

Abstract
We have investigated the involvement of nitric oxide and K+ channels in the vasorelaxant responses to physiologically-relevant concentrations of testosterone in the rat isolated mesenteric arterial bed.
Testosterone (100 pM – 10 μM) elicited concentration-dependent relaxations in the isolated mesenteric arterial bed (pEC50=9.47 (9.22 – 9.73, 95% CI), maximal relaxation, Rmax=62.8±2.0%, n=6). A nitric oxide synthase (NOS) inhibitor, NG-nitro-L-arginine methyl ester (L-NAME, 300 μM) or removal of the endothelium significantly inhibited maximal relaxations to testosterone (L-NAME: Rmax=51.4±1.1%, P<0.01, n=6; endothelium-denuded: Rmax=46.9±2.8%, P<0.001, n=5).
Raising the extracellular K+ concentration to 30 and 60 mM, or pre-treatment with 300 μM tetrabutylammonium chloride (TBA), a calcium-activated K+ channel inhibitor, abolished vasorelaxations induced by testosterone. A selective inhibitor of ATP-sensitive K+ (KATP) channels, glibenclamide (10 μM) and an inhibitor of voltage-sensitive K+ (KV) channels, 4-aminopyridine (4-AP, 1 mM) did not affect testosterone-induced responses. Vasorelaxation to 1 μM testosterone was significantly (P<0.05) inhibited by 100 nM charybdotoxin (ChTx), an inhibitor of large conductance calcium-activated K+ (BKCa) channels (control: 63.3±9.9%, n=6; ChTx: 11.9±12.7%, n=3).
Neither the testosterone receptor antagonist, flutamide (10 μM) nor an aromatase inhibitor, aminoglutethimide (10 μM) inhibited testosterone-induced responses.
In conclusion, the present findings demonstrate, in the rat isolated mesenteric arterial bed, that testosterone causes acute vasorelaxations at physiologically relevant concentrations which are, in part, mediated via NO- and endothelium-dependent pathways. However, the activation of BKCa channels plays a substantial role in testosterone-induced vasorelaxation.
Keywords: Testosterone, endothelium, nitric oxide, K+ channels, charybdotoxin
Introduction
The incidence of cardiovascular diseases (CVD) and myocardial infarction is higher in men than women of a similar age (Wild & Bartholomew, 1988; Adams et al., 1995). Indeed, testosterone is thought to be a risk factor for CVD and atherosclerosis (Wild et al., 1988; Adams et al., 1995; McCredie et al., 1998). A previous study by Adams et al. (1995) of hyperandrogenism induced in female monkeys demonstrated that testosterone induced progression of atheroma and an increased level of serum low-density lipoprotein. Recently, McCredie et al. (1998) have shown that high-density lipoprotein was decreased in female to male transsexuals after long-term administration of androgen. However, Rosano et al. (1999) demonstrated that, in men with coronary artery disease, short-term administration of testosterone improved tolerance to exercise-induced myocardial ischaemia, perhaps suggesting that testosterone-induced vasorelaxation may be involved in any cardiovascular protective effects of testosterone.
Several studies have shown that short-term administration of testosterone causes vasorelaxation in a range of species including human (Yue et al., 1995; Chou et al., 1996; Costarella et al., 1996; Perusquia et al., 1996; Crews & Khalil, 1999; Honda et al., 1999). However, the mechanisms of testosterone-induced vasorelaxation are unclear. Costarella et al. (1996) demonstrated, in the rat thoracic aorta, that vasorelaxation to testosterone at pharmacological concentrations (25 – 300 μM) was inhibited by a nitric oxide synthase (NOS) inhibitor. Similar findings have also been reported in canine coronary arteries (Chou et al., 1996). These results indicate that testosterone-induced vasorelaxation is mediated via activation of NOS activity (Chou et al., 1996; Costarella et al., 1996). Moreover, Honda et al. (1999) showed, in thoracic aorta from spontaneous hypertensive rats, that the vasorelaxation to testosterone is endothelium-dependent. In contrast, other studies have demonstrated that testosterone causes vasorelaxation in de-endothelialized rabbit coronary artery and rat thoracic aorta (Yue et al., 1995; Costarella et al., 1996; Perusquia et al., 1996). Furthermore, Yue et al. (1995) have shown, in rabbit coronary artery and aorta, that vasorelaxation induced by testosterone was unaffected by a NOS inhibitor or methylene blue, an inhibitor of NO-activated guanylyl cyclase, and mediated via receptor-independent pathway.
The activation of K+ channels on vascular smooth muscle cells is thought to be involved in the vasorelaxant effects of testosterone (Yue et al., 1995; Chou et al., 1996; Honda et al., 1999). Yue et al. (1995) demonstrated, in the rabbit coronary artery, that vasorelaxation to testosterone (1 and 10 μM) was sensitive to barium chloride, a non-selective K+ channel inhibitor, but not to glibenclamide, a selective inhibitor of KATP channels. Similarly, Chou et al. (1996) also showed in canine coronary arteries that vasorelaxation to testosterone (1 μM) was not affected by glibenclamide. In contrast, Honda et al. (1999) have recently demonstrated that vasorelaxation induced by testosterone at pharmacological concentrations was inhibited by glibenclamide in aortic rings from Wistar-Kyoto and spontaneously hypertensive rats.
In the context of vascular effects of sex hormones, we have recently shown that 17β-oestradiol (10 pM – 10 μM) causes acute and potent vasorelaxation, which were not mediated via NO. However, high extracellular K+ inhibited vasorelaxation to 17β-oestradiol, suggesting, therefore, that 17β-oestradiol induces vasorelaxation by enhancing K+ efflux. Furthermore, we found that 17β-oestradiol-induced responses were sensitive to TBA, an inhibitor of calcium-activated K+ channels (Tep-Areenan et al., 2001).
On the basis of previous studies, the precise mechanisms of testosterone-induced vasorelaxation are still controversial. Therefore, the aim of the present investigation was to investigate the involvement of NO, the endothelium, K+ channels, testosterone receptors, and aromatase in testosterone-induced vasorelaxation in the rat isolated mesenteric arterial bed.
Methods
Preparation of the rat isolated mesenteric arterial bed
Male Wistar rats (250 – 350 g) were anaesthetized with sodium pentobarbitone (60 mg kg−1, i.p.) and killed by cervical dislocation. The superior mesenteric artery was cannulated, and the arterial bed was dissected away from the guts and placed in a jacketed organ bath as previously described (McCulloch et al., 1997). The arterial vasculature was perfused with oxygenated (95%O2/5%CO2) Krebs-Henseleit solution (composition, mM: NaCl 118, KCl 4.7, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25, CaCl2 2, D-glucose 10 plus 10 μM indomethacin) at a constant flow rate of 5 ml min−1 and maintained at 37°C. In the experiments involving high extracellular K+, 30 mM and 60 mM KCl-containing isotonic Krebs-Henseleit solution was prepared by substituting an equimolar concentration of NaCl with KCl (McCulloch et al., 1997).
Experimental protocol
The perfusion pressure in the superior mesenteric arterial bed was continuously monitored by a pressure transducer coupled to a Maclab 4e recording system (AD instruments, New South Wales, Australia). Flow rate was maintained at 5 ml min−1 and alterations in perfusion pressure represent changes in resistance of the mesenteric arterial bed.
Following a 30-min equilibration period, methoxamine (3 – 14 μM) was added to perfusion fluid to increase tone by approximately 70 – 100 mmHg. Once stable tone was established, testosterone was added cumulatively to the perfusion fluid (100 pM – 10 μM).
The involvement of NO in testosterone-induced vasorelaxation was assessed by using 300 μM NG-nitro-L-arginine methyl ester (L-NAME), a NOS inhibitor (Randall & Griffith, 1991). In the presence of L-NAME, a low concentration of methoxamine (1 – 3 μM) was used to achieve equivalent tone, as the responses to methoxamine were enhanced.
In order to investigate the role of the endothelium in responses to testosterone, the endothelium was removed in some preparations by perfusion with distilled water for 10 min (Harris et al., 2000). The preparation was deemed to be endothelium-denuded when the response to 55 nmol carbachol was less than 15%.
To examine the involvement of K+ channels in vasorelaxation to testosterone, tone was established by KCl at concentrations of 30 mM (Yue et al., 1995) and 60 mM (McCulloch et al., 1997). The high extracellular K+ concentration causes vasoconstriction by depolarization and inhibiting K+ channel activity and thus preventing hyperpolarization and relaxation (Adeagbo & Triggle, 1993). In subsequent experiments, 300 μM tetrabutylammonium chloride (TBA), an inhibitor of calcium-activated K+ channels (Randall & Kendall, 1997), 10 μM glibenclamide, an inhibitor of KATP channels (Randall & Griffith, 1993), and 1 mM 4-aminopyridine, an inhibitor of voltage-sensitive potassium (KV) channels (Honda et al., 1999) were independently used to investigate the types of K+ channels involved in vasorelaxation to testosterone (10 pM – 10 μM).
To investigate the effects of a BKCa channel inhibitor, charybdotoxin (ChTx) at a concentration of 100 nM (Randall & Kendall, 1998), a test concentration of 1 μM testosterone was chosen and the effects of the toxin were examined against the response at this concentration.
The involvement of testosterone receptors and aromatase (to metabolize testosterone) in vasorelaxation to testosterone were examined by pre-treatment with 10 μM flutamide, a testosterone receptor antagonist (Teoh et al., 2000), and 50 μM aminoglutethimide, an aromatase inhibitor (Yue et al., 1995). In each case, the appropriate inhibitors were added to the perfusion fluid to establish the desired concentrations and allowed to equilibrate for 30 min. Then, testosterone was added in a cumulative fashion (100 pM – 10 μM).
Data and statistical analysis
The concentration of vasorelaxant giving the half-maximal relaxation (EC50) was obtained from the concentration-response curve. The data were best-fitted to 1- or 2- site models; the most appropriate model was as determined by regression analysis and the r2 values are stated. The EC50 values of curves fitted to a 1-site model were determined by the logistic equation:
Where R is the reduction in tone, A is the concentration of the relaxant, Rmax is the maximum relaxation of the established tone, nH is the slope function and EC50 is the concentration of vasorelaxant giving half-maximal relaxation.
Maximal responses are expressed as mean±s.e.mean, and pEC50 values (-ve log of EC50 values) are expressed as means with 95% confidence intervals (CI). The number of animals in each group is represented by n. The data were compared, as appropriate, by the Student's unpaired t-test or analysis of variance (ANOVA) with statistically significant differences between groups being determined by Bonferroni's post-hoc test. The curve-fitting and graph drawing were carried out using the graphical package GraphPad Prism.
Drugs and chemicals
All drugs and chemicals were purchased from Sigma Chemical Company (U.K.), except charybdotoxin, which was from Latoxan (France). Testosterone, flutamide, and indomethacin were dissolved in absolute ethanol. Testosterone, at a stock concentration of 10 mM in absolute ethanol, was diluted to working concentrations in Krebs-Henseleit solution; glibenclamide was dissolved in dimethyl sulphoxide; 4-aminopyridine was dissolved in distilled water; aminoglutethimide was dissolved in hydrochloric acid. The remaining drugs were dissolved in the perfusion fluid. All drugs were made upon the day of the experiment.
Results
Basal perfusion pressure and established tone
In the 60 experiments carried out, basal perfusion pressure was 15.4±0.7 mmHg. Addition of methoxamine (3 – 25 μM) increased perfusion pressure by 76.2±4.2 mmHg (n=22). In the presence of TBA, 4-AP, and aminoglutethimide, a higher concentration of methoxamine (45 – 178 μM) was used to increase perfusion pressure by 67.0±8.1 mmHg (n=15). In the experiments involving glibenclamide, tone was increased by adding methoxamine (45 – 100 μM) and 5-HT (1.5 – 5 μM) to 88.0±12.2 mmHg (n=6), as methoxamine alone was inadequate. Addition of a NOS inhibitor, L-NAME (300 μM) to the perfusion fluid did not affect basal perfusion pressure, but it was found necessary to use a lower concentration of methoxamine (1 – 3 μM) to induce equivalent tone (83.6±10.9 mmHg, n=5). Addition of 60 mM KCl to the Krebs-Henseleit solution increased the perfusion pressure by 75.2±7.4 mmHg (n=5) above basal level. After addition of 30 mM KCl to the perfusion solution, methoxamine was also added to induce tone (96.1±2.1 mmHg; n=7), as 30 mM KCl alone was not enough to establish tone (5.55±1.19 mmHg).
Effects of L-NAME and endothelial denudation on testosterone-induced vasorelaxation
Testosterone (100 pM – 10 μM) caused concentration-dependent relaxations of methoxamine-induced tone (pEC50=9.47(9.22 – 9.73), 95% CI), maximal relaxation, Rmax=62.8±2.0%, n=6). L-NAME (300 μM) significantly (P<0.01) reduced both the maximal relaxation to and potency of testosterone (pEC50=8.79(8.65 – 8.95), Rmax=51.4±1.1%, n=6; Figure 1). As shown in Figure 1, removal of the endothelium also significantly (P<0.001) opposed maximal relaxation induced by testosterone (Rmax=46.9±2.8%, n=5) with no change in the potency (pEC50=9.30(8.84 – 9.76), n=5).
Figure 1.

Effects of NG-nitro-L-arginine methyl ester (L-NAME, 300 μM) and removal of the endothelium on testosterone-induced vasorelaxation in the rat isolated mesenteric arterial bed. Data are shown as mean±s.e.mean.
Effects of high extracellular K+ concentrations and K+ channel inhibitors on testosterone-induced vasorelaxation
Vasorelaxation to testosterone was significantly (P<0.001) inhibited by 30 mM KCl (Rmax=9.12±4.86%, n=7). Similarly, vasorelaxant effects of testosterone were abolished by 60 mM KCl and a contractile response was uncovered (pEC50=7.66(7.23 – 8.10), Rmax=−19.8±2.4%, P<0.001, n=5, Figure 2).
Figure 2.
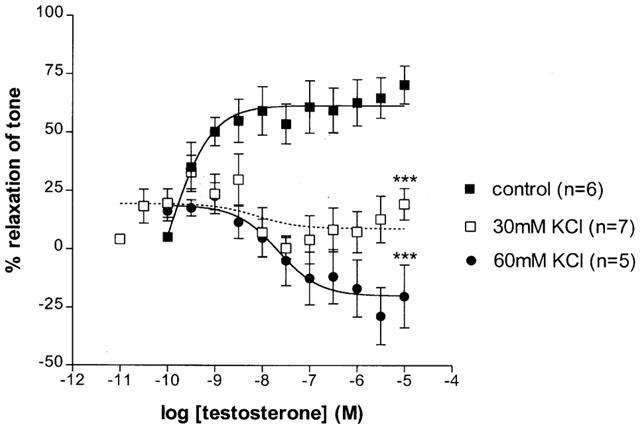
Effects of 30 and 60 mM KCl on testosterone-induced vasorelaxation in the rat isolated mesenteric arterial bed. Data are shown as mean±s.e.mean. ***Represents P<0.001 significantly different compared to control.
Testosterone-induced vasorelaxation was sensitive to TBA (300 μM), and once again vasoconstriction was uncovered (Rmax=−25.3±5.9%, P<0.001, n=5; Figure 3). Glibenclamide (10 μM) and 4-AP (1 mM) had no effect on testosterone-induced responses (glibenclamide: pEC50=9.39(6.22 – 9.57), Rmax=63.8±1.5%, n=6; 4-AP: pEC50=8.89(8.48 – 9.31), Rmax=86.6±3.7%, n=5, Figure 3).
Figure 3.

Effects of 4-aminopyridine (4-AP, 1 mM), tetrabutylammonium chloride (TBA, 300 μM), and glibenclamide (10 μM) on testosterone-induced vasorelaxation in the rat isolated mesenteric arterial bed. Data are shown as mean±s.e.mean. ***Represents P<0.001 significantly different compared to control.
The presence of ChTx (100 nM) significantly (P<0.05) inhibited vasorelaxation to 1 μM testosterone (control: 63.3±9.9%, n=6; ChTx: 11.9±12.7%, n=3, Figure 4).
Figure 4.

Effects of charybdotoxin (ChTx, 100 nM) on vasorelaxation to 1 μM testosterone in the rat isolated mesenteric arterial bed. Data are shown as mean±s.e.mean. *Represents P<0.05 significantly different compared to control.
Effects of testosterone receptor and aromatase inhibitors on testosterone-induced vasorelaxation
The addition of flutamide (10 μM), a testosterone receptor antagonist did not inhibit testosterone-induced vasorelaxation, but significantly (P<0.001) enhanced maximal relaxation to testosterone (pEC50=8.50(7.97 – 9.03), Rmax=90.1±4.2%, n=8).
Similarly, pre-treatment with aminoglutethimide (10 μM), an aromatase inhibitor, did not oppose testosterone-induced responses, but significantly (P<0.01) augmented maximal relaxation to testosterone (pEC50=8.27(7.83 – 8.70), Rmax=73.8±3.2%, n=5, Figure 5).
Figure 5.

Effects of flutamide (10 μM) and aminoglutethimide (50 μM) on testosterone-induced vasorelaxation in the rat isolated mesenteric arterial bed. Data are shown as mean±s.e.mean.
Discussion
The results of the present study demonstrate that testosterone (100 pM – 10 μM) causes potent and rapid vasorelaxations in the rat mesenteric arterial bed. The acute vasorelaxant effects of testosterone are partly mediated via endothelium- and NO-dependent, but not via receptor-dependent mechanisms. However, a substantial proportion of vasorelaxation to testosterone is mediated by increasing potassium efflux through BKCa channels, but not via KATP or KV channels.
In the present study, it was shown that testosterone caused acute vasorelaxations in the rat mesenteric arterial bed pre-contracted with methoxamine. These results are consistent with previous findings which show that testosterone induces vasorelaxation in contracted blood vessels, such as rat thoracic aorta (Costarella et al., 1996; Perusquia et al., 1996; Honda et al., 1999) and porcine (Chou et al., 1996), and human coronary arteries (Webb et al., 1999). Several studies also demonstrated that acute vasorelaxant effects of testosterone were observed at pharmacological concentrations (100 nM – 300 μM) (Yue et al., 1995; Chou et al., 1996; Costarella et al., 1996; Crews & Khalil, 1999; Honda et al., 1999). Interestingly, the present findings showed that testosterone, at low concentrations (100 pM – 1 nM), induced acute vasorelaxation, suggesting, therefore, that physiological (ca 20 nM; Johnson & Everitt, 1980) levels of testosterone may influence vascular tone.
Crews & Khalil (1999) demonstrated, in rat aortic strips, that vasorelaxation to relatively high concentrations of testosterone (100 nM – 10 μM) had a rapid time of onset between 30 s and 2 min. Moreover, acute vascular effects of testosterone were also previously reported in an in vivo study by Chou et al. (1996) showing that testosterone (0.1 – 1 μM) caused coronary relaxation within 90 to 120 s. Our results confirm that relaxant responses of the rat mesenteric arterial bed to testosterone were rapid in onset, occurring within 30 s to 20 min. Therefore, it is probable that testosterone-induced vasorelaxation is due to non-genomic mechanisms.
In contrast to the present study, others have demonstrated that testosterone enhanced vasoconstrictor responses to various agents, such as PGF2α, potassium chloride, endothelin-1 and 5-HT (Farhat et al., 1995; Teoh et al., 2000), and attenuated the effects of the endothelium-dependent vasorelaxants, bradykinin and the calcium ionophore, A23187 (Teoh et al., 2000). Clearly, in the present study testosterone caused relaxation rather than augmentation of methoxamine-induced tone. Although when K+ channels were blocked vasorelaxations to testosterone were abolished in the presence of 60 mM KCl or TBA, and that, under these conditions, contractile responses to testosterone were uncovered. Therefore, it appears that testosterone can cause mesenteric vasoconstriction, but only when K+ channels are inhibited.
Previous findings have demonstrated that vasorelaxation induced by testosterone was inhibited in the presence of NOS inhibitors in rat thoracic aorta (Costarella et al., 1996) and canine coronary artery (Chou et al., 1996). In the present investigation, we similarly found that testosterone-induced vasorelaxation was partly sensitive to L-NAME. Furthermore, we also found that removal of the endothelium opposed vasorelaxation to testosterone. These observations point to testosterone acting partly via endothelium- and NO-dependent pathways. Indeed, the effects of NOS inhibitor and removal of the endothelium had comparable effects, implying that NO is the principal endothelium-derived autacoid mediating these responses. The present findings are in agreement with the results of Chou et al. (1996), which showed that testosterone-induced vasorelaxation was inhibited in de-endothelialized canine coronary arteries. However, in our study there was a substantial endothelium-independent component. Indeed, other studies have demonstrated that neither removal of the endothelium nor the inclusion of NOS inhibitors affected vasorelaxation induced by testosterone in aortae from rat and rabbit, and coronary arteries from pig and rabbit (Yue et al., 1995; Perusquia et al., 1996; Crews & Khalil, 1999). Yue et al. (1995) also showed that methylene blue, an inhibitor of NO-activated guanylyl cyclase had no effects on testosterone-induced vasorelaxation in rabbit coronary artery and aorta. Furthermore, testosterone did not affect eNOS activity in the cultured bovine aortic endothelial cells (Goetz et al., 1999).
Clearly endothelial-derived NO contributes modestly to testosterone-induced vasorelaxation, therefore, we then sought to investigate the role of K+ channels. We found that vasorelaxation to testosterone was completely inhibited by raising the extracellular K+ concentrations to 30 and 60 mM, indicating that testosterone acutely causes vasorelaxation mainly by increasing K+ efflux (Adeagbo & Triggle, 1993). Furthermore, we also showed that testosterone-induced vasorelaxation was reduced by TBA, a selective inhibitor of calcium-activated K+ channels and ChTx, a selective BKCa channel inhibitor, but not by glibenclamide, a KATP channel inhibitor or 4-AP, a voltage-sensitive K+ channel inhibitor. From these results, we suggest that, in the rat mesenteric arterial bed, testosterone causes vasorelaxation via selective activation of BKCa channels. In agreement with our observations, Honda et al. (1999) recently demonstrated that tetraethylammonium, an inhibitor of calcium-activated potassium channels, inhibited vasorelaxation to testosterone in aortic rings from spontaneous hypertensive rats. In addition, previous studies also showed that glibenclamide had no effects on testosterone-induced vasorelaxation in coronary artery from rabbit (Yue et al., 1995) and dog (Chou et al., 1996). In contrast, neither glibenclamide or 4-AP inhibited vasorelaxation induced by testosterone in aortic rings from both Wistar-Kyoto rats and spontaneous hypertensive rats (Honda et al., 1999).
One possibility is that the endothelial-derived NO may act via activation of BKCa channels (Tare et al., 1990). However, there was a substantial endothelium-dependent component of vasorelaxation to testosterone. This suggests that endothelial-derived NO cannot account for all of the BKCa channel activation, which must occur by some other mechanism.
The present findings showed that testosterone-induced vasorelaxation was not inhibited by a testosterone receptor antagonist, flutamide, but potentiated. From these results, it is indicated that vasorelaxation induced by testosterone is mediated via testosterone receptor-independent pathway. Our results are consistent with a previous study in rabbit coronary artery and aorta which showed that vasorelaxation to testosterone was unaffected by flutamide (Yue et al., 1995). However, Murphy et al. (1999) demonstrated that testosterone inhibited contractile responses to prostaglandin F2α and KCl in pig coronary smooth muscle cells, and this was sensitive to flutamide. This may point to species and/or regional differences in the involvement of testosterone receptors. The potentiated relaxation in the presence of flutamide might actually point to testosterone receptors being coupled to vasoconstriction. Blocking these receptors with flutamide might leave the non-receptor mediated vasorelaxation unopposed, leading to enhanced relaxation.
Based on the possibility that testosterone could be metabolized to oestradiol by an aromatase enzyme, we investigated the possibility that testosterone was metabolized to a vasoactive mediator by using an aromatase inhibitor, aminoglutethimide. Our results showed that aminoglutethimide had no inhibitory effects on testosterone-induced vasorelaxation. The present findings are in agreement with a previous study by Yue et al. (1995) which demonstrated that, in rabbit coronary artery, an aromatase inhibitor, aminoglutethimide had no effect on vasorelaxation induced by testosterone. In addition, Chou et al. (1996) also showed that pre-treatment with an oestrogen receptor antagonist, ICI 182,780 had no effects on vasorelaxation of canine coronary artery to testosterone (1 μM). Taken together, it is suggested that vasorelaxation to testosterone is unlikely to be due to vasorelaxant effects of oestrogen (Yue et al., 1995; Chou et al., 1996). The small potentiation due to aminoglutethimide might be explained by reduced local metabolism of testosterone.
In summary, our findings demonstrate that, in the rat mesenteric arterial bed, testosterone causes acute vasorelaxations at physiologically relevant concentrations, which are partly mediated by endothelial NO via receptor-independent pathways. The inhibitory effects of TBA and ChTx on testosterone-induced responses suggest that vasorelaxation to testosterone involves mainly BKCa channel activation.
Abbreviations
- 4-AP
4-aminopyridine
- BKCa
large conductance calcium-activated potassium channels
- ChTx
charybdotoxin
- CVD
cardiovascular disease
- DMSO
dimethyl sulphoxide
- eNOS
endothelial nitric oxide synthase KATP, ATP-sensitive potassium channels
- Kv
voltage-sensitive K+ channels
- L-NAME
NG-nitro-L-arginine methyl ester
- NO
nitric oxide
- NOS
nitric oxide synthase
- TBA
tetrabutylammonium
References
- ADEAGBO A.S., TRIGGLE C.R. Varying extracellular [K+]: a functional approach to separating EDHF- and EDNO-related mechanisms in perfused rat mesenteric arterial bed. J. Cardiovasc. Pharmacol. 1993;21:423–429. [PubMed] [Google Scholar]
- ADAMS M.R., WILLIAMS J.K., KAPLAN J.R. Effects of androgens on coronary atheroslcerosis and atherosclerosis-related impairment of vascular responsiveness. Arterioscler. Thromb. Vasc. Biol. 1995;15:562–570. doi: 10.1161/01.atv.15.5.562. [DOI] [PubMed] [Google Scholar]
- CHOU T.M., SUDHIR K., HUTCHISON S.J., KO E., AMIDON T.M., COLLINS P., CHATTERJEE K. Testosterone induces dilation of canine coronary conductance and resistance arteries in vivo. Circulation. 1996;94:2614–2619. doi: 10.1161/01.cir.94.10.2614. [DOI] [PubMed] [Google Scholar]
- COSTARELLA C.E., STALLONE J.N., RUTECKI G.K., WHITTIER F.C. Testosterone causes direct relaxation of rat thoracic aorta. J. Pharmacol. Exp. Ther. 1996;277:34–39. [PubMed] [Google Scholar]
- CREWS J.K., KHALIL R.A. Gender-specific inhibition of Ca2+ entry mechanisms of arterial vasoconstriction by sex hormones. Clin. Exp. Pharmacol. Physiol. 1999;26:707–715. doi: 10.1046/j.1440-1681.1999.03110.x. [DOI] [PubMed] [Google Scholar]
- FARHAT M.Y., WOLFE R., VARGAS R., FOEGH M.L., RAMWELL P.W. Effects of testosterone treatment on vasoconstrictor response of left anterior descending coronary artery in male and female pigs. J. Cardiovasc. Pharmacol. 1995;25:495–500. doi: 10.1097/00005344-199503000-00023. [DOI] [PubMed] [Google Scholar]
- GOETZ R.M., THATTE H.S., PRABHAKAR P., CHO M.R., MICHEL T., GOLAN D.E. Estradiol induces the calcium-dependent translocation of endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. U.S.A. 1999;96:2788–2793. doi: 10.1073/pnas.96.6.2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARRIS D., MARTIN P.E.M., EVAN H., KENDALL D.A., GRIFFITH T.M., RANDALL M.D. Role of gap junctions in endothelium-derived hyperpolarizing factor responses and mechanisms of K+-induced relaxation. Eur. J. Pharmacol. 2000;402:119–128. doi: 10.1016/s0014-2999(00)00512-4. [DOI] [PubMed] [Google Scholar]
- HONDA H., UNEMOTO T., KOGO H. Different mechanisms for testosterone-induced relaxation of aorta between normotensive and spontaneously hypertensive rats. Hypertension. 1999;34:1232–1236. doi: 10.1161/01.hyp.34.6.1232. [DOI] [PubMed] [Google Scholar]
- JOHNSON M.H., EVERITT B.J. Essential Reproduction. Blackwell Scientific Publications; 1980. [Google Scholar]
- MCCREDIE R.J., MCCROHON J.A., TURNER L., GRIFFITHS K.A., HANDELSMAN D.J., CELERMAAJER D.A. Vascular reactivity is impaired in genetic females taking high-dose androgens. J. Am. Coll. Cardiol. 1998;32:1331–1335. doi: 10.1016/s0735-1097(98)00416-1. [DOI] [PubMed] [Google Scholar]
- MCCULLOCH A.I., BOTTRILL F.E., RANDALL M.D., HILEY C.R. Characterization and modulation EDHF-mediated relaxations in the rat isolated superior mesenteric arterial bed. Br. J. Pharmacol. 1997;120:1431–1438. doi: 10.1038/sj.bjp.0701066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURPHY J.G., KHALIL Decreased [Ca2+]i during inhibition of coronary smooth muscle concentraction by 17β-estradiol, progesterone, and testosterone. J. Pharmacol. Exp. Ther. 1999;291:44–52. [PubMed] [Google Scholar]
- PERUSQUIA M., HERNANDEZ R., MORALES M.A., CAMPOS M.G., VILLALON Role of endothelium in the vasodilating effect of progestins and androgens on the rat thoracic aorta. Gen. Pharmacol. 1996;27:181–185. doi: 10.1016/0306-3623(95)00091-7. [DOI] [PubMed] [Google Scholar]
- RANDALL M.D., GRIFFITH T.M. Differential effect of L-arginine on the inhibition by NG-nitro-L-arginine methyl ester of basal and agonist-stimulated EDRF activity. Br. J. Pharmacol. 1991;104:743–749. doi: 10.1111/j.1476-5381.1991.tb12498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RANDALL M.D., GRIFFITH T.M. Modulation of vasodilatation to levcromakalim by hypoxia and EDRF in the rabbit isolated ear: a comparison with pinacidil, sodium nitroprusside and verapamil. Br. J. Pharmacol. 1993;109:386–393. doi: 10.1111/j.1476-5381.1993.tb13581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RANDALL M.D., KENDALL D.A. Involvement of a cannabinoid in endothelium-derived hyperpolarizing factor-mediated coronary vasorelaxation. Eur. J. Pharmacol. 1997;335:205–209. doi: 10.1016/s0014-2999(97)01237-5. [DOI] [PubMed] [Google Scholar]
- RANDALL M.D., KENDALL D.A. Anandamide and endothelium-derived hyperpolarizing factor act via a common vasorelaxant mechanism in rat mesentery. Eur. J. Pharmacol. 1998;346:51–53. doi: 10.1016/s0014-2999(98)00003-x. [DOI] [PubMed] [Google Scholar]
- ROSANO G.M.C., LEONADO F., PAGNOTTA P., PELLICCIA F., PANINA G., CERQUETANI E., DELLA MONICA P., BONFIGLI B., VOLPE M., CHIERCHIA S. Acute anti-ischemic effect of testosterone in men with coronary artery disease. Circulation. 1999;99:1666–1670. doi: 10.1161/01.cir.99.13.1666. [DOI] [PubMed] [Google Scholar]
- TARE M., PARKINGTON H.C., COLEMAN H.A., NEILD T.O., DUSTING G.J. Hyperpolarization and relaxation of arterial smooth muscle caused by nitric oxide derived from the endothelium. Nature. 1990;346:69–71. doi: 10.1038/346069a0. [DOI] [PubMed] [Google Scholar]
- TEOH H., QUAN A., LEUNG S.W.S., MAN R.Y.K. Differential effects of 17beta-estradiol and testosterone on the contractile responses of porcine coronary arteries. Br. J. Pharmacol. 2000;129:1301–1308. doi: 10.1038/sj.bjp.0703164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TEP-AREENAN P., KENDALL D.A., RANDALL M.D. Vasorelaxation to 17β-oestradiol in the rat isolated mesenteric arterial bed. Br. J. Pharmacol. 2001;133:112P. doi: 10.1038/sj.bjp.0704522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEBB C., MCNEIL J., HAYWARD C.S., ZEIGLER D., COLLINS P. Effects of testosterone on coronary vasomotor regulation in men with coronary heart disease. Circulation. 1999;100:1690–1696. doi: 10.1161/01.cir.100.16.1690. [DOI] [PubMed] [Google Scholar]
- WILD R.A., BARTHOLEMEW M.J. The influence of body weight on lipoprotein lipids in patients with polycystic ovary syndrome. Am. J. Obstet. Gynecol. 1988;159:423–427. doi: 10.1016/s0002-9378(88)80099-1. [DOI] [PubMed] [Google Scholar]
- YUE P., CHATTERJEE K., BEALE C., POOLE-WILSON P.A., COLLINS P. Testosterone relaxes rabbit coronary arteries and aorta. Circulation. 1995;91:1154–1160. doi: 10.1161/01.cir.91.4.1154. [DOI] [PubMed] [Google Scholar]