

Abstract
Since agmatine has been identified as a clonidine displacing substance (CDS), the aim of this study was to investigate whether agmatine can mimic CDS-induced cardiovascular reactions in organ bath experiments, pithed spontaneously hypertensive rats (SHR) and anaesthetized SHR.
Intravenously-administered agmatine significantly reduced the blood pressure and heart rate of anaesthetized SHR at doses higher than 1 and 3 mg kg−1, respectively. These effects are probably mediated via central mechanisms, since there was an approximate 8 fold rightward shift of the dose-response curve in the pithed SHR (indicating a weakened cardiovascular effect). Moreover, in organ bath experiments, agmatine failed to alter the contractility of intact or endothelium-denuded aortal rings. When agmatine was administered i.c.v. to anaesthetized SHR, blood pressure was increased without any alteration of heart rate, whereas blood pressure was unchanged and heart rate was increased after injection into the 4th brain ventricle. This suggests that haemodynamic reaction patterns after central application are related to distinct influences on central cardiovascular mechanisms.
Agmatine reduces noradrenaline release in pithed SHR while α2-adrenoceptors are irreversibly blocked with phenoxybenzamine, but not while I1-binding sites are selectively blocked with AGN192403. This suggests that agmatine may modulate noradrenaline release in the same way that clonidine does, i.e. via imidazoline binding sites; this involves a reduction in sympathetic tone which in turn reduces blood pressure and heart rate.
Finally, CDS-like cardiovascular activity appears not to be due to agmatine, since (i) blood pressure in anaesthetized SHR is decreased by agmatine and clonidine, and (ii) agmatine did not antagonize the blood pressure reaction to clonidine in pithed or anaesthetized SHR.
Keywords: Agmatine, blood pressure, heart rate, imidazoline binding sites, clonidine, clonidine displacing substance, noradrenaline release, spontaneously hypertensive rats, sympathetic tone
Introduction
In the mid-1980s, Bousquet et al. (1984) introduced the concept that imidazoline binding sites, demonstrated in the central nervous system (Ernsberger et al., 1987), are involved in central blood pressure regulation. Two major subtypes of imidazoline binding sites (I1 and I2) were characterized on the basis of their affinity towards clonidine and idazoxan (Michel & Insel, 1989; Parini et al., 1996). Atlas & Burstein (1984a, 1984b) and Atlas et al. (1987) isolated a substance from rat and calf brains which was able to bind specifically to α2-adrenoceptors and displace clonidine. For this reason it was christened ‘clonidine displacing substance' (CDS). Later on, CDS could be characterized by radioligand binding studies as being 30 times more selective towards imidazoline binding sites than α2-adrenoceptors, strengthening the hypothesis that CDS might be an endogenous ligand for the imidazoline binding site (Ernsberger et al., 1988; 1990). After central application of CDS, arterial blood pressure increases significantly without any change in heart rate both in the cat and the rat (Bousquet et al., 1986; 1987); this is opposite to the effects seen after central administration of clonidine (Bousquet et al., 1984). Furthermore, CDS was found to antagonize clonidine-stimulated hypotension directly (Bousquet et al., 1986).
In 1994, Li et al. succeeded in identifying and characterizing mammalian agmatine as a candidate for CDS. Radioligand binding studies on chromaffin cells and membranes of bovine cerebral cortex and the ventrolateral medulla revealed a moderate affinity towards α2-adrenoceptors, I1- and I2-binding sites (Li et al., 1994). Agmatine arises enzymatically from the activity of arginine decarboxylase on arginine and is not supplied to the mammalian organism from nutritional components or the products of bacterial colonization (Li et al., 1994; Regunathan & Reis, 2000). Moreover, agmatine has been demonstrated in nearly all organs of the rat; it is also distributed in various regions of the rat brain, and is released and inactivated either by uptake or enzymatic degradation via agmatinase, diamine oxidase or monoamine oxidase (Raasch et al., 2001).
After a centrally-mediated regulation of blood pressure was described for CDS in addition to its binding properties towards imidazoline binding sites (Bousquet et al., 1986; 1987), it was considered how much agmatine (as the potential CDS) influences the cardiovascular system. For this reason we studied the effects of agmatine: (1) by injecting agmatine intravenously or directly into the central nervous system of pithed or anaesthetized spontaneously hypertensive rats (SHR); (2) by determining the cardiovascular interaction between agmatine and clonidine; and (3) by identifying a mechanism which might be responsible for the agmatine-induced regulation of blood pressure and heart rate. The use of SHR in our study was based on experience gained with this model from earlier studies; clonidine and agmatine were both shown to influence blood pressure and/or catecholamine overflow in pithed SHR (Häuser & Dominiak, 1995; Häuser et al., 1995). Furthermore, CDS was shown to be increased in plasma of hypertensive patients and was positively correlated with blood pressure (Dontenwill et al., 1993). This suggests that agmatine may also have a special impact in hypertension, which justifies the use of SHR.
Methods
Animals
The present study was conducted according to the Declaration of Helsinki, following the guidelines for the care and use of laboratory animals as adopted by the ‘Ministerium für Natur und Umwelt des Landes Schleswig Holstein, Deutschland', animal protocols No: 9/A4/91 and 9/s/96. Male, spontaneously hypertensive rats (SHR, Charles River, Sulzfeld, Germany) weighing 200 – 250 g were used in all subsequent protocols.
Organ bath experiments
SHR were anaesthetized with pentobarbitone (60 mg kg−1, i.p.) and the thoracic aorta was removed. Aortal rings of 4 – 5 mm length, all originating from the same segment of the thoracic aorta, were mounted in an organ bath, incubated with Krebs solution (mM: NaCl 122, KCl 4.7, MgSO4 1.2, KH2PO4 12.2, glucose 11.1, NaHCO3 20.8, CaCl2 2.5), aerated with carbogen (95% O2/5% CO2), and maintained at a resting tension of 10 mN. The viability of the smooth muscle and endothelium of the aortal rings was ascertained before each experiment by measuring the contractile response to KCl (20 mM) and the subsequent relaxation response to acetylcholine (10 μM). After washout of drugs from the organ chamber with Krebs solution, and restoration of baseline, aortal rings were constricted again with KCl (20 mM) and allowed to equilibrate for 60 min. Although 20 mM KCl appears to evoke an incomplete response compared to the more commonly used 50 – 80 mM KCl, the response still amounts to at least 95% of the maximal KCl response (Soltis & Katovich, 1985). Concentration-response curves were then established for agmatine (10−6 – 10−3 M), clonidine (10−9 – 10−4 M), and phenylephrine (10−9 – 10−4 M). In a second set of experiments, dose-response curves were generated for agmatine (10−6 – 10−3 M) using endothelium-denuded aortic rings (a coarse steel rod was employed to remove the endothelium). The effectiveness of endothelium removal was checked prior to agmatine application by ensuring that vessels would not relax in the presence of 10 μM acetylcholine. A third set of concentration-response curves was generated for clonidine and phenylephrine (same concentration range) in the presence of agmatine (1 mM) using intact aortal segments. After each experiment, vessel rings were washed with Krebs solution so that baseline was restored, and viability was determined again as described above. For analysis, we only included those experiments for which similar results were yielded in the functional tests at the beginning and end of each examination. In parallel experiments, serving as time controls, equivalent volumes of saline were added at the appropriate times after the KCl-induced contraction.
Preparation of animals
For measuring blood pressure and heart rate in anaesthetized SHR, animals were tracheotomized under thiobutabarbitone anaesthesia (100 mg kg−1; i.p.) to allow drainage of bronchial secretion and to facilitate recovery from interruption of spontaneous respiration. Blood pressure was monitored via polyethylene catheters (PE-50) inserted into the carotid arteries which were connected to a Statham P23 Db pressure transducer (Hellige, Freiburg, Germany). Blood pressure and heart rate were recorded continuously during the whole experimental period using a Gould Brush recorder (Gould, Dietzenbach, Germany). Polyethylene catheters (PE-10) were also inserted into both femoral veins for drug administration.
The pithed rat preparation was carried out according to the method of Gillespie & Muir (1967) under ether anaesthesia. After insertion of a tracheal cannula, the SHR were pithed by introducing a blunt steel rod (1.5 mm diameter, coated with enamel except for the length of vertebral segments (C5 – L3) into the spinal canal via the orbit. Animals were then immediately subjected to positive pressure artificial respiration with room air at a rate of 50 – 60 strokes min−1. Bilateral vagotomy was then performed in the cervical region. Polyethylene catheters were inserted into both femoral veins (PE-10) for drug administration and into both carotid arteries (PE-50) for measuring blood pressure and heart rate (see above), and collecting blood samples. For determining noradrenaline overflow in the pithed SHR, a steel cannula was inserted as an indifferent electrode into the dorsal subcutis located near the lumbar vertebral column. Neuromuscular junctions were blocked using d-tubocurarine (3 mg kg−1). Global sympathetic outflow (C7 – L3) was induced by preganglionic electrical stimulation (20 V, 1 ms pulse duration at 0.5 Hz for 0.5 min) before blood was sampled for catecholamine analysis (1 ml, stabilized with 4 mM reduced glutathione and 5 mM EDTA).
For determining haemodynamic parameters after a central application of agmatine, canullae were positioned under pentobarbitone anaesthesia (60 mg kg−1, i.p.) in SHR after their heads had been fixed in a stereotaxic frame (David Kopf, Tojunga, U.S.A.) (Qadri et al., 1999). When agmatine was applied to the 4th (or central lateral) ventricle, a polypropylene cannula (pp-20, outer diameter=1.09 mm, inner diameter=0.38 mm) was lowered to 5.0 mm (or 7.07 mm) below the surface of the skull through a burr hole placed 1 mm (or 1.3 mm) laterally and 11.3 mm (or 0.6 mm) posterior to the bregma at an angle of 81.86° (or 90°). At the end of the experiments, rats were sacrificed and the brain was removed. The placement of the cannulae in the central lateral or the 4th ventricle was verified macroscopically using Pontamine Sky Blue. Only data obtained from animals in which the cannulae were positioned correctly were included in the results.
Experimental protocols
Pithed SHR
Dose-response curves for blood pressure and heart rate were established for clonidine (0.01 – 1000 μg kg−1) and agmatine (0.01 – 100 mg kg−1) by cumulative bolus injections. Animals were rested for at least 10 min before the injection of a higher dose. Pithed SHR receiving the same volume (1 μl per g bodyweight) of saline served as appropriate controls. In a further set of experiments which served to investigate the interaction between agmatine and clonidine, agmatine (10 mg kg−1, i.v.) was injected into pithed SHR 1 min prior to each clonidine bolus injection (0.3 – 1000 μg kg−1). Controls received saline 1 min prior to clonidine.
Anaesthetized SHR
In order to generate dose-response curves for blood pressure and heart rate, individual doses of agmatine (0.01 – 100 mg kg−1) were injected intravenously as boli (1 μl per g bodyweight), whereas individual clonidine doses (0.03 – 100 μg kg−1) were infused over 5 min. Two additional groups of rats received bolus injections of agmatine (0, 10, 30, 100, 300, 1000 μmol, each dose infused over 15 – 20 s) dissolved in 0.9% saline either in the central lateral (1st group) or the 4th ventricle (2nd group). Each injection volume of 5 μl was followed by a 3 μl flush with saline. Animals were rested for 20 min before a higher dose was injected. For controls, saline was injected intravenously or into the central lateral or 4th ventricle in place of the agmatine or clonidine. When the influence of agmatine on clonidine effects (or vice versa) was examined, clonidine (3.3 μg kg−1 min−1) was infused into anaesthetized SHR for 8 min before a constant clonidine infusion of 1 μg kg−1 30 min−1 was carried out. After stabilization of the haemodynamic parameters within 30 min, agmatine was injected either intravenously (0.3 – 3 mg kg−1) or into the 4th ventricle (10 – 1000 μmol) as described above. Controls received saline.
Determination of catecholamine overflow
After preparation, pithed SHR were allowed to recover until blood pressure and heart rate were constant. In the first protocol, agmatine (0, 1, 3.3 10 and 33 mg kg−1) was injected cumulatively with each individual injection lasting 2 min. Electrical stimulation was performed 90 s thereafter via the pithing rod at 20 V (1 ms pulse duration at 0.5 Hz for 0.5 min). Blood was sampled for catecholamine analysis only after the 33 mg kg−1 agmatine injection. Time-matched controls received saline instead of agmatine. The second set of experiments was similar to the first, except that pithed SHR were pretreated with the selective α2-adrenoceptor antagonist rauwolscine (3 mg kg−1) 10 min prior to agmatine infusions. In all the following protocols, pithed SHR were also pretreated with desipramine (0.5 mg kg−1) to inhibit neuronal catecholamine uptake. In the third set of experiments, the non-selective but irreversible α1/2-adrenoceptor antagonist phenoxybenzamine (10 mg kg−1) was injected 10 min before the agmatine (6/60 mg kg−1) or saline (controls) infusion. Blood was sampled before and after electrical stimulation for catecholamine analysis. In the fourth protocol, the selective I1-ligand AGN192403 (10 mg kg−1) was injected prior to agmatine (6/60 mg kg−1) or saline (controls) infusion. Blood was sampled before and after electrical stimulation for catecholamine analysis.
Determination of adrenaline and noradrenaline
Blood samples were centrifuged at 4°C for 10 min at 5000 g to obtain plasma. Plasma adrenaline and noradrenaline (as indices of sympathetic overflow) were measured by high pressure liquid chromatography (HPLC) and electrochemical detection after plasma (350 μl) was adsorbed onto alumina in a TRIS-buffer system (700 μl, consisting of 1.5 M Tris[hydroxy-methyl]aminomethane hydrochloride, 68 mM EDTA and 3.6 mM glutathione (reduced) and eluted with 100 μl perchloric acid (0.1 M) using dihydroxybenzylamine (500 pg) as an internal standard (Eriksson & Persson, 1982).
Determination of agmatine's half life and lethal dose
The half-life of agmatine was determined in anaesthetized SHR. Agmatine (50 mg kg−1) was injected intravenously and blood was withdrawn from the carotid artery at different times. Agmatine was measured by HPLC and fluorometric detection after derivatization according to Raasch et al. (1999). The half-life was calculated by non-linear curve fitting based on a first order kinetic (Graph Pad Prism®, San Diego, U.S.A.).
Drugs
Phenoxybenzamine was obtained from RBI (Natick, Massachusetts, U.S.A.), AGN192403 from Tocris (Bristol, U.K.) and agmatine, clonidine, D-tubocurarine and desipramine were from Sigma (Deisenhofen, Germany). All other chemicals (HPLC or analytical grade) were purchased either from Sigma (Deisenhofen, Germany) or Merck (Darmstadt, Germany). Stock solutions of agmatine (50 mg kg−1 for in-vivo studies, 0.1 M for in-vitro studies), AGN192403 (10 mg kg−1), clonidine (1 mg ml−1 for in-vivo studies, 10 mM for in-vitro studies), phenylephrine (10 mM), and rauwolscine (10 mg ml−1) were dissolved in water. The stock solution of phenoxybenzamine (10 mg ml−1) was prepared by dissolving 10 mg in 200 μl propylene glycol, 10 μl 1N HCl and 790 μl water. These stock solutions were stored at −20°C for no more than 1 month before use. Before each experiment, stock solutions were diluted with saline to form their final concentrations. The injection volume of each drug solution was 1 μl per g bodyweight.
Statistical analysis
Data shown in tables and figures are expressed as means±s.e.mean. EC50, ED50 Emax, Emin and Hill slopes (nHill) were calculated by non-linear curve fitting (Graph Pad Prism®). Statistical analysis was performed either by Students t-test (paired or unpaired), or by one-way analysis of variance (ANOVA, paired or unpaired) followed by Bonferroni's Multiple Comparison test wherever three or more random samples were involved. A significance level of 0.05 or less was considered to be significant.
Results
In-vitro effects of agmatine on the contractility of the isolated thoracic rat aorta
The basal constriction of thoracic aortal rings was 10.2±0.3 mN. Vasoconstriction was doubled to 20.2±0.8 mN by administering 20 mM KCl. At the end of the control experiments (after 30 min) the vascular tone was not altered compared to that seen after the initial 20 mM KCl (18.7±1.0 mN, P=0.299, n=5) application. Agmatine (n=6) up to a concentration of 1 mM failed to alter vascular contractility irrespective of whether endothelium was present or not (Figure 1A). In contrast, the vascular tone of aortal segments with intact endothelium was significantly enhanced by clonidine (n=6) at concentrations higher than 0.1 μM so that an EC50 of 223.8±10.1 nM and an Emax of 8.9±1.5%KCl-preconstriction resulted. When aortal rings were pre-incubated with agmatine (1 mM), clonidine's concentration-response curve was not substantially influenced; the EC50 (104±30.3 nM, n=6) and Emax (9.2±0.6%KCl-preconstriction) were both similar (Figure 1B). Phenylephrine (n=6) induced a larger dose-dependent contraction of aortal rings in the presence of intact endothelium. The Emax of phenylephrine (34.6±0.8%KCl-preconstriction) was four times larger than that of clonidine, although the EC50 of 213.7±23.2 nM was similar. As with clonidine, agmatine (1 mM, n=6) did not influence the phenylephrine concentration-response curve (EC50=223.7±27.3, Emax=35.9±1.1%KCl-preconstriction, Figure 1C). Our results clearly indicate that preconstriction of aortal segments by KCl (20 mM) was appropriate for detecting a potential contractile or relaxation response to agmatine, since 20 mM KCl doubled the resting tension of 10 mN and phenylephrine induced a 35% additional increase in contractility.
Figure 1.
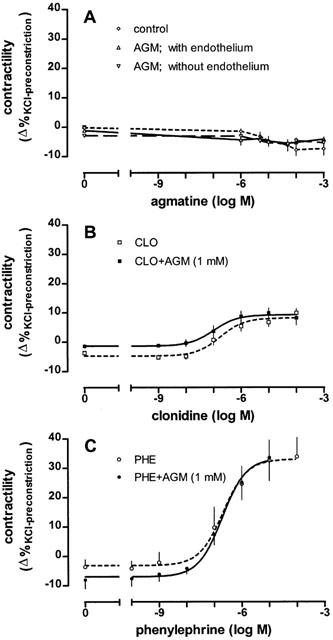
Effects of various drugs on the KCl (20 mM)-induced contraction of isolated thoracic aortal rings. (A) Influence of agmatine (AGM) on the contractility of aortal rings in the presence or absence of endothelium compared to controls. (B) Influence of clonidine (CLO) on vascular tone in intact aortal rings in the absence or presence of agmatine (1 mM). (C) Influence of phenylephrine (PHE) on contractility in intact aortal rings in the absence or presence of agmatine (1 mM). Values are expressed as means±s.e.mean of 5 – 6 experiments.
Effects of peripherally or centrally applied agmatine and clonidine on blood pressure and heart rate in pithed and anaesthetized SHR
In the following experiments both diastolic and systolic blood pressure were monitored via a carotid catheter. Changes induced by various drugs were similar for diastolic and systolic blood pressure. Since changes in blood pressure of pithed SHR are related more to peripheral resistance (which is reflected more by the diastolic blood pressure), only this parameter is depicted in the following figures; this also helped to clarify the presentation of the results. In pithed SHR (n=9), diastolic blood pressure and heart rate were decreased significantly (P<0.05) by agmatine alone at concentrations higher than 33 mg kg−1 (Figure 2C,D). In contrast, clonidine increased diastolic blood pressure dose-dependently so that an ED50 of 3.26±0.57 μg kg−1 (n=8) and a maximal increase of 121±3 mmHg resulted (Figure 2C). The heart rate was minimally affected by clonidine and at most revealed only a trend towards a reduction over the dose range tested (Figure 2D).
Figure 2.
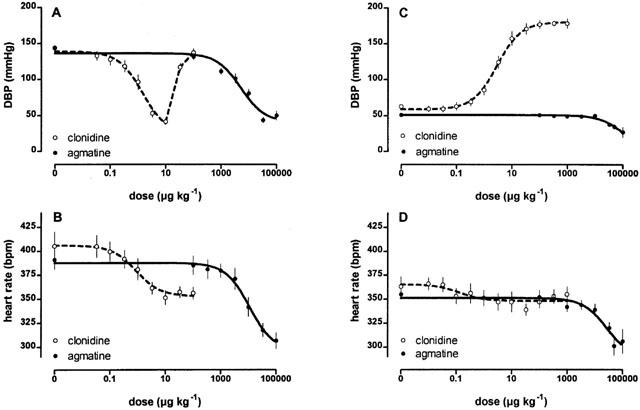
The influence of agmatine and clonidine on diastolic blood pressure (DBP) and heart rate of anaesthetized (A,B) or pithed (C,D) SHR. Agmatine decreased DPB and heart rate significantly (P<0.05) in anaesthetized and pithed SHR, respectively at doses >> and >33 mg kg−1 respectively. Values are expressed as means±s.e.mean of 8 – 9 experiments.
Unlike the pithed SHR model, clonidine was administered to anaesthetized SHR not as a bolus injection (but as a 5 min infusion). In this way direct vascular constriction could be avoided. Clonidine reduced blood pressure by a maximum of 103±8 mmHg (ED50=1.59±0.36 μg kg−1; n=8) at doses of up to 10 μg kg−1, while at higher doses the blood pressure returned to initial values (Figure 2A). Heart rate was dose-dependently diminished by maximal 67±9 beats per minute (b.p.m; ED50=0.85±0.30 μg kg−1; Figure 2B). Agmatine (n=9) also decreased diastolic blood pressure (P<0.05) in anaesthetized SHR at doses higher than 1 mg kg−1 by a maximum of 98±7 mmHg. An ED50 of 6507±1367 μg kg−1 indicated that agmatine's dose-response curve was displaced 4100 fold rightward compared to that of clonidine (Figure 2A). The blood pressure reaction at lower doses lasted only about 30 s after a latent period of 10 s, but at doses >10 mg kg−1 blood pressure effects persisted for much longer (up to 25 min). In parallel to the decrease in blood pressure, heart rate was also reduced dose-dependently by agmatine (ED50=22.9±5.3 mg kg−1, Emax=94±11 b.p.m., Figure 2B).
For determining central cardiovascular effects, agmatine was microinjected directly into the brain. When injected into the central lateral ventricle, agmatine increased the diastolic blood pressure dose-dependently (ED50=201±33 nmol; n=8) by a maximum of 24.2±1.8 mmHg without affecting heart rate (Figure 3A). In contrast, blood pressure was not altered but heart rate was enhanced (ED50=53±26 nmol, Emax=36.8±2.6 b.p.m., n=7) after injection of agmatine into the 4th brain ventricle (Figure 3B).
Figure 3.
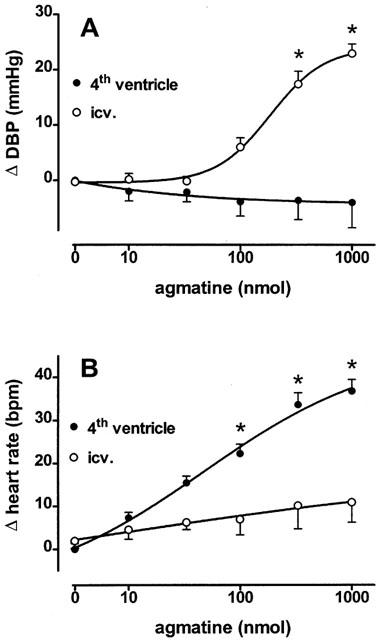
The influence of agmatine on diastolic blood pressure (DBP, A) and heart rate (B) in anaesthetized SHR after microinjection into the central lateral (i.c.v.) or 4th ventricle. Each agmatine dose was applied within a volume of 5 μl. Values are expressed as means±s.e.mean of 7 – 8 experiments. *P<0.05, versus control injection.
Experiments on the antagonism between agmatine and clonidine
The cardiovascular interaction between agmatine and clonidine was determined using pithed and anaesthetized SHR. Firstly, in pithed SHR, a blood-pressure-ineffective dose of agmatine (10 mg kg−1) was applied 1 min prior to each clonidine bolus injection. Agmatine failed to affect the blood pressure dose-response curve to clonidine since the parameters of clonidine's dose response curve remained unaltered (clonidine vs agmatine+clonidine; ED50: 3.26±0.57 vs 3.81±0.63 mg kg−1, P=0.5492; Emax: 122±3 vs 116±4 mmHg, P=0.2446; nhill: 1.203±0.01 vs 1.277±0.03, P=0.797; n=7, Figure 4A). Minimal clonidine effects on heart rate (Emax=31±6 b.p.m.) were enhanced by agmatine pretreatment (Emax=70±9 b.p.m., P=0.0018).
Figure 4.
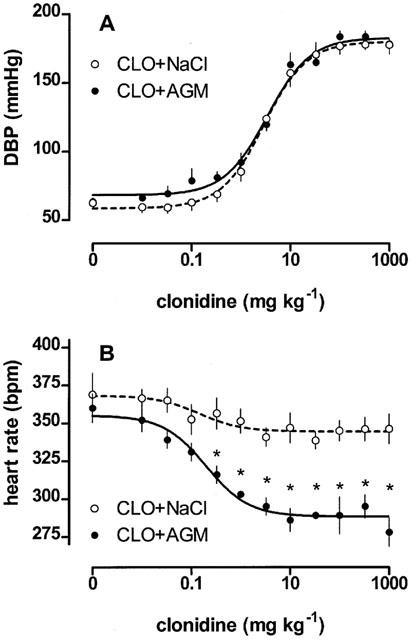
The influence of agmatine (10 mg kg−1) on the clonidine-induced increase of diastolic blood pressure (DBP) and heart rate reduction in pithed SHR. Values are expressed as means±s.e.mean of 7 experiments. *P<0.05, versus clonidine/NaCl.
Secondly, a constant infusion of clonidine into anaesthetized SHR (initial diastolic blood pressure=148.6±6.8 mmHg, initial heart rate: 385.7±14.6 b.p.m., n=7) led to significant reductions in blood pressure (64.0±4.9 mmHg) and heart rate (99.6±9.8 b.p.m.). The incremental and cumulative injection of agmatine after a 30 min period of haemodyanmic stabilization further reduced blood pressure (Figure 5A) and heart rate (Figure 5B) by a maximum of 24.6±4.4 mmHg (at >0.3 mg kg−1 agmatine) and 7.8±2.1 b.p.m. (at >10 mg kg−1 agmatine), compared to controls.
Figure 5.
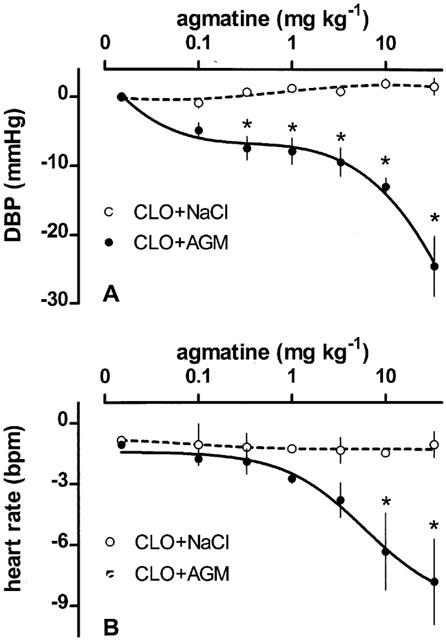
The influence of agmatine on the clonidine-induced decrease of diastolic blood pressure (DBP) and heart rate reduction in anaesthetized SHR. Values are expressed as means±s.e.mean of 7 experiments. *P<0.05 versus clonidine/NaCl.
In a further set of experiments on anaesthetized and clonidine-pretreated SHR (administered as described above, n=5), in which diastolic blood pressure dropped from 134.8±7.9 mmHg to 75.3±5.2 mmHg and heart rate dropped from 366.5±8.4 b.p.m. to 305.1±11.5, agmatine was injected centrally into the 4th ventricle. In contrast to peripheral agmatine application, blood pressure and heart rate were not affected between the dose range of 10 – 1000 nmol (not depicted).
Determination of agmatine's plasma half-life and lethal dose
Using HPLC, the half-life of agmatine in the blood of anaesthetized SHR after a bolus injection of agmatine (50 mg kg−1) was found to be 5.07±0.5 min (n=6, not depicted); this resulted in plasma concentrations of 148 and 13.5 μg ml−1 after 2 and 35 min, respectively.
Lethal doses of agmatine were determined in anaesthetized SHR by infusing agmatine (10.66 mg kg−1 min−1). All animals (n=4) died within 29.8±1.5 min, from which a lethal dose of 317.6±16.3 mg kg−1 could be calculated.
Effects of agmatine on electrically stimulated noradrenaline release
The last experiments addressed the electrically stimulated release of noradrenaline in pithed SHR. Diastolic blood pressure (79.5±6.0 mmHg, n=6) in pithed SHR was increased significantly following electrical stimulation (132.4±11.2 mmHg, P<0.001). This effect was dose-dependently reduced by agmatine (ED50 8.3±1.4 mg kg−1), whereby the stimulated blood pressure increase was completely eradicated by 33 mg kg−1 agmatine (93.5±7.4 mmHg; P>0.05). Heart rate and the stimulation-dependent plasma concentrations of noradrenaline (187±35 pg ml−1) or adrenaline (122±46 pg ml−1) were not affected by agmatine (33 mg kg−1) under these conditions.
After pretreatment with rauwolscine (3 mg kg−1, n=6) for blocking α2-adrenoceptors, the stimulation-dependent noradrenaline (but not adrenaline) plasma levels were significantly reduced by 33 mg kg−1 agmatine (211±10 pg ml−1) compared to controls (273±16 pg ml−1). Diastolic blood pressure (69.0±4.6 mmHg) under rauwolscine was increased following electrical stimulation to 84.0±6.4 mmHg (P<0.05). However, this stimulation-dependent increase was significantly lower compared to that seen without rauwolscine pretreatment due to its property to block vascular postjunctional α2-adrenoceptors; it was also dose-dependently lowered by agmatine (ED50 7.1±1.8 mg kg−1) to values that did not differ from these seen under unstimulated conditions (76.5±5.9 mmHg at 33 mg kg−1; P<0.05, not depicted).
When pithed SHR were pretreated with phenoxybenzamine (10 mg kg−1, n=6) combined with desipramine (0.5 mg kg−1), basal noradrenaline and adrenaline plasma concentrations were 595±47 and 40±7 pg ml−1, respectively. These plasma catecholamine concentrations were not significantly altered by an agmatine dose of 6 mg kg−1 (656±106 and 46±6 pg ml−1, respectively). In parallel, the mean arterial pressure (MAP) without electrical stimulation (80.8±1.7 mmHg) was not affected by agmatine (−0.8±1.4 mmHg, P>0.05). Electrical stimulation enhanced noradrenaline (4.7 fold) and adrenaline (4.1 fold) plasma concentrations significantly. Stimulated noradrenaline plasma concentrations under phenoxybenzamine and desipramine were much higher than those seen with rauwolscine. However, both phenoxybenzamine and rauwolscine block α2-adrenoceptors and should therefore result in comparable plasma catecholamine concentrations. Differences in plasma levels of noradrenaline seen when rauwolscine or phenoxybenzamine were applied can be explained more by a desipramine-induced blockade of uptake-1 than differences in their potencies regarding the stimulation of noradrenaline release. This was confirmed by the fact that the stimulated noradrenaline plasma concentrations in pithed SHR after desipramine (0.5 mg kg−1) plus rauwolscine (3 mg kg−1) pre-treatment were 6 fold higher (Häuser et al., 1995) than those seen in our rauwolscine experiments; this emphasises the importance of uptake-1 and indicates a comparable efficacy of rauwolscine regarding release. The stimulated noradrenaline plasma concentrations were significantly reduced at agmatine doses of 6 and 60 mg kg−1 by 37 and 52%, respectively, while blood pressure, heart rate and adrenaline plasma concentrations following electrical stimulation were unaffected (Table 1).
Table 1.
The influence of agmatine on stimulation-dependant cardiovascular parameters and plasma catecholamine concentrations in pithed SHR pretreated with phenoxybenzamine (10 mg kg−1) and desipramine (0.5 mg kg−1)

The basal noradrenaline plasma concentration under AGN192403 (10 mg kg−1, n=6) combined with desipramine (0.5 mg kg−1) was 194±23 pg ml−1 and significantly lower compared to that (595±48 pg ml−1) seen in phenoxybenzamine (10 mg kg−1) pretreated SHR. Moreover, such plasma levels were not altered by agmatine (6 mg kg−1; 170±6 mg kg−1). Similarly, basal plasma adrenaline concentrations under AGN192403 and desipramine were not altered by 6 mg kg−1 agmatine (39±5 vs 33±6 pg ml−1). The MAP (80.8±1.7 mmHg) without electrical stimulation was slightly reduced by agmatine (−4.2±1.6 mmHg, P<0.05). In contrast to the results following phenoxybenzamine pretreatment, stimulated noradrenaline plasma levels were not influenced by agmatine when pithed SHR were pretreated with AGN192403, whereas the stimulation-dependent increase in MAP showed signs of being reduced with low dose agmatine and was significantly diminished by 73% with the higher dose (Table 2).
Table 2.
The influence of agmatine on stimulation-dependant cardiovascular parameters and plasma catecholamine concentrations in pithed SHR pretreated with AGN192403 (10 mg kg−1) and desipramine (0.5 mg kg−1)

Discussion
Cardiovascular effects of agmatine
The most prominent effect of agmatine on cardiovascular reaction was achieved using the anaesthetized SHR model. After intravenous administration, agmatine reduces blood pressure slightly, although significantly, at doses higher than 1 mg kg−1. These hypotensive effects of agmatine endured for less than 30 s, possibly due to its very short half-life in plasma. Since the half-life of agmatine in mitochondria (40 min; due mainly to metabolic breakdown; Raasch et al., 1999) is much longer than its half-life in plasma (5.5 min), the disappearance of agmatine in plasma is related mainly to vascular reuptake, as hypothesized by Regunathan et al. (1996). At concentrations between 10 and 100 mg kg−1, agmatine induced a more pronounced and longer lasting blood pressure decrease. No reflex tachycardia was evident, despite the fact that the baroreflex arch was maintained. Heart rate was diminished and showed a similar ED50 compared to the blood pressure reduction. Analogous results on blood pressure reduction were acquired using anaesthetized normotensive rats (Sun et al., 1995; Gao et al., 1995). Our observation that 10 mg kg−1 agmatine induced a 65 mmHg drop in blood pressure was comparable to results obtained by Gao et al. (1995). Thus, the blood pressure lowering doses of agmatine were extremely high compared to those of other endogenous vasodilatators, since bradykinin (30 μg kg−1) or acetylcholine (1 μg kg−1) evoke comparable effects at doses 330 – 10,000 times lower (Gao et al., 1995). Furthermore, the lethal doses of agmatine (317.6±16.3 mg kg−1) in anaesthetized SHR are not substantially higher than those exerting pronounced blood pressure effects, a fact which clearly underlines its cardiovascular impotence.
Comparing the agmatine-induced decrease in blood pressure, the EC50's of the dose response curves of pithed SHR were about eight times higher than those of anaesthetized animals. This may at least in part be due to the fact that the tone is much lower in pithed rats, although this would imply that agmatine influences vascular tone via a direct mechanism. However, agmatine failed to alter contractility in intact aortal rings, which is consistent with the opinions of others who have shown that agmatine does not influence the contraction of the palmar lateral vein, thoracic aorta, tail artery (Pinthong et al., 1995; Gonzalez et al., 1996) or the left atria (Raasch et al., 2000). Since rat aorta is mainly populated by α1- and less by α2-adrenoceptors (Macia et al., 1984), clonidine's effect on isolated aorta is related to an α1-agonistic effect, as confirmed by the similar ED50's of clonidine and phenylephrine. Moreover, potential alterations of vascular tone due to agmatine must be related to imidazoline binding sites. Since agmatine shows no affinity towards α1-adrenoceptors (Li et al., 1994) and fails to antagonize the dose-response curves of phenylephrine or clonidine (also showing affinity towards α1-adrenoceptors; Armah, 1988), its lack of efficacy towards α1-adrenoceptors is functionally confirmed and an imidazoline binding site-mediated on vascular tone is also rendered doubtful. Although I2-binding sites have been reported on vascular smooth muscle cells (Regunathan et al., 1995), the likelihood that these imidazoline binding sites mediate vascular tone is reduced by the fact that the I2-binding site has already been characterized as an allosteric binding site on MAO (Parini et al., 1996; Raasch et al., 1999). Recently, an I3-binding site was shown to regulate vasoconstriction in the porcine isolated rectal artery (Minyan et al., 2001). However, agmatine shows no activity towards this I3-binding site, which is pharmacologically similar to the I3-binding site on islet cells (Raasch et al., 2001). This reduces the likelihood that agmatine is an endogenous ligand for an imidazoline binding site. Moreover, a regulation of vascular tone by agmatine via α2 adrenoceptors in vessels expressing these receptors is also unlikely, since agmatine exerts neither agonistic nor antagonistic activity towards the α2-adrenoceptor (Pinthong et al., 1995). This was shown at the subcellular level, since agmatine failed to alter [3H]-cyclic AMP concentration or forskolin-stimulated [3H]-cyclic AMP accumulation in the palmar lateral vein. However, it is also possible, albeit unlikely, that a null effect of agmatine on isolated vessels may represent the sum of a contractile and a relaxing effect, despite the fact that agmatine failed to alter contractility in endothelium-denuded aortal rings. Regarding potential interactions with relaxing neurotransmitters, agmatine inhibits NOS (Galea et al., 1996). Concerning interactions with vasoconstrictive neurotransmitters, agmatine suppresses catecholamine release (this study; Molderings & Göthert, 1995).
Since blood pressure effects in anaesthetized SHR were more pronounced than those seen in pithed SHR, and since CDS increased blood pressure via a central mechanism (Bousquet et al., 1986; 1987), studies on the blood pressure reaction after central agmatine application were performed. After an i.c.v. injection into SHR, agmatine caused a dose-dependent, significant increase in blood pressure after its injection into the 4th ventricle, although the heart rate increased dose-dependently. This was somehow contrary to our expectations, since agmatine applied intravenously to anaesthetized SHR causes hypotension, previously thought to be mediated via a central mechanism. Pressor effects of agmatine have also been shown in the anaesthetized Sprague Dawley rat after intracisternal injection (Sun et al., 1995), as well as in conscious rabbits (Szabo et al., 1995). In contrast, Head et al. (1997) saw no increase in heart rate in conscious rabbits, except at high doses which caused agitation, tachypnoea, an increase in blood pressure and a reversal of the dose-dependent bradycardia effect. In another paper, however, no effects of an i.c.v. application of agmatine on blood pressure and heart rate were observed (Penner & Smyth, 1996). It must therefore be emphasized that cardiovascular reaction patterns following central administration of agmatine have revealed inconsistent effects in different studies, possibly due to species differences or differences in the sites of application.
Identification of a mechanism that might be responsible for the agmatine-induced regulation of blood pressure and heart rate
Since agmatine (i.v.) decreased arterial pressure and sympathetic nerve activity, it has been suggested that agmatine probably blocks transmission through sympathetic ganglia (Sun et al., 1995). Moreover, another mechanism, i.e. agmatine-induced suppression of noradrenergic neurotransmission in rat tail arteries, was thought to be due to agmatine's blood pressure reduction, since agmatine inhibits contraction after transmural nerve stimulation. However, contraction by exogenous noradrenaline was not inhibited, so that a presynaptic effect of agmatine was suggested that could not be attributed to an inhibition of noradrenaline reuptake into the sympathetic nerve endings (Gonzalez et al., 1996). Even in this study there is strong evidence that agmatine inhibits sympathetic tone, since blood pressure and heart rate are decreased in a manner similar to that brought about by clonidine. Agmatine reduced noradrenaline release when α2-adrenoceptors were blocked reversibly and irreversibly by rauwolscine and phenoxybenzamine, respectively, but not under selective blockade of I1-binding sites by AGN192403 (Munk et al., 1996). Similar effects of agmatine on noradrenaline release modulated by presynaptic imidazoline binding sites were also demonstrated in various vascular preparations (Molderings & Göthert, 1995). The present study suggests that the presynaptic activity of agmatine is probably related to a regulation of noradrenaline release by presynaptic I1-binding sites as hypothesized by Heemskerk et al. (1998). However, a ganglionic mechanism can not be excluded since, (1) the presynaptic imidazoline binding site was pharmacologically characterized as belonging to a nonI1-/nonI2-subclass (Molderings & Göthert, 1999); (2) I1-binding sites are present at the cell bodies of sympathetic ganglia and adrenal medulla (Molderings et al., 1993); and (3) agmatine blocks nicotinic-cholinergic transmission in sympathetic ganglia (Loring, 1990). Furthermore, since activation of I1-binding sites releases prostaglandins (Ernsberger et al., 1995) and histamine (Molderings et al., 1999), which have both been shown to diminish noradrenaline release (Wennmalm & Junstad, 1976; Göthert et al., 1999), it also seems likely that agmatine might be able to reduce noradrenaline overflow via such an indirect mechanism.
Does agmatine influence haemodynamics in the way that CDS does?
Since 1994, numerous papers have been published concerning the physiological and/or pathophysiological functions of agmatine; these have discussed the hypothesis that agmatine might be a neurotransmitter or neuromodulator. Against the background of all the results published for CDS, i.e. a clear blood pressure-increasing effect and an antagonism to clonidine (Bousquet et al., 1984; 1986; 1987), our results emphatically indicate that agmatine cannot be CDS on its own, since (1) agmatine does not influence the contractility of thoracic aortal rings, whereas CDS increases it. (2) Agmatine (although only at very high concentrations) reduces blood pressure in anaesthetized SHR. A comparable decrease in blood pressure regarding the maximal effect was also observed under clonidine, but only at low doses at which central effects of clonidine dominate over direct vascular effects, and at which such direct agmatine effects on vascular contractility are excluded as discussed above. (3) Clonidine's dose-response curve regarding blood pressure in pithed SHR is not antagonized by agmatine. A minor clonidine-induced reduction of heart rate was enlarged by agmatine, which may have been due to agmatine's own effect at 10 mg kg−1. (4) The drop of blood pressure in anaesthetized SHR due to clonidine infusion was increased rather than reduced by agmatine. Similarly to the pithed rat model, the reduction in heart rate induced by clonidine was increased by agmatine. Pinthong et al. (1995) and Berdeu et al. (1996) also tended to dispute a CDS-like activity for agmatine; they found no agmatine-induced alterations of contractility in isolated organs and no insulin releasing effect for agmatine. (5) The plasma concentrations of endogenous agmatine (Raasch et al., 1995) are approximately 30,000 – 300,000 fold lower compared to plasma levels seen after an injected bolus of agmatine (50 mg kg−1) required to induce cardiovascular effects (this study). (6) Agmatine was shown to reduce catecholamine release, whereas CDS induced the opposite effect (Regunathan et al., 1991).
In conclusion, the blood pressure reduction following agmatine in anaesthetized SHR is not due to direct vascular dilatation, but rather to an inhibition of imidazoline binding site-mediated noradrenaline release. Furthermore, it seems doubtful that agmatine regulates cardiovascular function as a neurotransmitter because of the heroic doses that must be applied in order to induce any relevant effects. Ultimately, agmatine cannot be responsible for the biological activity of CDS, since it does not antagonize the cardiovascular effects of clonidine. Therefore, other substances with CDS-like properties still await identification.
Acknowledgments
We would like to thank Mrs A. Kaiser for expert technical assistance and Dr J.P. Keogh for editorial assistance in preparing the manuscript.
Abbreviations
- AGM
agmatine
- b.p.m.
beats per minute
- cyclic AMP
adenosine 3′,5′ cyclic monophosphate
- CDS
clonidine displacing substance
- CLO
clonidine
- DBP
diastolic blood pressure
- EDTA
ethylendiaminetetraacetic acid
- HPLC
high performance liquid chromatography
- NOS
nitric oxide synthase
- PHE
phenylephrine
- SHR
spontaneously hypertensive rats
- TRIS
Tris[hydroxy-methyl]aminomethane hydrochloride
References
- ARMAH B.I. Unique presynaptic alpha 2-receptor selectivity and specificity of the antihypertensive agent moxonidine. Arzneimittelforschung. 1988;38:1435–1442. [PubMed] [Google Scholar]
- ATLAS D., BURSTEIN Y. Isolation and partial purification of a clonidine-displacing endogenous brain substance. Eur. J. Biochem. 1984a;144:287–293. doi: 10.1111/j.1432-1033.1984.tb08462.x. [DOI] [PubMed] [Google Scholar]
- ATLAS D., BURSTEIN Y. Isolation of an endogenous clonidine-displacing substance from rat brain. FEBS Lett. 1984b;170:387–390. doi: 10.1016/0014-5793(84)81350-2. [DOI] [PubMed] [Google Scholar]
- ATLAS D., DIAMANT S., FALES H.M., PANNELL L. The brain's own clonidine: purification and characterization of endogenous clonidine displacing substance from brain. J. Cardiovasc. Pharmacol. 1987;10 Suppl 12:S122–S127. [PubMed] [Google Scholar]
- BERDEU D., PUECH R., LOUBATIERES-MARIANI M.M., BERTRAND G. Agmatine is not a good candidate as endogenous ligand for imidazoline sites of pancreatic B cells and vascular bed. Eur. J. Pharmacol. 1996;308:301–304. doi: 10.1016/0014-2999(96)00329-9. [DOI] [PubMed] [Google Scholar]
- BOUSQUET P., FELDMAN J., ATLAS D. An endogenous, non-catecholamine clonidine antagonist increases mean arterial blood pressure. Eur. J. Pharmacol. 1986;124:167–170. doi: 10.1016/0014-2999(86)90138-x. [DOI] [PubMed] [Google Scholar]
- BOUSQUET P., FELDMAN J., ATLAS D. Central cardiovascular effects of a noncatecholamine endogenous ligand for clonidine receptors. J. Cardiovasc. Pharmacol. 1987;10 Suppl 12:S167–S171. [PubMed] [Google Scholar]
- BOUSQUET P., FELDMAN J., SCHWARTZ J. Central cardiovascular effects of alpha adrenergic drugs: differences between catecholamines and imidazolines. J. Pharmacol. Exp. Ther. 1984;230:232–236. [PubMed] [Google Scholar]
- DONTENWILL M., MOLINES A., VERDUN A., BRICCA G., LAURENT S., BOUSQUET P. A circulating substance cross-reacting with antiimidazoline antibodies. Detection in serum in relation to essential hypertension. J. Clin. Invest. 1993;92:1068–1072. doi: 10.1172/JCI116611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ERIKSSON B.M., PERSSON B.A. Determination of catecholamines in rat heart tissue and plasma samples by liquid chromatography with electrochemical detection. J. Chromatogr. 1982;228:143–154. doi: 10.1016/s0378-4347(00)80427-2. [DOI] [PubMed] [Google Scholar]
- ERNSBERGER P., FEINLAND G., MEELEY M.P., REIS D.J. Characterization and visualization of clonidine-sensitive imidazoline sites in rat kidney which recognize clonidine-displacing substance. Am. J. Hypertens. 1990;3:90–97. doi: 10.1093/ajh/3.2.90. [DOI] [PubMed] [Google Scholar]
- ERNSBERGER P., GRAVES M.E., GRAFF L.M., ZAKIEH N., NGUYEN P., COLLINS L.A., WESTBROOKS K.L., JOHNSON G.G. I1-imidazoline receptors. Definition, characterization, distribution, and transmembrane signaling. Ann. N.Y. Acad. Sci. 1995;763:22–42. doi: 10.1111/j.1749-6632.1995.tb32388.x. [DOI] [PubMed] [Google Scholar]
- ERNSBERGER P., MEELEY M.P., MANN J.J., REIS D.J. Clonidine binds to imidazole binding sites as well as alpha 2-adrenoceptors in the ventrolateral medulla. Eur. J. Pharmacol. 1987;134:1–13. doi: 10.1016/0014-2999(87)90125-7. [DOI] [PubMed] [Google Scholar]
- ERNSBERGER P., MEELEY M.P., REIS D.J. An endogenous substance with clonidine-like properties: selective binding to imidazole sites in the ventrolateral medulla. Brain Res. 1988;441:309–318. doi: 10.1016/0006-8993(88)91409-6. [DOI] [PubMed] [Google Scholar]
- GALEA E., REGUNATHAN S., ELIOPOULOS V., FEINSTEIN D.L., REIS D.J. Inhibition of mammalian nitric oxide synthases by agmatine, an endogenous polyamine formed by decarboxylation of arginine. Biochem. J. 1996;316:247–249. doi: 10.1042/bj3160247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAO Y., GUMUSEL B., KOVES G., PRASAD A., HAO Q., HYMAN A., LIPPTON H. Agmatine: a novel endogenous vasodilator substance. Life Sci. 1995;57:L83–L86. doi: 10.1016/0024-3205(95)02011-7. [DOI] [PubMed] [Google Scholar]
- GILLESPIE J.S., MUIR T.C. A method of stimulating the complete sympathetic outflow from the spinal cord to blood vessels in the pithed rat. Br. J. Pharmacol. 1967;30:78–87. doi: 10.1111/j.1476-5381.1967.tb02114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GONZALEZ C., REGUNATHAN S., REIS D.J., ESTRADA C. Agmatine, an endogenous modulator of noradrenergic neurotransmission in the rat tail artery. Br. J. Pharmacol. 1996;119:677–684. doi: 10.1111/j.1476-5381.1996.tb15726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GÖTHERT M., BRÜSS M., BÖNISCH H., MOLDERINGS G.J. Presynaptic imidazoline receptors. New developments in characterization and classification. Ann. N.Y. Acad. Sci. 1999;881:171–184. doi: 10.1111/j.1749-6632.1999.tb09356.x. [DOI] [PubMed] [Google Scholar]
- HÄUSER W., DOMINIAK P. Effects of agmatine on catecholamine release and blood pressure of spontaneously hypertensive rats. A possible role for imidazoline receptors. Pharm. Pharmacol. Lett. 1995;2:87–89. [Google Scholar]
- HÄUSER W., GÜTTING J., NGUYEN T., DOMINIAK P. Influence of imidazolines on catecholamine release in pithed spontaneously hypertensive rats. Ann. N.Y. Acad. Sci. 1995;763:573–579. doi: 10.1111/j.1749-6632.1995.tb32452.x. [DOI] [PubMed] [Google Scholar]
- HEAD G.A., CHAN C.K., GODWIN S.J. Central cardiovascular actions of agmatine, a putative clonidine-displacing substance, in conscious rabbits. Neurochem. Int. 1997;30:37–45. doi: 10.1016/s0197-0186(96)00044-7. [DOI] [PubMed] [Google Scholar]
- HEEMSKERK F.M., DONTENWILL M., GRENEY H., VONTRHRON C., BOUSQUET P. Evidence for the existence of imidazoline-specific binding sites in synaptosomal plasma membranes of the bovine brainstem. J. Neurochem. 1998;71:2193–2202. doi: 10.1046/j.1471-4159.1998.71052193.x. [DOI] [PubMed] [Google Scholar]
- LI G., REGUNATHAN S., BARROW C.J., ESHRAGHI J., COOPER R., REIS D.J. Agmatine: an endogenous clonidine-displacing substance in the brain. Science. 1994;263:966–969. doi: 10.1126/science.7906055. [DOI] [PubMed] [Google Scholar]
- LORING R.H. Agmatine acts as an antagonist of neuronal nicotinic receptors. Br. J. Pharmacol. 1990;99:207–211. doi: 10.1111/j.1476-5381.1990.tb14680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACIA R.A., MATTHEWS W.D., LAFFERTY J., DEMARINIS R.M. Assessment of alpha-adrenergic receptor subtypes in isolated rat aortic segments. Naunyn Schmiedebergs Arch. Pharmacol. 1984;325:306–309. doi: 10.1007/BF00504373. [DOI] [PubMed] [Google Scholar]
- MICHEL M.C., INSEL P.A. Are there multiple imidazoline binding sites. Trends Pharmacol. Sci. 1989;10:342–344. doi: 10.1016/0165-6147(89)90002-3. [DOI] [PubMed] [Google Scholar]
- MINYAN W., DUNN W.R., BLAYLOCK N.A., CHAN S.L., WILSON V.G. Evidence for a non-adrenoceptor, imidazoline-mediated contractile response to oxymetazoline in the porcine isolated rectal artery. Br. J. Pharmacol. 2001;132:1359–1363. doi: 10.1038/sj.bjp.0703949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOLDERINGS G.J., GÖTHERT M. Inhibitory presynaptic imidazoline receptors on sympathetic nerves in the rabbit aorta differ from I1- and I2-imidazoline binding sites. Naunyn Schmiedebergs Arch. Pharmacol. 1995;351:507–516. doi: 10.1007/BF00171042. [DOI] [PubMed] [Google Scholar]
- MOLDERINGS G.J., GÖTHERT M. Imidazoline binding sites and receptors in cardiovascular tissue. Gen. Pharmacol. 1999;32:17–22. doi: 10.1016/s0306-3623(98)00070-6. [DOI] [PubMed] [Google Scholar]
- MOLDERINGS G.J., MENZEL S., GÖTHERT M. Imidazoline derivatives and agmatine induce histamine release from the rat stomach. Naunyn Schmiedebergs Arch. Pharmacol. 1999;360:711–714. doi: 10.1007/s002109900129. [DOI] [PubMed] [Google Scholar]
- MOLDERINGS G.J., MOURA D., FINK K., BÖNISCH H., GÖTHERT M. Binding of [3H]clonidine to I1-imidazoline sites in bovine adrenal medullary membranes. Naunyn Schmiedebergs Arch. Pharmacol. 1993;348:70–76. doi: 10.1007/BF00168539. [DOI] [PubMed] [Google Scholar]
- MUNK S.A., LAI R.K., BURKE J.E., ARASASINGHAM P.N., KHARLAMB A.B., MANLAPAZ C.A., PADILLO E.U., WIJONO M.K., HASSON D.W., WHEELER L.A., GARST M.E. Synthesis and pharmacologic evaluation of 2-endo-amino-3-exo-isopropylbicyclo[2.2.1]heptane: a potent imidazoline 1 receptor specific agent. J. Med. Chem. 1996;39:1193–1195. doi: 10.1021/jm960012o. [DOI] [PubMed] [Google Scholar]
- PARINI A., MOUDANOS C.G., PIZZINAT N., LANIER S.M. The elusive family of imidazoline binding sites. Trends Pharmacol. Sci. 1996;17:13–16. doi: 10.1016/0165-6147(96)81564-1. [DOI] [PubMed] [Google Scholar]
- PENNER S.B., SMYTH D.D. Natriuresis following central and peripheral administration of agmatine in the rat. Pharmacology. 1996;53:160–169. doi: 10.1159/000139427. [DOI] [PubMed] [Google Scholar]
- PINTHONG D., WRIGHT I.K., HANMER C., MILLNS P., MASON R., KENDALL D.A., WILSON V.G. Agmatine recognizes alpha 2-adrenoceptor binding sites but neither activates nor inhibits alpha 2-adrenoceptors. Naunyn Schmiedebergs Arch. Pharmacol. 1995;351:10–16. doi: 10.1007/BF00169058. [DOI] [PubMed] [Google Scholar]
- QADRI F., BÄURLE L., HÄUSER W., RASCHER W., DOMINIAK P. Centrally bradykinin B2-receptor-induced hypertensive and positive chronotropic effects are mediated via activation of the sympathetic nervous system. J. Hypertens. 1999;17:1265–1271. doi: 10.1097/00004872-199917090-00005. [DOI] [PubMed] [Google Scholar]
- RAASCH W., CHUN K.R.J., DENDORFER A., DOMINIAK P. Positive inotropic effects of imidazoline derivatives are not mediated via imidazoline binding sites by alpha1-adrenergic receptors. Jpn. J. Pharmacol. 2000;84:1–6. doi: 10.1254/jjp.84.1. [DOI] [PubMed] [Google Scholar]
- RAASCH W., MUHLE H., DOMINIAK P. Modulation of MAO activity by imidazoline and guanidine derivatives. Ann. N.Y. Acad. Sci. 1999;881:313–331. doi: 10.1111/j.1749-6632.1999.tb09376.x. [DOI] [PubMed] [Google Scholar]
- RAASCH W., REGUNATHAN S., LI G., REIS D.J. Agmatine, the bacterial amine, is widely distributed in mammalian tissues. Life Sci. 1995;56:2319–2330. doi: 10.1016/0024-3205(95)00226-v. [DOI] [PubMed] [Google Scholar]
- RAASCH W., SCHÄFER U., CHUN J., DOMINIAK P. Biological significance of agmatine, an endogenous ligand at imidazoline binding sites. Br. J. Pharmacol. 2001;133:755–780. doi: 10.1038/sj.bjp.0704153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REGUNATHAN S., MEELEY M.P., REIS D.J. Clonidine-displacing substance from bovine brain binds to imidazoline receptors and releases catecholamines in adrenal chromaffin cells. Mol. Pharmacol. 1991;40:884–888. [PubMed] [Google Scholar]
- REGUNATHAN S., REIS D.J. Characterization of arginine decarboxylase in rat brain and liver: distinction from ornithine decarboxylase. J. Neurochem. 2000;74:2201–2208. doi: 10.1046/j.1471-4159.2000.0742201.x. [DOI] [PubMed] [Google Scholar]
- REGUNATHAN S., YOUNGSON C., RAASCH W., WANG H., REIS D.J. Imidazoline receptors and agmatine in blood vessels: a novel system inhibiting vascular smooth muscle proliferation. J. Pharmacol. Exp. Ther. 1996;276:1272–1282. [PubMed] [Google Scholar]
- REGUNATHAN S., YOUNGSON C., WANG H., REIS D.J. Imidazoline receptors in vascular smooth muscle and endothelial cells. Ann. N.Y. Acad. Sci. 1995;763:580–590. doi: 10.1111/j.1749-6632.1995.tb32453.x. [DOI] [PubMed] [Google Scholar]
- SOLTIS E.E., KATOVICH M.J. Phentolamine and rat aortic smooth muscle responsiveness to potassium chloride, isoproterenol and norepinephrine. Pharmacology. 1985;31:67–71. doi: 10.1159/000138100. [DOI] [PubMed] [Google Scholar]
- SUN M.K., REGUNATHAN S., REIS D.J. Cardiovascular responses to agmatine, a clonidine-displacing substance, in anesthetized rat. Clin. Exp. Hypertens. 1995;17:115–128. doi: 10.3109/10641969509087059. [DOI] [PubMed] [Google Scholar]
- SZABO B., URBAN R., LIMBERGER N., STARKE K. Cardiovascular effects of agmatine, a ‘clonidine-displacing substance', in conscious rabbits. Naunyn Schmiedebergs Arch. Pharmacol. 1995;351:268–273. doi: 10.1007/BF00233246. [DOI] [PubMed] [Google Scholar]
- WENNMALM A., JUNSTAD M. Prostaglandin-mediated inhibition of noradrenaline release: its resistance to variations in rate of prostaglandine synthesis. Acta Physiol. Scand. 1976;97:60–65. doi: 10.1111/j.1748-1716.1976.tb10235.x. [DOI] [PubMed] [Google Scholar]