBAN E. Molecular basis of electrocardiographic ST-segment elevation. Circ. Res. 2000;87:837–839. doi: 10.1161/01.res.87.10.837. [DOI] [PubMed] [Google Scholar]
- NEUBAUER S., HORN M., PABST T., GODDE M., LUBKE D., JILLING B., HAHN D., ERTL G. Contributions of 31P-magnetic resonance spectroscopy to the understanding of dilated heart muscle disease. Eur. Heart J. 1995;16 Suppl O:115–118. doi: 10.1093/eurheartj/16.suppl_o.115. [DOI] [PubMed] [Google Scholar]
- O'ROURKE B., KASS D.A., TOMASELLI G.F., KÄÄB S., TUNIN R., MARBAN E. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure. Circ. Res. 1999;84:562–570. doi: 10.1161/01.res.84.5.562. [DOI] [PubMed] [Google Scholar]
- PAK P.H., NUSS H.B., TUNIN R., KÄÄB S., TOMASELLI G.F., MARBAN E., KASS D.A. Repolarization abnormalities, arrhythmia, and sudden death in canine tachycardia-induced cardiomyopathy. J. Am. Coll. Cardiol. 1997;30:576–584. doi: 10.1016/s0735-1097(97)00193-9. [DOI] [PubMed] [Google Scholar]
- SENZAKI H., ISODA T., PAOLOCCI N., EKELUND U., HARE J.M., KASS D.A. Improved mechanoenergetics and cardiac rest and reserve function of in vivo failing heart by calcium sensitizer EMD-57033. Circulation. 2000;101:1040–1048. doi: 10.1161/01.cir.101.9.1040. [DOI] [PubMed] [Google Scholar]
- TOMASELLI G.F., BEUCKELMANN D.J., CALKINS H.G., BERGER R.D., KESSLER P.D., LAWRENCE J.H., KASS D., FELDMAN A.M., MARBAN E. Sudden cardiac death in heart failure. The role of abnormal repolarization. Circulation. 1994;90:2534–2539. doi: 10.1161/01.cir.90.5.2534. [DOI] [PubMed] [Google Scholar]
- TOMASELLI G.F., MARBAN E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc. Res. 1999;42:270–283. doi: 10.1016/s0008-6363(99)00017-6. [DOI] [PubMed] [Google Scholar]
- WILDE A.A., ESCANDE D., SCHUMACHER C.A., THURINGER D., MESTRE M., FIOLET J.W., JANSE M.J. Potassium accumulation in the globally ischemic mammalian heart. A role for the ATP-sensitive potassium channel. Circ. Res. 1990;67:835–843. doi: 10.1161/01.res.67.4.835. [DOI] [PubMed] [Google Scholar]
- WIRTH K.J., KLAUS E., ENGLERT H.G., SCHOLKENS B.A., LINZ W. HMR 1883, a cardioselective K(ATP) channel blocker, inhibits ischaemia- and reperfusion-induced ventricular fibrillation in rats. Naunyn Schmiedebergs Arch. Pharmacol. 1999a;360:295–300. doi: 10.1007/s002109900084. [DOI] [PubMed] [Google Scholar]
- WIRTH K.J., ROSENSTEIN B., UHDE J., ENGLERT H.C., BUSCH A.E., SCHOLKENS B.A. ATP-sensitive potassium channel blocker HMR 1883 reduces mortality and ischemia-associated electrocardiographic changes in pigs with coronary occlusion. J. Pharmacol. Exp. Ther. 1999b;291:474–481. [PubMed] [Google Scholar]
- YAMAMOTO M., EGASHIRA K., ARIMURA K., TADA H., SHIMOKAWA H., TAKESHITA A. Coronary vascular K+ATP channels contribute to the maintenance of myocardial perfusion in dogs with pacing-induced heart failure. Jpn. Circ. J. 2000;64:701–707. doi: 10.1253/jcj.64.701. [DOI] [PubMed] [Google Scholar]
- YOKOSHIKI H., SUNAGAWA M., SEKI T., SPERELAKIS N. ATP-sensitive K+ channels in pancreatic, cardiac, and vascular smooth muscle cells. Am. J. Physiol. 1998;274:C251998–C371998. doi: 10.1152/ajpcell.1998.274.1.C25. [DOI] [PubMed] [Google Scholar]