

Abstract
The effects of aconitine, an Aconitum alkaloid, on spontaneous inhibitory and excitatory postsynaptic currents (IPSCs and EPSCs respectively) were investigated in the mechanically dissociated rat ventromedial hypothalamic (VMH) neurons in which native presynaptic nerve terminals remained intact.
Under current-clamp conditions, aconitine (3×10−6 M) depolarized the neuron with generating the action potentials. The aconitine-induced depolarization was markedly suppressed in the presence of CNQX but it was facilitated in the presence of bicuculline, suggesting that release of excitatory and inhibitory neurotransmitters may be involved in the aconitine action in addition to its direct action on postsynaptic membrane.
Under the voltage-clamp conditions, aconitine reversibly increased the frequency of spontaneous IPSC and EPSC frequency, but it did not alter their amplitude distribution.
Tetrodotoxin (TTX, 3×10−7 M) completely abolished the aconitine action on spontaneous IPSC frequency. Likewise removal of extracellular Na+ completely suppressed the aconitine action.
Both Ca2+-free external solution or addition of 10−4 M Cd2+ to normal solutions eliminated the facilitatory effect of aconitine on the IPSC frequency.
Overall these results suggest that aconitine depolarizes the presynaptic membrane by activating voltage-dependent Na+ channels. Increase of intraterminal Ca2+ concentration via an activation of voltage-dependent Ca2+ channels in turn enhances the spontaneous transmitter release from presynaptic nerve terminals. The presynaptic action of aconitine may play a crucial role for membrane excitability of rat VMH neurons.
Keywords: Aconitine, EPSC, IPSC, synaptic bouton preparation, ventromedial hypothalamic neuron, voltage-dependent sodium channel
Introduction
Certain alkaloids isolated from plant species Aconitum have hypotensive and bradycardiac actions which are thought to be due to an activation of autonomic reflexes (Chiao et al., 1995; Kimura et al., 1997). Aconitine, the main Aconitum alkaloid, is known as a potent neurotoxin while it also produces antinociception in rodents (Hikino et al., 1979; Oyama et al., 1994). The symptoms of aconitine intoxication include systemic paralysis, nausea and vomiting, followed by dizziness, palpitation, hypotention, arrhythmia, shock and coma (Ameri, 1998). These actions of aconitine are thought to be due to its interaction with voltage-dependent Na+ channels. Aconitine causes a negative shift in the voltage-dependence of Na+ channel activation (Naumov, 1983), and binds to the open state of Na+ channels (Catterall, 1980; 1987). Such persistent activation of Na+ channels arises from inhibition of inactivation and produces a sustained Na+ influx (Catterall, 1980). On the other hand, aconitine suppresses the stimulus-evoked population spike in rat hippocampal slices (Ameri & Peters, 1997). In addition, Onur et al. (1995) reported that aconitine at low concentrations increased electrically evoked ACh release, while higher concentration decreased the evoked and increased spontaneous ACh release in the rat neuromuscular junction.
The ventromedial nucleus of hypothalamus (VMH) plays an important role in controlling the activity of autonomic nervous system (Itoi et al., 1991; Narita et al., 1994). An activation of VMH neurons has also been reported to be responsible for suppression of the circulatory system in rats (Hirasawa et al., 1998). Interestingly, the aconitine-induced cardiac irregularities are thought to be attributed to an activation of the neurons of hypothalamic regions, since the aconitine-induced bradycardia and hypotention were prevented by bilateral lesions of the whole hypothalamus (Kimura et al., 1997). However, little information is available about the effect of aconitine on VMH neurons. In the present study, therefore, we investigated the effect of aconitine on membrane excitability of acutely dissociated rat VMH neurons attached with native inhibitory and excitatory presynaptic nerve terminals by using the conventional whole-cell recording configuration (Koyama et al., 1999; Rhee et al., 1999).
Methods
Preparation
Neurons with synaptic boutons were acutely isolated from ventromedial hypothalamus (VMH) of the rat. Ten- to 14-day-old Wistar rats were decapitated under pentobarbital sodium anaesthesia (50 mg kg−1). The brains were quickly removed and immersed in an ice-cold incubation medium, well oxygenated with 95% O2 and 5% CO2. Coronal slices at a thickness of 400 μm containing the VMH region were prepared with a tissue slicer (DTK-1000, Dosaka, Kyoto, Japan). The brain slices were then incubated for at least 1 h at room temperature (21 – 24°C). Thereafter, the slices were transferred into a 35 mm culture dish (Primaria 3801, Falcon, NJ, U.S.A.) and the VMH region was identified under a binocular microscope (SMZ-1, Nikon, Tokyo, Japan). A fire-polished glass pipette was touched lightly onto the surface of the VMH region and was vibrated horizontally at 3 – 5 Hz for about 2 min using an apparatus developed in our laboratory (Rhee et al., 1999). The slices were then removed from the dish and the mechanically dissociated neurons settled and adhered to the bottom of the dish. These neurons were dissociated without using any enzymes or requiring trituration and, thus, portions of their original proximal dendritic processes retained.
External solutions
The ionic composition of the incubation medium was (mM): NaCl 124, KCl 5, KH2PO4 1.2, NaHCO3 24, CaCl2 2.4, MgSO4 1.3 and glucose 10. The ionic composition of the standard external solution was (mM): NaCl 150, KCl 5, CaCl2 2, MgCl2 1, N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES) 10 and glucose 10. Na+-free external solution was made by replacing all Na+ in the standard external solution with equimolar N-methyl-D-glucamine (NMG). Ca2+-free external solution contained (mM): NaCl 150, KCl 5, MgCl2 3, glucose 10, HEPES 10 and EGTA 2. The pH of these external solutions was adjusted to 7.4 with tris(hydroxymethyl)aminomethane (Tris-OH). In recording the spontaneous IPSCs, all extracellular solutions routinely contained 10−6 M 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and DL-2-amino-5-phosphonovaleric acid (DL-AP5) to block glutamatergic responses. In recording spontaneous EPSCs (Figure 6), the extracellular solution contained 3×10−6 M bicuculline to block the spontaneous GABAergic IPSCs.
Figure 6.
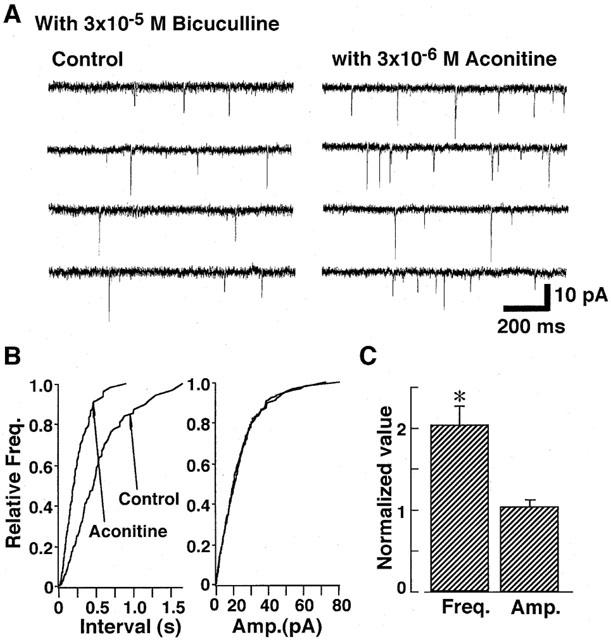
Effect of aconitine on spontaneous EPSCs in the presence of 3×10−5 M bicuculline. (A) Example of consecutive traces of spontaneous EPSCs in control conditions (left) and with 3×10−6 M aconitine (right). (B) Cumulative frequency distribution (left) and amplitude (right) distributions of spontaneous EPSCs. (C) Pooled data of the aconitine action on the mean frequency (left) and amplitude (right) of spontaneous EPSCs. Each column is average of four neurons.
Electrical measurements
Electrical measurements were performed using conventional whole-cell patch recording configuration under current- and voltage-clamp conditions. Patch-pipettes were made from borosilicate glass tubes in two stages on a vertical pipette pullar (PB-7, Narishige, Tokyo, Japan). The ionic composition of the internal (patch-pipette) solution for current-clamp experiments was (in mM): K-methanesulfonate 140, ATP-Mg 5, KCl 10, ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) 2 and HEPES 10. The composition of internal solution for voltage-clamp experiments was (in mM): Cs-methanesulfonate 70, ATP-Mg 5, CsCl 80, tetraethylammonium chloride 5, EGTA 2 and HEPES 10. The pH of these pipette solutions was adjusted to 7.2 with Tris-OH. Under these recording conditions, both inhibitory and excitatory postsynaptic responses apparently generate inward currents under the voltage-clamp conditions. Thus, effects of aconitine on inhibitory or excitatory postsynaptic currents were investigated in the presence of CNQX and AP-5 or bicuculline, respectively. The resistance between the recording electrode filled with the internal solution and reference electrode was 5 – 7 MΩ. After rupture the patch membrane, the series resistance ranged from 12 to 20 MΩ, which was compensated as previously described (Rhee et al., 1999). Ionic currents and voltages were measured with a patch-clamp amplifier (EPC-7, List-Electric, Darmstadt, Germany), monitored on both an oscilloscope (Tektronix 5111A, Sony, Tokyo, Japan) and a pen recorder (Recti-Horiz 8K, Nippondenki San-ei, Tokyo, Japan). The signals were also filtered at 3 kHz and digitized at 10 kHz using Laboview software on a Macintosh computer. All experiments were carried out at room temperature (21 – 24°C).
Data analysis
The digitized periods of synaptic events were analysed and counted using DETECTiVENT, a spontaneous event analysis software (Ankri et al., 1994) and IGOR PRO software (Wavemetrics, Lake Oswego, OR, U.S.A.). Analysis of spontaneous synaptic currents was performed with cumulative probability plots, and the cumulative histograms were compared using the Kolmogorov-Smirnov test for significant differences (P<0.05). Data were presented as mean±standard error of the mean. Differences in amplitude and frequency distributions were tested by the Wilcoxon signed rank test.
Drugs
Drugs used in the present study were aconitine, DL-AP5, ATP, bicucullline, CNQX and EGTA [Sigma], and tetrodotoxin (TTX) [Sankyo].
Results
Effect of aconitine on membrane excitability of the mechanically dissociated rat VMH neurons attached with native presynaptic nerve endings was investigated under the current-clamp conditions. Aconitine produced the depolarization which was accompanied by action potential discharges (Figure 1A). As shown in Figure 1B,C, on the other hand, the aconitine-induced depolarization was markedly inhibited in the presence of 3×10−6 M CNQX which blocks excitatory synaptic inputs and was facilitated in the presence of 3×10−5 M bicuculline which suppresses inhibitory synaptic inputs. Aconitine induced only slight depolarization in the presnce of both CNQX and bicuculline.
Figure 1.
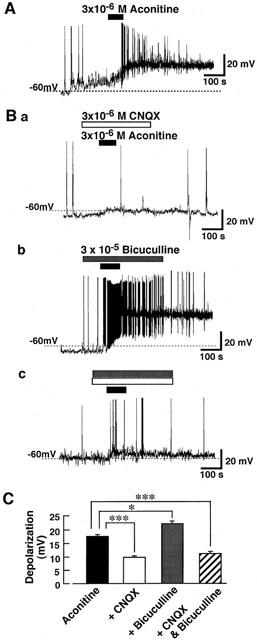
Effect of aconitine on the mechanically isolated rat ventromedial hypothalamic (VMH) neurons under current-clamp conditions. Recordings were performed with patch-pipette solution containing 10 mM Cl−. (A) Aconitine-induced depolarization in a VMH neuron. Aconitine was applied during time indicated by closed bar above the trace. The figure is representative of six reproducible experiments. (B) Effects of aconitine (3×10−6 M) in the presence of 3×10−6 M CNQX (a) or 3×10−5 M bicuculline (b). In the presence of both CNQX and bicuculline, aconitine produced slight depolarization (c). Each trace is representative of six reproducible experiments. (C) The figure summarizes the aconitine-induced depolarization in control condition and in the presence of CNQX, bicuculline or both. Each column represents the mean±s.e. of six neurons. The statistical significance was determined by one-way analysis of variance (ANOVA) test for multiple comparisons. *P<0.05, ***P<0.001.
In order to reveal how aconitine increases the transmitter release, effects of aconitine on spontaneous IPSCs and EPSCs were investigated under the voltage-clamp conditions at a holding potential (VH) of -80 mV. The effect of aconitine on the spontaneous IPSCs was firstly investigated in the presence of 3×10−6 M CNQX and 10−5 M AP-5. The spontaneous IPSCs were fully blocked by 10−5 M bicuculline in a reversible manner (not shown), indicating that they were mediated by GABAA receptors. As shown in Figure 2, addition of aconitine reversibly increased the rate of spontaneous GABAergic IPSCs. Aconitine acted rapidly and typically recovered within 2 min following its removal (Figure 2B). Mean IPSC frequency was increased significantly by adding 10−6 M aconitine while the mean amplitude was unaffected at any tested concentrations (Figure 2C).
Figure 2.
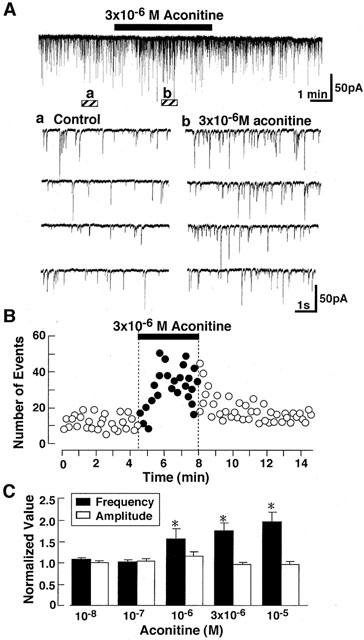
Effect of aconitine on spontaneous IPSCs. (A) Representative current traces in the absence and presence of aconitine. Current recordings were performed with patch-pipette solution containing 80 mM Cl−. Holding potential (VH) was -80 mV. Lower panel shows the four consecutive current traces at expanded time scale at the symbols. (a,b) indicating in the upper panel. (B) Effects of 3×10−6 M aconitine on spontaneous IPSCs. Aconitine was applied during a period shown by horizontal bar. Number of events in every 10 s was plotted. (C) Summary of the effect of various concentration of aconitine on the mean amplitude (white) and frequency (black) of spontaneous IPSCs. Each column is average of six neurons.
Application of 3×10−7 M TTX markedly reduced the spontaneous IPSC frequency to 29±5% (n=5) of control while TTX produced no significant effect on their amplitude distribution. In the presence of TTX, aconitine (3×10−6 M) failed to increase the rate of spontaneous IPSC. Cumulative inter-event intervals and cumulative amplitude histograms of spontaneous IPSCs were unaltered by adding aconitine in the presence of TTX (Figure 3B). Figure 3C shows the mean frequency and amplitude of spontaneous IPSCs in the presence of TTX.
Figure 3.
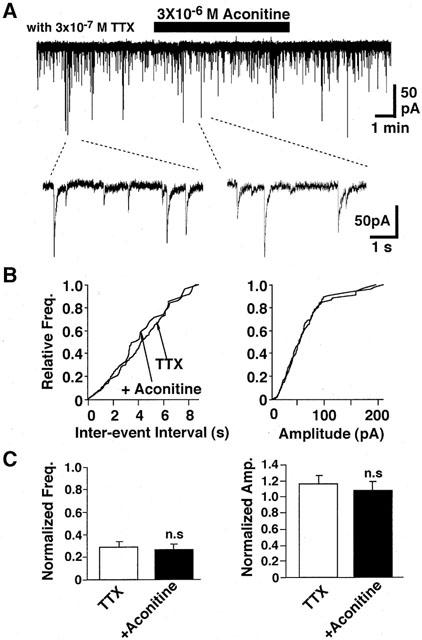
Effect of aconitine on spontaneous IPSCs in the presence of TTX. (A) Spontaneous IPSCs before, during and after application of 3×10−6 M aconitine in the presence of TTX (3×10−7 M) at a VH of −80 mV. (B) Cumulative frequency distribution (left) and cumulative amplitude distribution (right) of spontaneous IPSCs. The number of events used for cumulative distributions was 243 for TTX and 114 for TTX plus aconitine. (C) Summary of the effects of aconitine on spontaneous IPSCs in the presence of TTX. Pooled data of aconitine action on the mean frequency (left) and amplitude (right) of spontaneous IPSCs. Each column is average of five neurons.
Since TTX specifically blocks fast voltage dependent Na+ channels, we tested whether the Na+ influx contributed to the aconitine action. Replacement of extracellular Na+ with NMG+ rapidly reduced the frequency of spontaneous IPSCs by 45±5% (Figure 4A, n=4). During Na+-free perfusion, the addition of aconitine did not facilitate the rate of occurrence of spontaneous IPSC. Consequently, aconitine produced no significant effect on the cumulative inter-event interval and cumulative amplitude distributions (Figure 4B). Likewise, the mean amplitude and frequency were not affected by aconitine in the absence of extracellular Na+ (Figure 4C).
Figure 4.
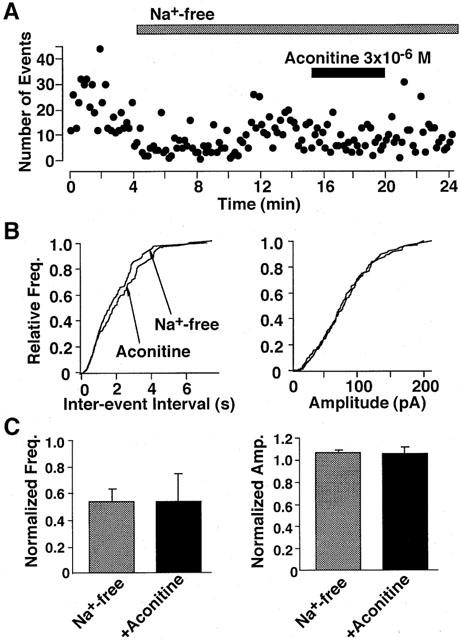
Effect of aconitine on spontaneous IPSCs in a Na+-free external solution. (A) Effect of aconitine on spontaneous IPSCs. Number of events in every 10 s was plotted. (B) Cumulative frequency (left) and distribution (left) and amplitude (right) distributions of spontaneous IPSCs. The number of events used for cumulative distribution was 273 for Na+-free and 161 for Na+-free plus aconitine. (C) Pooled data of the aconitine action on the mean frequency (left) and amplitude (right) of spontaneous IPSCs. Each column is average of four neurons.
Ca2+-influx into presynaptic nerve terminal through voltage-dependent Ca2+ channels plays a crucial role for neurotransmitter release (Stanley, 1997). A possible contribution of presynaptic voltage-dependent Ca2+ channels in the facilitatory effect of aconitine on the spontaneous IPSCs was evaluated. Omission of Ca2+ from external solution markedly decreased the frequency of spontaneous IPSCs (Figure 5A). In the absence of extracellular Ca2+, aconitine failed to alter either the mean frequency or amplitude of spontaneous IPSCs (n=6; Figure 5B). Additionally, Cd2+ (10−4 M), a non-selective blocker of voltage-dependent Ca2+ channels, prevented aconitine actions on spontaneous IPSC frequency (n=4; Figure 5C). Together, the results suggest that aconitine action is mediated by the presynaptic Ca2+ influx through voltage-dependent Ca2+ channels to facilitate spontaneous IPSC frequency.
Figure 5.
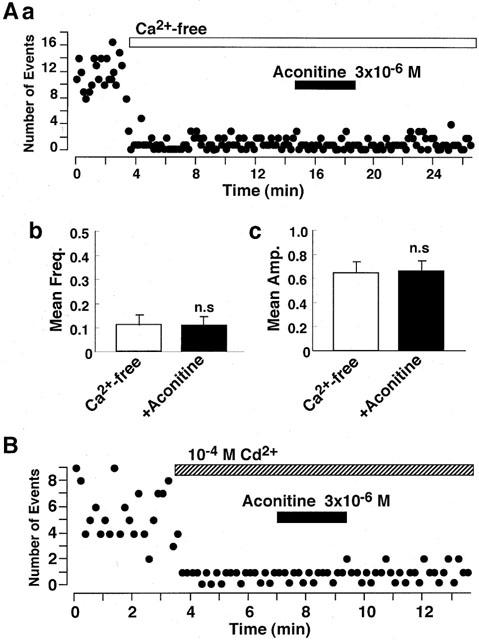
Effect of aconitine on spontaneous IPSCs in Ca2+-free external solution. (A) Effect of aconitine on spontaneous IPSCs. Number of events in every 10 s was plotted. (B) Summary of the effect of aconitine on spontaneous IPSCs in the Ca2+-free external solution. Pooled data of the aconitine action on the mean frequency (left) and amplitude (right) of spontaneous IPSCs. Each column is average of six neurons. (C) Effect of aconitine on spontaneous IPSCs in the normal external solution containing 10−4 M Cd2+. Number of events in every 10 s was plotted.
Effect of aconitine on spontaneous EPSCs was also investigated at a VH of −80 mV. In the presence of 3×10−5 M bicuculline, the spontaneous postsynaptic currents were completely inhibited by 3×10−6 M CNQX (n=4, not shown), suggesting that they were EPSCs mediated by non-NMDA receptors. As shown in Figure 6, aconitine also increased the frequency of spontaneous EPSCs to 203±18% (n=4) without altering their amplitude, suggesting that aconitine also increases glutamate release from excitatory presynaptic nerve terminals.
Discussion
It has been reported that intracerebroventricular and intraperitoneal administrations of aconitine produce cardiovascular changes including bradycardia with bigeminy and hypotension (Bhargava & Srivastava, 1972; Kimura et al., 1997). The hypothalamus plays an important role in cardiovascular regulation, and neuronal activity in the VMH is associated with suppression of the circulatory system (Hirasawa et al., 1998). In the present study, we demonstrated that aconitine depolarized the mechanically isolated rat VMH neurons with native excitatory and inhibitory synaptic boutons attached. The depolarizing action of aconitine was markedly inhibited in the presence of CNQX and enhanced in the presence of bicuculline (Figure 1). In the presence of both CNQX and bicuculline, on the other hand, aconitine produced only slight depolarization (Figure 1Bc and C). These observations suggest that the presynaptic action of aconitine may be stronger than its direct action on postsynaptic membrane. Therefore, it seems that the cardiovascular responses to aconitine may result from an activation of VMH neurons through the increase in extracellular concentrations of excitatory or inhibitory neurotransmitters.
The present study also demonstrated that aconitine increases the frequency of both spontaneous GABAergic IPSCs and glutamatergic EPSCs in rat VMH neurons. Comparable neurotransmitter release response have been described for the spontaneous acetylcholine release in the mouse (Okazaki et al., 1994) and rat (Onur et al., 1995) neuromuscular junctions. Since the mean amplitude of spontaneous IPSCs was not significantly changed (Figure 2), aconitine does not appear to alter the sensitivity of postsynaptic GABAA-receptor-channel complexes to transmitter released from presynaptic nerve terminals. Thus, facilitation of spontaneous IPSC frequency by aconitine is due to the increased GABA release from presynaptic nerve terminals. Similarly, the mean amplitude of spontaneous EPSCs was not significantly changed by aconitine (Figure 6), indicating that aconitine also increases glutamate release from excitatory nerve endings.
Aconitine acts at the site II of toxin-binding sites on the voltage-dependent Na+ channel to produce a negative shift of the voltage-dependent Na+ channel activation and an inhibition of the channel inactivation (Catterall, 1980; Strichartz et al., 1987). Consequently, aconitine not only depolarizes the neuronal membrane but also increases the intracellular Na+ concentration with prolonged Na+ channel activation. Since the Na+ concentration of rat cortical synaptosomes is increased by aconitine (Friese et al., 1997), aconitine is thought to increase the intraterminal Na+ concentration. The sodium ionophore, monensin, increases GABA release from mouse synaptosome preparations (Sitges, 1989). This releasing response to monensin was dependent on extracellular Na+, but was insensitive to either external TTX or removal of extracellular Ca2+. Thus, the increase in intraterminal Na+ concentration itself is thought to be sufficient to enhance the GABA release (Sitges, 1989). However, this was unlikely in the case of the aconitine-induced enhancement of GABA release. In the present study, aconitine-induced facilitation of GABA release was fully inhibited in the presence of 3×10−7 M TTX (Figure 3). Furthermore, failure of aconitine to enhance the spontaneous GABA release in the Ca2+-free external solution (Figure 5) suggests that the facilitatory action of aconitine finally depends on Ca2+ influx into presynaptic nerve terminals and the Ca2+-influx is induced by the activation of voltage-dependent Ca2+ channels triggered by activation of Na+ channels. Aconitine also increases the Ca2+ concentration in rat cortical synaptosomes (Friese et al., 1997).
Aconitine may alter the ionic selectivity of the voltage-dependent Na+ channels by increasing the relative permeability of Na+ channel to NH4+, K+ and Cs+ (Campbell, 1982; Rao & Sikdar, 2000). It remains unclear whether such aconitine-modified Na+ channels pass Ca2+. However, since aconitine-induced enhancement of the IPSC frequency was not observed in the absence of extracellular Na+ (Figure 4), influx of Ca2+ ions passing through aconitine-modified Na+ channels seems unlikely to contribute.
Calcium ions may enter terminals via Na+/Ca2+ exchangers or voltage-dependent Ca2+ channels which could be triggered by aconitine. The Na+/Ca2+ exchanger exchanges 3 Na+ for 1 Ca2+ and leads to Ca2+ accumulation (reverse mode) or to Ca2+ extrusin (forward mode), depending on the concentration of each ion on either side across plasma membrane and on membrane potential (Hoyt et al., 1998; Yu & Choi, 1997). Intracellular accumulation of Na+ induced by veratridine, a Na+ channel activator, leads to a rise in intracellular Ca2+ concentration most likely via Na+/Ca2+ exchange and in turn causes a large increase in frequency of spontaneous excitatory postsynaptic current in cultured hippocampal neurons (Bouron & Reuter, 1996). The reverse mode of Na+/Ca2+ exchanger also increases GABA release from the GABAergic presynaptic nerve terminals projecting to rat Meynert neurons (our unpublished data). In the present study, however, the aconitine-induced enhancement of spontaneous GABA release was fully inhibited in the presence of 10−4 M Cd2+ (Figure 4). Cd2+ at this concentration is known to produce complete block of high-voltage-activated Ca2+ channels but has no effect on the Na+/Ca2+ exchanger in reverse mode (Bouron & Reuter, 1996). The Na+/Ca2+ exchanger does not contribute to the aconitine-induced transmitter release in neuromuscular junction (Onur et al., 1995). Thus, reversed Na+/Ca2+ exchange is unlikely to participate in aconitine actions, and the facilitatory action of aconitine on spontaneous GABA release is most likely mediated by Ca2+ influx through voltage-dependent Ca2+ channels that are presumably activated by the aconitine-induced depolarization of presynaptic nerve terminals.
Aconitine also increased the spontaneous glutamatergic EPSC frequency (Figure 6), indicating that the agent increases the glutamate release from excitatory synaptic nerve terminals. Thus, the present results suggest that aconitine induces the presynaptic depolarization by activating the voltage-dependent Na+ channels in both inhibitory and excitatory presynaptic nerve terminals. The presynaptic depolarization, in turn, increases cytoplasmic Ca2+ concentrations at GABAergic and glutamatergic presynaptic boutons and results in the increase of GABA and glutamate release, respectively. The increase in transmitter release from both excitatory and inhibitory presynaptic nerve terminals modulates the excitability of postsynaptic neuronal membrane in addition to the direct depolarizing action of aconitine on postsynaptic membrane.
Abbreviations
- ACh
acetylcholine
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione
- CNS
central nervous system
- DL-AP5
DL-2-amino-5-phosphonovaleric acid
- EGTA
ethylene glycol-bis(β-aminoethylether)-N,N,N′,N′-tetraacetic acid
- EPSC
excitatory postsynaptic current
- HEPES
N-2-hydroxyethylpiperazine-N′-2-ethanesulphonie acid
- IPSC
inhibitory postsynaptic current
- NMG
N-methyl-D-glucamine
- Tris-OH
tris(hydroxymethyl)aminomethane
- TTX
tetrodotoxin
- VMH
ventromedial hypothalamic
References
- AMERI A. The effect of Aconitum alkaloids on the central nervous system. Prog. Neurobiol. 1998;56:211–235. doi: 10.1016/s0301-0082(98)00037-9. [DOI] [PubMed] [Google Scholar]
- AMERI A., PETERS T. Calcium-dependent, sustained enhancement of excitability during washout of aconitine in rat hippocampal slices. Exp. Brain Res. 1997;114:518–524. doi: 10.1007/pl00005661. [DOI] [PubMed] [Google Scholar]
- ANKRI N., LEGENDRE P., DABER D.S., KORN H. Automatic detection of spontaneous synaptic responses in central neurons. J. Neurosci. Meth. 1994;52:87–100. doi: 10.1016/0165-0270(94)90060-4. [DOI] [PubMed] [Google Scholar]
- BHARGAVA K.P., SRIVASTAVA R.K. Analysis of the central receptors concerned in the cardiovascular response induced by intracerebroventricular aconitine. Neuropharmacology. 1972;11:123–135. doi: 10.1016/0028-3908(72)90063-9. [DOI] [PubMed] [Google Scholar]
- BOURON A., REUTER H. A role of intracellular Na+ in the regulation of synaptic transmission and turnover of the vesicular pool in cultured hippocampal cells. Neuron. 1996;17:969–978. doi: 10.1016/s0896-6273(00)80227-5. [DOI] [PubMed] [Google Scholar]
- CAMPBELL D.T. Modified kinetics and selectivity of sodium channels in frog skeletal muscle fibers treated with aconitine. J. Gen. Physiol. 1982;80:713–731. doi: 10.1085/jgp.80.5.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CATTERALL W.A. Neurotoxins that act voltage-sensitive sodium channels in excitable membranes. Annu. Rev. Pharmacol. Toxicol. 1980;20:15–43. doi: 10.1146/annurev.pa.20.040180.000311. [DOI] [PubMed] [Google Scholar]
- CATTERALL W.A. Common modes of drug action on Na+ channels: local anesthetics, antiarrhythmics and anticonvulsants. Trends Pharmacol. Sci. 1987;8:57–65. [Google Scholar]
- CHIAO H., PELLETIER S.W., DESAI H.K., REBAGAY W.R., CALDWELL R.W. Effect of diterpenoid alkaloids on cardiac sympathetic efferent and vagal afferent nerve activity. Eur. J. Pharmacol. 1995;283:103–106. doi: 10.1016/0014-2999(95)00290-2. [DOI] [PubMed] [Google Scholar]
- FRIESE J., GLEITZ J., GUTSER U.T., HEUBACH J.F., MATTHIESEN T., WILFFERT B., SELVE N. Aconitum sp. alkaloids: the modulation of voltage-dependent Na+ channels, toxicity and antinociceptive properties. Eur. J. Pharmacol. 1997;337:165–174. doi: 10.1016/s0014-2999(97)01268-5. [DOI] [PubMed] [Google Scholar]
- HIKINO H., ITO T., YAMADA C., SATO H., KONNO C., OHIZUMI Y. Analgesic principles of Aconitum roots. J. Pham. Dyn. 1979;2:78–83. [Google Scholar]
- HIRASAWA M., NISHIHARA M., TAKAHASHI M. Activity of ventromedial hypothalamic neurons suppressing heart rate is associated with paradoxical sleep in the rat. Brain Res. 1998;797:103–108. doi: 10.1016/s0006-8993(98)00339-4. [DOI] [PubMed] [Google Scholar]
- HOYT K.R., ARDEN S.R., AIZENMAN E., REYNOLDS I.J. Reverse Na+/Ca2+ exchange contributes to glutamate-induced intracelluar Ca2+ concentration increases in cultured rat forebrain neurons. Mol. Pharmacol. 1998;53:724–749. [PubMed] [Google Scholar]
- ITOI K., JOST N., BADOER E., TSCHOPE C., CULMAN C., UNGER T. Locarization of the substance P-induced cardiovascular responses in the rat hypothalamus. Brain Res. 1991;558:123–126. doi: 10.1016/0006-8993(91)90727-d. [DOI] [PubMed] [Google Scholar]
- KIMURA I., TAKADA M., NOJIMA H. Aconitine induces bradycardia through transmission pathway including the anterior hypothalamus in conscious mice. Biol. Pharm. Bull. 1997;20:856–860. doi: 10.1248/bpb.20.856. [DOI] [PubMed] [Google Scholar]
- KOYAMA S., KUBO C., RHEE J.S., AKAIKE N. Presynaptic serotonergic inhibition of GABAergic synaptic transmission in mechanically dissociated rat basolateral amygdala neurons. J. Physiol. (Lond.) 1999;518:525–538. doi: 10.1111/j.1469-7793.1999.0525p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAUMOV A.P.Modification of sodium channel with scorpion toxins and alkaloids Toxins As Tools in Neurochemistry 1983Berlin: de Gruyter; 13–23.(ed) Hucho F, Ovachinnikov Y. pp [Google Scholar]
- NARITA K., NISHIHARA M., TAKAHASHI M. Concomitant regulation of running activity and metabolic change by the ventromedial nucleus of the hypothalamus. Brain Res. 1994;642:290–296. doi: 10.1016/0006-8993(94)90933-4. [DOI] [PubMed] [Google Scholar]
- OKAZAKI M., KIMURA I., KIMURA M. Aconitine-induced increase and decrease of acetylcholine release in the mouse phrenic nerve-hemidiaphragm muscle preparation. Jpn. J. Pharmacol. 1994;66:421–426. doi: 10.1254/jjp.66.421. [DOI] [PubMed] [Google Scholar]
- ONUR R., BOZDAGI O., AYATA C. Effects of aconitine on neurotransmitter release in the rat neuromuscular junction. Neuropharmacology. 1995;34:1139–1145. doi: 10.1016/0028-3908(95)00050-g. [DOI] [PubMed] [Google Scholar]
- OYAMA T., ISONO T., SUZUKI Y., HAYAKAWA Y. Anti-nociceptive effects of Aconiti tuber and its alkaloids. Am. J. Chinese Med. 1994;22:175–182. doi: 10.1142/S0192415X94000218. [DOI] [PubMed] [Google Scholar]
- RAO S., SIKDAR S.K. Modification of alpha subunit of RIIA sodium channels by aconitine. Pflügers Arch. 2000;439:349–355. doi: 10.1007/s004249900121. [DOI] [PubMed] [Google Scholar]
- RHEE J.H., ISHIBASHI H., AKAIKE N. Calcium channels in the GABAergic presynaptic nerve terminals projecting to Meynert neurons of the rat. J. Neurochem. 1999;72:800–807. doi: 10.1046/j.1471-4159.1999.0720800.x. [DOI] [PubMed] [Google Scholar]
- SITGES M. Characterization of the effects of monensin on γ-amino-n-butyric acid release from isolated nerve terminals. J. Neurochem. 1989;53:442–447. doi: 10.1111/j.1471-4159.1989.tb07354.x. [DOI] [PubMed] [Google Scholar]
- STANLEY E.F. The calcium channels and the organization of the presynaptic transmitter release face. Trends Neurosci. 1997;20:404–409. doi: 10.1016/s0166-2236(97)01091-6. [DOI] [PubMed] [Google Scholar]
- STRICHARTZ G., RANDO T., WANG G.K. An integrated view of the molecular toxicology of sodium channel gating in excitable cells. Annu. Rev. Neurosci. 1987;10:237–267. doi: 10.1146/annurev.ne.10.030187.001321. [DOI] [PubMed] [Google Scholar]
- YU S.P., CHOI D.W. Na+-Ca2+-exchange currents in cortical neurons: concomitant forward and reverse operation and effect of glutamate. Eur. J. Neurosci. 1997;9:1273–1281. doi: 10.1111/j.1460-9568.1997.tb01482.x. [DOI] [PubMed] [Google Scholar]