

Abstract
The maintenance of renal function in decompensated cirrhosis is highly dependent on prostaglandins (PGs). Since PG synthesis is mediated by cyclooxygenase-1 and -2 (COX-1 and COX-2), the present study was designed to examine which COX isoform is involved in this phenomenon.
Renal COX-1 and COX-2 protein expression and distribution were analysed by Western blot and immunohistochemistry in nine rats with carbon tetrachloride-induced cirrhosis and ascites and 10 control animals. The effects of placebo and selective COX-1 (SC-560) and COX-2 (celecoxib) inhibitors on urine flow (V), urinary excretion of sodium (UNaV) and PGE2 (UPGE2V), glomerular filtration rate (GFR), renal plasma flow (RPF), the diuretic and natriuretic responses to furosemide and renal water metabolism were assessed in 88 rats with cirrhosis and ascites.
COX-1 protein levels were found to be unchanged in kidneys from cirrhotic rats. In contrast, these animals showed enhanced renal COX-2 protein expression which was focally increased in the corticomedullary region. Although UPGE2V was equally reduced by SC-560 and celecoxib, only SC-560 produced a significant decrease in UNaV, GFR and RPF and a pronounced impairment in the diuretic and natriuretic responses to furosemide in rats with cirrhosis and ascites. Neither SC-560 nor celecoxib affected renal water metabolism in cirrhotic rats.
These results indicate that despite abundant renal COX-2 protein expression, the maintenance of renal function in cirrhotic rats is mainly dependent on COX-1-derived prostaglandins.
Keywords: Experimental cirrhosis, renal COX expression, selective COX inhibition, renal function
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are among the most widely prescribed class of pharmaceutical agents world-wide, having broad clinical utility in treating pain, fever and inflammation (Payan & Katzung, 1995). Despite a relatively low incidence of renal side effects in healthy subjects, administration of NSAIDs to patients with unbalanced effective arterial blood volume, such as decompensated cirrhosis, represents a major clinical problem since renal function in these patients is critically dependent on prostaglandins (PGs) (Arroyo et al., 1983; 1986; Dunn, 1984). In fact, acute PG inhibition with NSAIDs in patients with cirrhosis and ascites is associated with a significant impairment in renal hemodynamics, sodium excretion, free water clearance and renal response to furosemide and spironolactone (Boyer et al., 1979; Zipser et al., 1979; Arroyo et al., 1983; 1986; Planas et al., 1983; Mirouze et al., 1983; Pérez-Ayuso et al., 1984; Dunn, 1984). Thus, in clinical practice, patients with decompensated cirrhosis cannot be treated with NSAIDs on a long term basis because of the high risk of developing renal failure and refractory ascites.
Cyclooxygenase (COX) is the key enzyme in the formation of PGs from arachidonate and is the major therapeutic target for NSAIDs (Vane, 1971). Two isoforms of COX, designated COX-1 and COX-2, have been identified (Kujubu et al., 1991, Masferrer et al., 1992; Hla & Neilson, 1992). COX-1 is ubiquitous and has been previously linked to the cytoprotective effects of PGs in the gut as well as to the integrity of platelet function. Conversely, COX-2 is undetectable in most tissues, but its expression can be induced by a variety of stimuli related to inflammatory response (Herschman, 1996). Conventional NSAIDs inhibit COX-1 and COX-2 and this feature accounts for both therapeutic and side effects (Warner et al., 1999). Recently, selective COX-2 inhibitors have been developed to effectively inhibit COX-2-dependent inflammation while sparing physiologic PG production (Seibert et al., 1994; Marnett & Kalgutkar, 1999). These novel compounds could potentially be the NSAIDs of choice in patients in whom, as occurs in cirrhosis with ascites, renal function is critically dependent on PGs.
To address the renal-damaging effects of NSAIDs in liver disease, we have previously used rats with carbon tetrachloride (CCl4)-induced cirrhosis, an experimental model that closely reproduces the systemic and renal abnormalities seen in human cirrhosis (Clària & Jiménez, 1999). In fact, CCl4-induced cirrhotic rats also show increased renal PG synthesis and develop renal failure after receiving conventional NSAIDs such as aspirin (Ros et al., 1995) and ketorolac (Bosch-Marcé et al., 1999). Interestingly, we have recently shown that renal function in these animals remains unaffected by the administration of SC-236 (a research pre-clinical compound with high selectivity for COX-2 (Bosch-Marcé et al., 1999)), suggesting that the renal side-effects of conventional NSAIDs in cirrhosis are secondary to COX-1 inhibition. Given that a selective COX-1 inhibitor was not available in that study, the current investigation was aimed to unequivocally establish which COX isoform is responsible for the synthesis of PGs involved in the maintenance of renal function in decompensated cirrhosis. To this end, we first analysed COX-1 and COX-2 protein expression and distribution by Western blot and immunohistochemistry, respectively, in kidneys from rats with CCl4-induced cirrhosis and ascites. In these animals, we next compared the effects of a selective COX-1 inhibitor (SC-560) with those of a selective COX-2 inhibitor (celecoxib) on renal hemodynamics and renal sodium handling. Subsequently, we assessed the effects of these COX inhibitors on the diuretic and natriuretic response to furosemide, and finally characterized which COX isoform is involved in the regulation of renal water metabolism in cirrhotic rats.
Methods
Materials
125I-Iothalamate, specific PGE2 EIA and the ECL system were from Amersham Pharmacia (Buckinghamshire, U.K.). PAH was obtained from Merck & Co (West Point, PA, U.S.A.). Sep-Pack C18 cartridges and HPLC columns were from Waters (Milford, MA, U.S.A.). Furosemide was from Hoechst Marion Roussel (Seguril®, Barcelona, Spain). MBF was from Strecks Laboratories (Omaha, NE, U.S.A.). Immobilon-P PVDF membranes were from Millipore (Bedford, MA, U.S.A.). Tween 80, aprotinin, leupeptin, pepstatin A and phenylmethylsulphonyl fluoride were purchased from Sigma Chemical Co (St Louis, MO, U.S.A.). Ketorolac, COX-1 and COX-2 protein standards, COX-1 (murine) monoclonal antibody and COX-2 (murine) polyclonal antibody (Western blot) were from Cayman Chemical Company (Ann Arbor, MI, U.S.A.). Polyclonal rabbit anti-human COX-2 serum and anti-ovine COX-1 monoclonal antibody were from Oxford Biomedical Research (Oxford, MI, U.S.A.). Selective COX-1 (SC-560) and COX-2 (celecoxib) inhibitors were provided by Pharmacia Research and Development (St Louis, MO, U.S.A.).
Induction of cirrhosis in rats
The study was performed in 97 rats with cirrhosis and ascites. Cirrhosis was induced by CCl4 inhalation in adult male Wistar rats (Charles-River, Saint Aubin les Elseuf, France) initially weighing 160 – 180 g, following a method described elsewhere (Clària & Jiménez, 1999). Cirrhotic rats were obtained from a group of 136 animals submitted to the cirrhosis induction protocol. Thirty-nine of these animals could not be included in the study for several reasons: 24 rats died before the development of ascites and/or impairment of renal water excretion, seven rats died before completing the experimental procedures and eight rats did not develop ascites and/or impairment to renal water excretion. Animals were maintained at the University of Barcelona animal facility and were fed ad libitum with standard chow and distilled water containing phenobarbital (0.3 g l−1) as drinking fluid. Cirrhotic rats were studied after the development of ascites, usually between 12 and 18 weeks of CCl4 administration.
Western blot analysis of COX-1 and COX-2 protein expression in renal tissue
Protein expression for COX-1 and COX-2 was assessed in renal tissue samples isolated from six cirrhotic rats with ascites and from seven control animals. Animals were anaesthetized with an i.m. injection of ketamine (50 mg kg−1) and the kidneys were rapidly dissected and stored in liquid nitrogen. Frozen tissues (150 – 180 mg) were homogenized in 30 mM Tris/HCl pH 7.4, 100 μM phenylmethylsulphonyl fluoride, containing protease inhibitors (1 μg ml−1 each): leupeptin, pepstatin A and aprotinin. The homogenate was centrifuged at 1000×g for 10 min and the supernatant was taken and subjected to a final centrifugation step at 100,000×g for 70 min. Total protein concentration was determined by the Bradford Protein Assay. Aliquots from each sample containing equal amounts of protein (30 – 50 μg) were resuspended in SDS-containing Laemmli sample buffer and heated at 100°C for 5 min, electrophoresed on 10 – 12% SDS-polyacrylamide gels and transferred overnight to PVDF membranes. The efficiency of the transfer was visualized by Ponceau staining. The blots were subsequently blocked for 6 h with 50 mM Tris/HCl pH 7.4 and 100 mM NaCl (TBS) containing 5% nonfat dry milk and 0.5% Tween 20, followed by incubation (1 : 1000 dilution) for 16 h with antisera specific for either COX-1 or COX-2. After washing three times for 5 min each with 0.1% Tween 20 in TBS, the blots were incubated for 1 h at room temperature with 1 : 2000 dilution of sheep anti-mouse or donkey anti-rabbit secondary antibodies conjugated to horseradish peroxidase. Bands were visualized by an enhanced chemiluminescence (ECL) detection system.
Immunohistochemical analysis of COX-1 and COX-2 protein distribution in renal tissue
Localization of COX-1 and COX-2 expression was assessed in renal tissue isolated from three cirrhotic rats with ascites and three control rats. Animals were anaesthetized with an i.m. injection of ketamine (50 mg kg−1) and sacrificed. The kidneys were rapidly resected and cut to the appropriate size to facilitate efficient fixative penetration of the molecular biology fixative (MBF). MBF is an aqueous buffered solution containing organic and inorganic salts, supplemented with a bacteriostatic and fungistatic agent and provides excellent preservation of tissue structure and COX antigenicity. Tissues were fixed for 24 h at 4°C, transferred to 70% ethanol, embedded in paraffin, sectioned at 5 μm onto Fisher Gold SuperFrost Plus glass slides and finally, deparaffinized in xylene and re-hydrated in descending alcohols. Tissues were then blocked for endogenous peroxidase (3% H2O2 in MeOH) and avidin/biotin (Avidin Biotin Blocking Kit SP-2001, Vector Laboratories, Burlingame, CA, U.S.A.). Sections were permeabilized in TNB-BB (0.1 M Tris pH 7.5, 0.015 M NaCl, 0.5% blocking agent, 0.3% Triton-X, 0.2% saponin), and incubated overnight at 4°C with polyclonal rabbit anti-human COX-2 serum diluted to 2.5 μg ml−1 (Oxford Biomedical Research). COX-1 was immunolocalized with the anti-ovine monoclonal antibody (diluted to 1 μg ml−1) (Oxford Biomedical Research). Immunoreactive complexes were detected using tyramide signal amplification (TSA-indirect, NEN Life Science), and were visualized with the peroxidase substrates AEC or DAB. Control sections were treated with isotype-matched controls, or were pre-incubated with 100 fold excess of human recombinant COX-1 or COX-2 protein. To ensure rigid inter-slide consistency all slides were stained simultaneously on an autoimmunostainer. Pathological changes were described by a licensed pathologist following preparation of standard hematoxylin and eosin staining.
Drug administration studies
Study 1
The goal of this study was to investigate the effects of selective COX-1 (SC-560) and COX-2 (celecoxib) inhibitors on renal function in conscious rats with cirrhosis and ascites. For this purpose, 28 rats with cirrhosis and ascites were anaesthetized with an i.m. injection of ketamine (50 mg kg−1) and prepared with a PVC-50 catheter in the left femoral artery for hemodynamic measurements and blood collection, a double-lumen PVC-100 catheter in the right jugular vein for the infusion of substances, and a PVC-50 tube in the bladder for urine collection. The femoral artery catheter was connected to a highly sensitive transducer and a multichannel recorder (MX4P and MT4, Lectromed Ltd, Jersey, Channel Islands, U.K.) to register pulsatile mean arterial pressure (MAP) and heart rate (HR), while the jugular vein catheter was continuously perfused with Ringer solution (0.5 ml h−1). Catheters were connected to a swivel (Harvard Apparatus, South Natick, MA, U.S.A.) and animals were placed in rectangular cages with no restriction of movement. Twenty-four hours later, a priming dose of 125I-Iothalamate (0.37 μCi) and para-aminohippurate (PAH) (3 mg) was given through the jugular vein, followed by a constant infusion (2 ml h−1) of a Ringer solution containing 125I-Iothalamate (0.37 μCi ml−1) and PAH (3 mg ml−1). Animals were equilibrated for 1 h and after two baseline 20-min urine collection periods received 1 ml kg−1 of 2% Tween 80 in Dulbecco's Phosphate Buffered Saline (DPBS) containing the following: Group 1 (n=8): Placebo; Group 2 (n=10): Selective COX-1 inhibitor, SC-560 (20 mg kg−1, i.v.); Group 3 (n=10): Selective COX-2 inhibitor, celecoxib (20 mg kg−1, i.v.).
The dose of SC-560 was selected from previous studies demonstrating inhibition of constitutive PG synthesis in euvolemic rats (Smith et al., 1998; Wallace et al., 2000; Gretzer et al., 2001). The dose of celecoxib was selected from previous studies reporting complete inhibition of PG synthesis in the rat carrageenan footpad and airpouch models of inflammation (Smith et al., 1998; Wallace et al., 2000). Moreover, by using the zymosan-induced inflammation model in rats, Niederberger et al. (2001) have recently reported that celecoxib retains its anti-inflammatory efficacy at doses of up to 100 mg kg−1.
Subsequently, four 20-min urine collection periods were performed and changes in MAP, HR, urine volume (V), urinary sodium excretion (UNaV), glomerular filtration rate (GFR) and renal plasma flow (RPF) were recorded during the entire period. At the midpoint of each clearance period, a 0.5 ml blood sample was obtained. Urinary PGE2 excretion (UPGE2V) was measured under baseline conditions and during the fourth 20-min urine collection period following the administration of COX inhibitors.
Sodium and potassium concentrations in plasma and urine samples were measured by flame photometry (IL 943, Instrumentation Laboratory, Lexington, MA, U.S.A.) and serum osmolality by the osmometric depression of the freezing point in an Advanced Instruments Osmometer (model 3MO, Needham, MA, U.S.A.). GFR and RPF were estimated from 125I-Iothalamate and PAH clearance respectively, using the standard formula: CIothalamate=urinary 125I counts×V/plasma 125I counts and CPAH=urinary PAH concentration×V/plasma PAH concentration. The radioactivity of 125I-Iothalamate samples was counted in an automatic scintillation counter (Wallac, Turku, Finland). PAH was determined by RP – HPLC as described (Decosterd et al., 1997) with modifications. Briefly, 2 μl of plasma or urine in MeOH (final volume 25 μl) were injected into a RP – HPLC system which consisted of a Waters integrated system controller (model 600E) equipped with a 996 Photodiode Array Detector (DAD) and Millennium HPLC analysis software (Waters, Milford, MA, U.S.A.). For analysis of PAH, a HPLC column (either a Kromasil 100 C18 (5 μm, 4.6×250 mm) or a Tracer Extrasil ODS2 (5 μm, 4.6×250 mm) was eluted with a linear gradient of MeOH:citrate-citric acid pH 3.9 (from 1 : 99 to 15 : 85, v v−1 over 15 min) at a flow rate of 1.0 ml min−1. The spectrophotometric UV-VIS DAD was set at 273 nm. PGE2 levels in urine were measured by specific EIA after extraction of samples on Sep-Pack C18 cartridges.
Study 2
The aim of this study was to investigate the effect of SC-560 and celecoxib on the renal diuretic and natriuretic responses to furosemide in rats with cirrhosis and ascites. Twenty-six rats with cirrhosis and ascites were prepared as described in Study 1 and received placebo (n=6), the selective COX-1 inhibitor SC-560 (20 mg kg−1, i.v.) (n=10) or the selective COX-2 inhibitor celecoxib (20 mg kg−1, i.v.) (n=10). Sixty minutes later, they received an i.v. injection of furosemide (5 mg kg−1) and changes in V, UNaV, GFR and RPF were recorded during four 20-min urine collection periods. In an additional group of 13 rats with cirrhosis and ascites, the effects of increasing doses of SC-560 (10 and 30 mg kg−1, i.v) and celecoxib (10 and 30 mg kg−1, i.v,) on the diuretic and natriuretic response to furosemide were also evaluated.
Study 3
The aim of this study was to investigate the effect of SC-560 and celecoxib on renal water metabolism in rats with cirrhosis and ascites. Renal water metabolism was estimated in cirrhotic rats resting in metabolic cages, as follows: two hours after removing water and food, a water load (50 ml kg−1) was administered via a gastric tube inserted under light ether anaesthesia. Immediately afterwards, the animals were reintroduced into their metabolic cages where each spontaneous urine void was individually collected for a total period of 3 h. The renal ability of these animals to excrete free water was established by measuring the minimum urinary osmolality (mUOsm) from the osmometric depression of the freezing point of each spontaneously voided sample and by gravimetically measuring the percentage (%) of the water load excreted during the 3-h urine collection period. After a 24-h period, animals received 1 ml kg−1 of 2% Tween 80 in 0.5% carboxymethylcellulose containing the following treatments and doses selected from Smith et al. (1998), Wallace et al. (2000), Gretzer et al. (2001) and Niederberger et al. (2001). Group 1 (n=6): Placebo; Group 2 (n=7): SC-560 (30 mg kg−1, p.o.); Group 3 (n=8): celecoxib (30 mg kg−1, p.o.).
Twenty minutes after receiving the drug, the animals were submitted to a second oral water load of 50 ml kg−1 and the percentage of water load excreted and the mUOsm were again determined during the 3-h period following the water administration (see above).
At the end of each study, tissue specimens were obtained from the middle liver lobe of each animal, fixed in 10% buffered formalin and stained with hematoxylin-eosin, reticulin and Masson's trichrome for histological examination.
Statistical analysis of the results was performed by one-way ANOVA, Newman-Keuls and paired and unpaired Student's t-tests, as appropriate. Data are expressed as mean±s.e.m. and were considered significant at a P level of 0.05 or less.
All animal studies were conducted in accordance with the criteria of the Investigation and Ethics Committee of the Hospital Clínic and the European Community laws governing the use of experimental animals.
Results
Animals treated with CCl4 showed the characteristic features of micronodular cirrhosis (i.e. a stage of well-established cirrhosis with a combination of nodular regeneration of liver cell plates surrounded by thick connective tissue septa and proliferating bile ducts). The volume of ascites of cirrhotic rats ranged between 12 and 40 ml.
Constitutive COX-1 and COX-2 protein expression was detected by Western blot and immunohistochemistry analysis in renal tissue collected from rats with cirrhosis and ascites (Figures 1, 2 and 3). However, whereas COX-1 protein expression was found to be unchanged as compared to controls, COX-2 expression was consistently up-regulated in kidneys from cirrhotic rats (Figure 1). In these animals, COX-1 immunoreactivity was observed in the collecting ducts, renal vasculature, and papillary interstitial cells, being its expression intense in the papillary collecting ducts and low-to-moderate in the cortical collecting ducts (Figure 2). On the other hand, positive COX-2 immunoreactive protein was localized in the macula densa and outer medulla collecting ducts (Figure 3).
Figure 1.
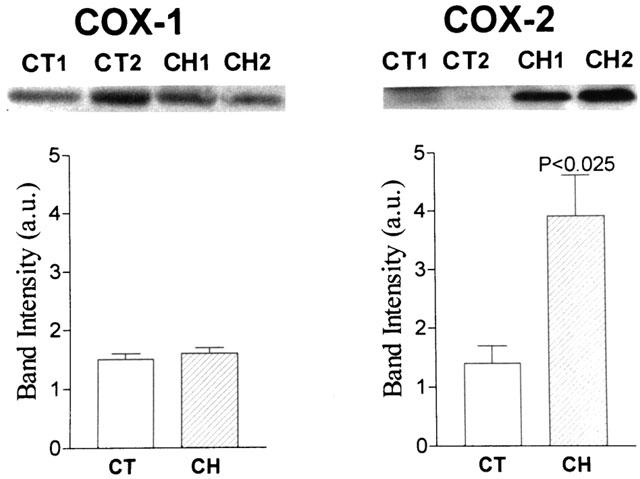
Renal COX-1 and COX-2 protein levels. Upper panels: representative Western blot analysis of COX-1 (Mr, 70 kDa) and COX-2 (Mr, 72 kDa) in renal tissue samples from two control rats (CT1 and CT2) and two cirrhotic rats with ascites (CH1 and CH2). Renal protein extracts were electrophoresed and probed with specific anti-COX-1 and anti-COX-2 antibodies. Lower panels: COX-1 and COX-2 band intensities were determined by scanning densitometry. Results show the mean±s.e.m. of six cirrhotic and seven control rats.
Figure 2.
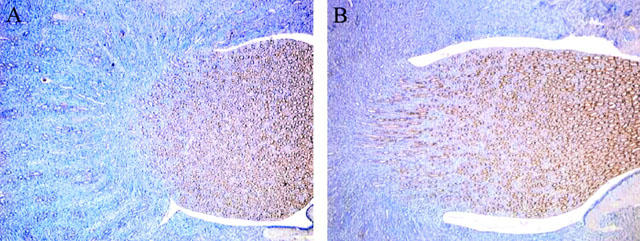
Localization of COX-1 protein expression in control rats (A) and rats with cirrhosis and ascites (B). COX-1 is observed in the collecting ducts, renal vasculature, and papillary interstitial cells. Intense COX-1 immunoreactivity is detected in the papillary collecting ducts, and low-to-moderate expression detected in the cortical collecting ducts.
Figure 3.
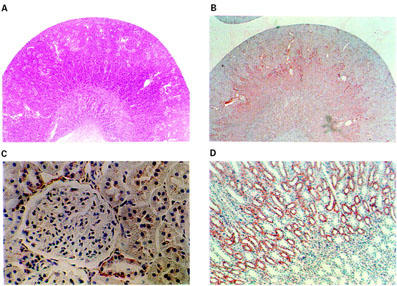
Localization of immunoreactive COX-2 protein expression in kidneys from rats with cirrhosis and ascites. Low-magnification micrograph showing a longitudinal section of the kidney stained with hematoxylin-eosin (A) or reacted with a polyclonal rabbit antimurine COX-2 (B). In the cortex, COX-2 staining was observed in the macula densa (C), whereas in the medulla, strong immunoreactivity for COX-2 was localized in tubular epithelial cells (D).
Study 1: Effect of selective COX inhibitors on renal hemodynamics and renal excretory function in rats with cirrhosis and ascites
No statistically significant differences were detected in the three groups of rats receiving placebo, SC-560 or celecoxib with respect to baseline body weight, MAP, HR, serum sodium and potassium concentrations, serum osmolality, V, UNaV, UPGE2V, GFR and RPF (Table 1).
Table 1.
Baseline values for the three groups of cirrhotic rats receiving placebo, SC-560 or celecoxib included in Study 1.
Figure 4 shows the effects of placebo, SC-560 or celecoxib on V, UNaV, GFR and RPF in cirrhotic rats with ascites. The i.v. administration of the selective COX-1 inhibitor SC-560 significantly decreased V, UNaV, GFR and RPF in cirrhotic rats. In these animals, renal function was impaired as early as 20 min following SC-560 administration and experienced a progressive decline throughout the entire study period, except for UNaV which reached a maximum plateau at 40 min after the administration of the drug (Figure 4). In contrast, the renal effects associated with selective COX-2 inhibition with celecoxib were similar to those of placebo except that celecoxib induced a reduction in V which reached statistical significance at 60 min after administration of the drug (Figure 4). This effect, however, was transient and rapidly reversed and after 80 min of receiving celecoxib, the V values in this group of animals did not differ from those of the placebo group. No changes in MAP and HR were observed throughout the entire course of the study in any group of cirrhotic animals (data not shown).
Figure 4.
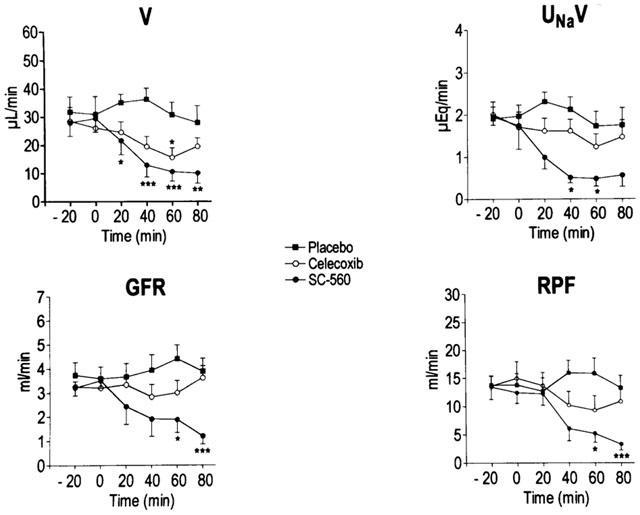
Effect of selective COX inhibitors on renal function in rats with cirrhosis and ascites. Urine flow (V), urinary sodium excretion (UNaV), glomerular filtration rate (GFR) and renal plasma flow (RPF) were measured before (−20 and 0 min) and up to 80 min after the i.v. administration of placebo (n=8), the selective COX-1 inhibitor (SC-560, 20 mg kg−1, n=10) or the selective COX-2 inhibitor (celecoxib, 20 mg kg−1, n=10) to rats with cirrhosis and ascites. Results are given as mean±s.e.m. Data are compared with the Student's t-test for paired data. *P<0.05; **P<0.025 and ***P<0.001 vs values at 0 min.
The administration of SC-560 and celecoxib to cirrhotic rats was associated with a 48% (from 312±82 to 163±54 pg min−1, P<0.05) and 47% (from 389±93 to 203±111 pg min−1), inhibition in UPGE2V, respectively.
Study 2: Effect of selective COX inhibitors on the renal response to furosemide in rats with cirrhosis and ascites
Figure 5 shows the results obtained in the three groups of animals included in this study. As expected, in cirrhotic rats receiving placebo, the i.v. injection of 5 mg kg−1 of furosemide was followed by a pronounced increase in V (diuretic effect) and UNaV (natriuretic effect) (Figure 5). This effect was rapid (reached a maximum after 20 min) and transient (disappeared in most cases 60 – 80 min after the administration of furosemide). Parallel changes in GFR and RPF were also observed in this group of animals following the administration of furosemide, although the duration of these changes was shorter than that elicited on V and UNa V (data not shown). The injection of furosemide to cirrhotic rats treated with the selective COX-1 inhibitor SC-560 was associated with reduced diuretic and natriuretic response to this drug (Figure 5). In sharp contrast, the diuretic and natriuretic efficacy of furosemide in cirrhotic rats was not affected by celecoxib (Figure 5). Consequently, rats treated with SC-560 showed a pronounced reduction in overall urine output and cumulative sodium excretion over the 80 min period after furosemide injection, whereas these parameters remained unaltered in those rats treated with celecoxib (Figure 6). The decrease in the response to furosemide in rats receiving SC-560 was dose-dependent and reached statistical significance at doses of 20 and 30 mg kg−1 (Figure 6).
Figure 5.
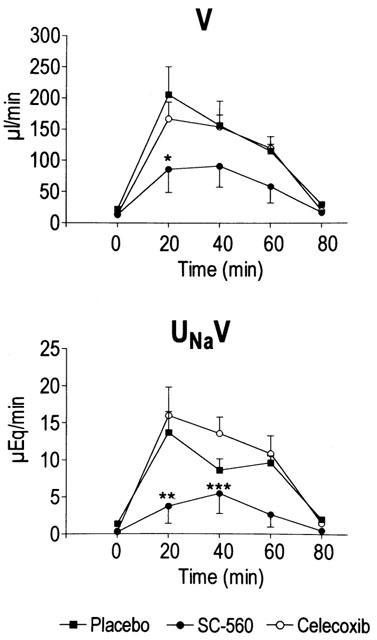
Effect of selective COX inhibitors on the renal response to furosemide in rats with cirrhosis and ascites. Animals received i.v. placebo (n=6), the selective COX-1 inhibitor (SC-560, 20 mg kg−1, n=10) or the selective COX-2 inhibitor (celecoxib, 20 mg kg−1, n=10) and 1 h later they were challenged with an i.v. injection of furosemide (5 mg kg−1) (time 0 min). Changes in urine flow (V) and urinary sodium excretion (UNaV) were recorded for four 20-min urine collection periods after the injection of furosemide. Results are given as mean±s.e.m. Data are compared by the Student's t-test for unpaired data. *P<0.05; **P<0.025 and ***P<0.005 vs placebo-treated group.
Figure 6.
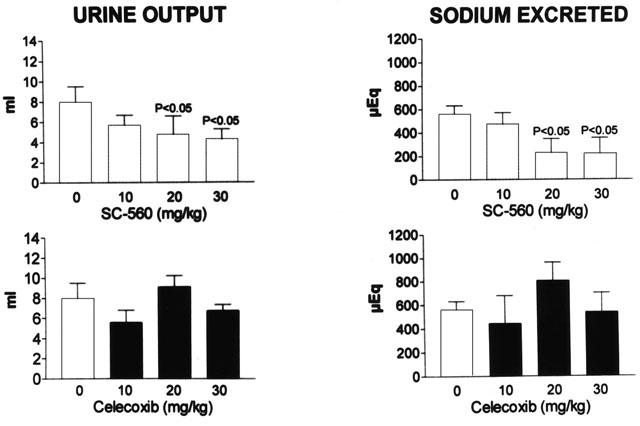
Diuretic and natriuretic responses to furosemide in rats with cirrhosis and ascites treated with increasing doses of selective COX inhibitors. Graph bars show the overall urine output and the cumulative sodium excretion over the 80 min after the administration of furosemide (5 mg kg−1) to different groups of rats treated with placebo (0 mg kg−1), SC-560 (10, 20 or 30 mg kg−1) or celecoxib (10, 20 or 30 mg kg−1). Results are given as mean±s.e.m. Data are compared by the Student's t-test for unpaired data and P values denote statistically significant differences with respect to the placebo-treated group.
Study 3: Effects of COX inhibitors on renal water metabolism in rats with cirrhosis and ascites.
Under baseline conditions, all three treatment groups exhibited similar renal ability to excrete free water, as estimated by the percentage of water load excreted (placebo: 82±11%; SC-560: 92±6% and celecoxib: 86±7%) and mUOsm (Placebo: 103±15 mOsm kg−1; SC-560: 69±5 mOsm kg−1 and celecoxib: 88±17 mOsm kg−1). Figure 7 shows the percentage of water load excreted and mUOsm under baseline conditions and following the oral administration of the drugs to rats with cirrhosis and ascites. In these animals, neither placebo, SC-560 or celecoxib produced any significant change in either the percentage of water load excreted or mUOsm (Figure 7).
Figure 7.
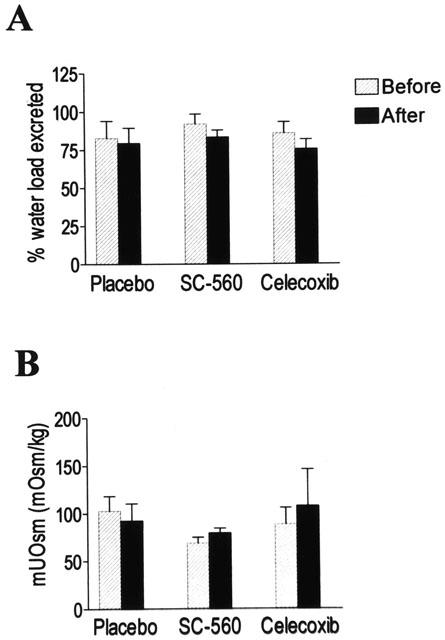
Effects of selective COX inhibitors on renal water metabolism in rats with cirrhosis and ascites. The percentage (%) water load excreted (A) and the mUOsm (B) were measured before and after the administration of placebo (n=6), SC-560 (30 mg kg−1, n=7) or celecoxib (30 mg kg−1, n=8) to cirrhotic rats with ascites. Results represent the mean±s.e.m. and data are compared by the Student's t-test for paired data.
Discussion
Renal PG synthesis is markedly increased in patients with cirrhosis and ascites as compared to patients with compensated cirrhosis (no ascites) and normal subjects (Arroyo et al., 1986; Dunn, 1984; Arroyo et al., 1983; Planas et al., 1983). In compensated cirrhosis, this increase is a homeostatic response to antagonize the exacerbated activity of the endogenous vasoconstrictor systems (i.e. the renin-angiotensin system, the sympathetic nervous system and the antidiuretic hormone (ADH)). Two lines of evidence support this contention. Firstly, acute PG inhibition with NSAIDs (indomethacin, ibuprofen, naproxen, sulindac or aspirin) in patients with cirrhosis and ascites is associated with a significant decrease in V, UNaV, GFR and RPF (Dunn, 1984; Arroyo et al., 1983; Boyer et al., 1979; Mirouze et al., 1983; Zipser et al., 1979). Patients with high plasma renin activity and plasma norepinephrine concentrations are particularly sensitive to these adverse effects. NSAIDs do not impair renal function in patients with compensated cirrhosis who do not show increased activity of the renin-angiotensin and sympathetic nervous systems. Secondly, acute PG inhibition with aspirin in patients with cirrhosis, ascites and increased plasma ADH levels is associated with a significant reduction in the renal ability to excrete free water (Pérez-Ayuso et al., 1984). In addition, PGs are also important in the renal response to diuretics since the administration of aspirin, naproxen, indomethacin and sulindac suppresses the renal hemodynamic effect and reduces the natriuretic efficacy of furosemide in cirrhotic patients with ascites (Planas et al., 1983).
Although the role of PGs in the maintenance of renal function in cirrhosis is well established, at present it is unknown which COX isoform (COX-1 or COX-2) is responsible for the synthesis of PGs involved in renal homeostasis in this disease. Under normal conditions, both COX-1 and COX-2 are constitutively expressed in the kidneys (O'neill & Ford-Hutchinson, 1993; Harris et al., 1994; Vio et al., 1997; Jensen & Kurtz, 1997; Khan et al., 1998; Bosch-Marcé et al., 1999). Specifically, in normal rats, COX-1 is mainly expressed in cells of the collecting duct and renal vasculature and in a small number of papillary interstitial cells, while COX-2 expression is consistently focal and limited to the macula densa of the juxtaglomerular apparatus, epithelial cells of the thick ascending limb and papillary interstitial cells (Khan et al., 1998; Vio et al., 1997; Harris et al., 1994; Jensen & Kurtz, 1997). Our results show that both COX-1 and COX-2 are also present in kidneys from rats with cirrhosis and ascites. Moreover, whereas the expression of COX-1 was found to be unchanged, the expression of COX-2 was up-regulated in kidneys of cirrhotic rats. These findings are consistent with the notion that COX-2, but not COX-1, is regulated in a cell-specific fashion in response to altered volume status. Indeed, increased COX-2 expression has been reported in the macula densa and peri-macula densa region of rats and dogs with chronic salt depletion and in the renal medulla of rats with a high-salt diet (Khan et al., 1998; Harris et al., 1994; Jensen & Kurtz, 1997; Yang et al., 1998). Furthermore, increased COX-2 expression has been reported in the renal medulla of water-deprived rats (Yang et al., 1999). Collectively, these results suggest that renal COX-2 up-regulation in cirrhosis is likely to be a consequence of changes in volume status, although the exact mechanism for this phenomenon is, at present, not understood.
Despite cirrhotic rats showing increased COX-2 protein immunoreactivity in the renal area, renal function in these animals appears to be mainly dependent on PGs derived from COX-1. In fact, in Study 1 of this investigation, we observed a significant inhibition in renal PGE2 synthesis concomitant with marked impairment in renal haemodynamics and renal sodium handling following the administration of a selective COX-1 inhibitor (SC-560) to CCl4-induced cirrhotic rats. In contrast, the selective COX-2 inhibitor celecoxib did not compromise renal function in cirrhotic rats. Interestingly, and although celecoxib administration was not associated with severe renal effects in cirrhotic rats, this compound reduced UPGE2V to levels equivalent to SC-560. McAdam et al. (1999) also found a similar suppression in urinary PG excretion in both healthy subjects treated with celecoxib and in those receiving ibuprofen. Taken together, these results suggest that urinary PGs are derived from both COX-1 and COX-2, yet only those from COX-1 are involved in the maintenance of renal function in cirrhosis. The physiological significance of PGs derived from COX-2 in the kidneys, not only in disease states, but also in healthy conditions, remains to be determined.
PGs are clearly involved in the renal natriuretic response to loop diuretics (Katayama et al., 1984). Loop diuretics, such as furosemide, are the most powerful diuretics currently available. These drugs inhibit sodium reabsorption in the ascending limb of the loop of Henle by acting on a specific co-transport system, the Na+-2Cl−K+ carrier (Puschett, 1981). Loop diuretics also increase GFR and renal blood flow and the renal production of PGE2 whereas NSAIDs can modulate the renal vasodilatory effect and the natriuretic efficiency of these drugs (Katayama et al., 1984). The results obtained in Study 2 demonstrate that response to furosemide in cirrhotic rats is mainly dependent on COX-1 derived PGs and that selective COX-2 inhibition spares furosemide-induced renal salt transport in cirrhosis. It is noteworthy to point out that in cirrhotic rats the effects of selective COX-1 inhibition with SC-560 on furosemide-induced diuretic and natriuretic response were dose-dependent. However, contrary to what was previously reported by Katayama et al. (1984), who found that indomethacin can either enhance or inhibit the effect of furosemide-induced natriuresis depending on the dose of this conventional NSAID used, we found that at low doses (10 mg kg−1), SC-560 induced no changes whereas higher doses (20 and 30 mg kg−1) significantly inhibited the response to furosemide.
PGs are also involved in renal water transport by modulating the tubular actions of ADH (Mattix & Badr, 2000). Specifically, PGE2 inhibits the hydroosmotic effect of ADH whereas PG inhibition with NSAIDs enhances the tubular effect of this hormone (Mattix & Badr, 2000; Hébert et al., 1990). In our study, neither SC-560 nor celecoxib modified renal water metabolism in rats with cirrhosis and ascites. These results are in sharp contrast with those previously obtained with the NSAID ketorolac which significantly impaired the ability to excrete free water in cirrhotic rats (Bosch-Marcé et al., 1999). Given that ketorolac is a mixed COX-1/COX-2 inhibitor, these findings suggest that renal water homeostasis in cirrhosis is dependent on PGs derived from both COX isoforms. Thus, a compensatory mechanism appears to be present in cirrhotic rat kidneys, in such a way that when only one isoform is inhibited, water metabolism can be maintained by PGs produced by the other isoform.
Controlled clinical studies have demonstrated that selective COX-2 inhibitors have equal therapeutic efficacy but with a lower incidence of gastrointestinal ulcers and erosions than conventional NSAIDs (Emery et al., 1999; Bombardier et al., 2000). The marketed selective COX-2 inhibitors rofecoxib and celecoxib have also been evaluated in randomized controlled trials in terms of their effects on renal PGs, renal function and occurrence of adverse renal events. Whereas in elderly subjects, rofecoxib did not produce any significant alteration in GFR after 2 weeks of treatment, it produced a reduction in urinary sodium excretion similar to that with indomethacin during the first 3 days of therapy (Catella-Lawson et al., 1999). In contrast, in elderly patients with mild renal impairment and stabilized on a low-sodium diet, rofecoxib produced decreases in GFR of similar magnitude to those observed in indomethacin-treated subjects, without affecting urinary sodium excretion (Swan et al., 2000). In elderly patients, celecoxib, like rofecoxib, failed to decrease GFR, but did produce transient reductions in urinary sodium excretion similar to those of naproxen (Whelton et al., 2000). Finally, in a trial in salt-depleted subjects, celecoxib not only promoted sodium retention but also produced significant dose-related decreases in GFR (Rossat et al., 1999). Therefore, the unwanted renal-side effects of selective COX-2 inhibitors still remain, at present, uncharacterized. This subject is particularly important in circumstances more susceptible to NSAID-induced renal failure, such as advanced liver disease, congestive heart failure and nephrotic syndrome. For this reason, our findings in the experimental model of cirrhosis warrant further studies in humans investigating the feasibility of selective COX-2 inhibitors possibly being the anti-inflammatory option of choice in patients with chronic liver disease.
Acknowledgments
These studies were supported in part by Ministerio de Ciencia y Tecnología (SAF 00/0043) and Fondo de Investigaciones Sanitarias (FIS 00/0616). M. López-Parra and E. Titos were supported by Ministerio de Ciencia y Tecnología and Ministerio de Sanidad y Consumo (BEFI 98/9314), respectively. A. Planagumà received a fellowship from IDIBAPS. The authors are indebted to M. Borrajo for expert technical assistance.
Abbreviations
- Celecoxib
4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]
- GFR
glomerular filtration rate
- NSAIDs
nonsteroidal anti-inflammatory drugs
- PG
prostaglandin
- RPF
renal plasma flow
- SC-560
[5-(4-chlorophenyl)-1-(4-methoxyphenyl)-3-trifluoromethylpyrazole)
- V
urine flow
- UPGE2V
urinary PGE2 excretion
- UNaV
urinary sodium excretion
References
- ARROYO V., PLANAS R., GAYA J., DEULOFEU R., RIMOLA A., PÉREZ-AYUSO R.M., RIVERA F., RODÉS J. Sympathetic nervous activity, renin-angiotensin system and renal excretion of prostaglandin E2 in cirrhosis. Relationship to functional renal failure and sodium and water excretion. Eur. J. Clin. Invest. 1983;13:271–278. doi: 10.1111/j.1365-2362.1983.tb00100.x. [DOI] [PubMed] [Google Scholar]
- ARROYO V., GINÉS P., RIMOLA A., GAYA J. Renal function abnormalities, prostaglandins, and effects of nonsteroidal anti-inflammatory drugs in cirrhosis with ascites. An overview with emphasis on pathogenesis. Am. J. Med. 1986;81:104–122. doi: 10.1016/0002-9343(86)90912-5. [DOI] [PubMed] [Google Scholar]
- BOMBARDIER C., LAINE L., REICIN A., SHAPIRO D., BURGOS-VARGAS R., DAVIS B., DAY R., FERRAZ M.B., HAWKEY C.J., HOCHBERG M.C., KVIEN T.K., SCHNITZER T.J. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. N. Engl. J. Med. 2000;343:1520–1528. doi: 10.1056/NEJM200011233432103. [DOI] [PubMed] [Google Scholar]
- BOSCH-MARCÉ M., CLÀRIA J., TITOS E., MASFERRER J.L., ALTUNA R., POO J.L., JIMÉNEZ W., ARROYO V., RIVERA F., RODÉS J. Selective inhibition of cyclooxygenase-2 spares renal function and prostaglandin synthesis in cirrhotic rats with ascites. Gastroenterology. 1999;116:1167–1175. doi: 10.1016/s0016-5085(99)70020-x. [DOI] [PubMed] [Google Scholar]
- BOYER T.D., ZIA P., REYNOLDS T.B. Effect of indomethacin and prostaglandin A1 on renal function and plasma renin activity in alcoholic liver disease. Gastroenterology. 1979;77:215–222. [PubMed] [Google Scholar]
- CATELLA-LAWSON F., MCADAM B., MORRISON B.W., KAPOOR S., KUJUBU D., ANTES L., LASSETER K.C., QUAN H., GERTZ B.J., FILTZGERALD G.A. Effects of specific inhibition on cyclooxygenase-2 on sodium balance, hemodynamics and vasoactive eicosanoids. J. Pharm. Exp. Ther. 1999;289:735–741. [PubMed] [Google Scholar]
- CLÀRIA J., JIMÉNEZ W.Renal dysfunction and ascites in carbon tetrachloride-induced cirrhosis in rats The Liver and the Kidney 1999Boston: Blackwell Science; 379–396.ed. Arroyo, V., Schrier, R.W., Rodés, J., Ginès, P., pp [Google Scholar]
- DECOSTERD L.A., KARAGIANNIS A., ROULET J.M., BÉLAZ N., APPENZELLER M., BUCLIN T., VOGEL P., BIOLLAZ J. High-performance liquid chromatography of the renal blood flow marker p-aminohippuric acid (PAH) and its metabolite N-acetyl PAH improves PAH clearance measurements. J. Chromatog. 1997;703:25–36. doi: 10.1016/s0378-4347(97)00396-4. [DOI] [PubMed] [Google Scholar]
- DUNN M.J. Role of eicosanoids in the control of renal function in severe hepatic disease. Gastroenterology. 1984;87:1392–1395. [PubMed] [Google Scholar]
- EMERY P., ZEIDLER H., KVIEN T.K., GUSLANDI M., NAUDIN R., STEAD H., VERBURG K.M., ISAKSON P.C., HUBBARD R.C., GEIS G.S. Celecoxib versus diclofenac in long-term management of rheumatoid arthritis: randomised double-blind comparison. Lancet. 1999;354:2106–2111. doi: 10.1016/S0140-6736(99)02332-6. [DOI] [PubMed] [Google Scholar]
- GRETZER B., MARICIC N., RESPONDEK M., SCHULIGOI R., PESKAR B.M. Effects of specific inhibition of cyclo-oxygenase-1 and cyclo-oxygenase-2 in the rat stomach with normal mucosa and after acid challenge. Br. J. Pharmacol. 2001;132:1565–1573. doi: 10.1038/sj.bjp.0703955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARRIS R.C., MCKANNA J.A., AKAI Y., JACOBSON H.R., DUBOIS R.N., BREYER M.D. Cyclooxygenase-2 is associated with the macula densa of rat kidney and increases with salt restriction. J. Clin. Invest. 1994;94:2504–2510. doi: 10.1172/JCI117620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HÉBERT R.L., JACOBSON H.R., BREYER M.D. PGE2 inhibits AVP-induced water flow in cortical collecting ducts by protein kinase C activation. Am. J. Physiol. 1990;259:F318–F325. doi: 10.1152/ajprenal.1990.259.2.F318. [DOI] [PubMed] [Google Scholar]
- HERSCHMAN H.R. Prostaglandin synthase 2. Biochim. Biophys. Acta. 1996;1299:125–140. doi: 10.1016/0005-2760(95)00194-8. [DOI] [PubMed] [Google Scholar]
- HLA T., NEILSON K. Human cyclooxygenase-2 cDNA. Proc. Natl. Acad. Sci. U.S.A. 1992;89:7384–7388. doi: 10.1073/pnas.89.16.7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JENSEN B.L., KURTZ A. Differential regulation of renal cyclooxygenase mRNA by dietary salt intake. Kidney Int. 1997;52:1242–1249. doi: 10.1038/ki.1997.449. [DOI] [PubMed] [Google Scholar]
- KATAYAMA S., ATTALLAH A.A., STAHL R.A., BLOCH D.L., LEE J.B. Mechanism of furosemide-induced natriuresis by direct stimulation of renal prostaglandin E2. Am. J. Physiol. 1984;247:F555–F561. doi: 10.1152/ajprenal.1984.247.4.F555. [DOI] [PubMed] [Google Scholar]
- KHAN K., VENTURINI C.M., BUNH R.T., BRASSARD J.A., KOKI A.T., MORRIS D.L., TRUMP B.F., MAZIASZ T.J., ALDEN C.L. Interspecies differences in renal localization of cyclooxygenase isoforms: implications in nonsteroidal antiinflammatory drug-related nephrotoxicity. Toxic. Pathol. 1998;26:612–620. doi: 10.1177/019262339802600504. [DOI] [PubMed] [Google Scholar]
- KUJUBU D.A., FLETCHER B.S., VARNUM B.C., LIM R.W., HERSCHMAN H.R. TIS10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J. Biol. Chem. 1991;266:12866–12872. [PubMed] [Google Scholar]
- MARNETT L.J., KALGUTKAR A.S. Cyclooxygenase 2 inhibitors: discovery, selectivity and the future. TIPS. 1999;20:465–469. doi: 10.1016/s0165-6147(99)01385-1. [DOI] [PubMed] [Google Scholar]
- MASFERRER J.L., SEIBERT K., ZWEIFEL B.S., NEEDLEMAN P. Endogenous glucocorticoids regulate an inducible cyclooxygenase enzyme. Proc. Natl. Acad. Sci. U.S.A. 1992;89:3917–3921. doi: 10.1073/pnas.89.9.3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATTIX H.J., BADR K.F.Arachidonic acid metabolites and the kidney The Kidney 2000Philadelphia: Saunders; 756–792.ed. Brenner, B.M., Rector, F.C. pp [Google Scholar]
- MCADAM B.F., CATELLA-LAWSON F., MARDINI I.A., KAPOOR S., LAWSON J.A., FITZGERALD G.A. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc. Natl. Acad. Sci. U.S.A. 1999;96:272–277. doi: 10.1073/pnas.96.1.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIROUZE D., ZIPSER R.D., REYNOLDS T.D. Effects of inhibitors of prostaglandin synthesis on induced diuresis in cirrhosis. Hepatology. 1983;3:50–55. doi: 10.1002/hep.1840030108. [DOI] [PubMed] [Google Scholar]
- NIEDERBERGER E., TEGEDER I., VETTER G., SCHMIDTKO A., SCHMIDT H., EUCHENHOFER C., BRÄUTIGAM L., GRÖSCH S., GEISSLINGER G. Celecoxib loses its anti-inflammatory efficacy at high doses through activation of NF-kB. FASEB J. 2001;15:1622–1624. doi: 10.1096/fj.00-0716fje. [DOI] [PubMed] [Google Scholar]
- O'NEILL G.P., FORD-HUTCHINSON A.W. Expression of mRNA for cyclooxygenase-1 and cyclooxygenase-2 in human tissues. FEBS Lett. 1993;330:156–160. doi: 10.1016/0014-5793(93)80263-t. [DOI] [PubMed] [Google Scholar]
- PAYAN D.G., KATZUNG B.G.Nonsteroidal anti-inflammatory drugs; nonopioid analgesics; drugs used in gout Basic & Clinical Pharmacology 1995Norwalk: Appleton & Lange; 536–559.ed. Katzung, B.G. pp [Google Scholar]
- PÉREZ-AYUSO R.M., ARROYO V., CAMPS J., RIMOLA A., GAYA J., COSTA J., RIVERA F., RODÉS J. Evidence that renal prostaglandins are involved in renal water metabolism in cirrhosis. Kidney Int. 1984;26:72–80. doi: 10.1038/ki.1984.136. [DOI] [PubMed] [Google Scholar]
- PLANAS R., ARROYO V., RIMOLA A., PÉREZ-AYUSO R.M., RODÉS J. Acetylsalicylic acid suppresses the renal hemodynamic effect and reduces the diuretic action of furosemide in cirrhosis with ascites. Gastroenterology. 1983;84:247–252. [PubMed] [Google Scholar]
- PUSCHETT J.B. Sites and mechanisms of diuretics in the kidney. J. Clin. Pharmacol. 1981;21:564–574. doi: 10.1002/j.1552-4604.1981.tb05665.x. [DOI] [PubMed] [Google Scholar]
- ROS J., CLÀRIA J., JIMÉNEZ W., BOSCH-MARCÉ M., ANGELI P., ARROYO V., RIVERA F., RODÉS J. Role of nitric oxide and prostacyclin in the control of renal perfusion in experimental cirrhosis. Hepatology. 1995;22:915–920. [PubMed] [Google Scholar]
- ROSSAT J., MAILLARD M., NUSSBERGER J., BRUNNER H.R., BURNIER M. Renal effects of selective cyclooxygenase-2 inhibition in normotensive salt-depleted subjects. Clin. Pharmacol. Ther. 1999;66:76–84. doi: 10.1016/S0009-9236(99)70056-1. [DOI] [PubMed] [Google Scholar]
- SWAN S., RUDY D.W., LASSETER K.C., RYAN C.F., BUECHEL K.L., LAMBRECHT L.J., PINTO M.B., DILZER S.C., OBRDA O., SUNDBLAD K.J., GUMBS C.P., EBEL D.L., QUAN H., LARSON P.J., SCHWARTZ J.I., MUSLINER T.A., GERTZ B.J., BRATER C., YAO S.-L. Effect of cyclooxygenase-2 inhibition on renal function in elderly persons receiving a low-salt diet. Ann. Intern. Med. 2000;133:1–9. doi: 10.7326/0003-4819-133-1-200007040-00002. [DOI] [PubMed] [Google Scholar]
- SEIBERT K., ZHANG Y., LEAHY K., HAUSER S., MASFERRER J., PERKINS W., LEE L., ISAKSON P. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc. Natl. Acad. Sci. U.S.A. 1994;91:12013–12017. doi: 10.1073/pnas.91.25.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMITH C.J., ZHANG Y., KOBOLDT C.M., MUHAMMAD J., ZWEIFEL B.S., SHAFFER A., TALLEY J.J., MASFERRER J.L., SEIBERT K., ISAKSON P.C. Pharmacological analysis of cyclooxygenase-1 in inflammation. Proc. Natl. Acad. Sci. U.S.A. 1998;95:13313–13318. doi: 10.1073/pnas.95.22.13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VANE J.R. Inhibition of prostaglandin synthesis as a mechanism of action for the aspirin-like drugs. Nature. 1971;231:232–235. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]
- VIO C.P., CESPEDES C., GALLARDO P., MASFERRER J.L. Renal identification of cyclooxygenase-2 in a subset of thick ascending limb cells. Hypertension. 1997;30:687–692. doi: 10.1161/01.hyp.30.3.687. [DOI] [PubMed] [Google Scholar]
- WALLACE J.L., MCKNIGHT W., REUTER B.K., VERGNOLLE N. NSAID-induced gastric damage in rats: requirement for inhibition of both cyclooxygenase 1 and 2. Gastroenterology. 2000;199:706–714. doi: 10.1053/gast.2000.16510. [DOI] [PubMed] [Google Scholar]
- WARNER T.D., GIULIANO F., VOJNOVIC I., BUKASA A., MITCHELL J.A., VANE J.R. Nonsteroid drug selectivities for cyclooxygenase-1 rather than cyclooxygenase-2 are associated with human gastrointestinal toxicity: a full in vitro analysis. Proc. Natl. Acad. Sci. U.S.A. 1999;96:7563–7568. doi: 10.1073/pnas.96.13.7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHELTON A., SCHULMAN G., WALLEMARK C., DROWER E.J., ISAKSON P.C., VERBURG K.M., GEIS G.S. Effects of celecoxib and naproxen on renal function in the elderly. Arch. Intern. Med. 2000;160:1465–1470. doi: 10.1001/archinte.160.10.1465. [DOI] [PubMed] [Google Scholar]
- YANG T., SCHNERMANN J.B., BRIGGS J.P. Regulation of cyclooxygenase-2 expression in renal medulla by tonicity in vivo and in vitro. Am. J. Physiol. 1999;277:F1–F9. doi: 10.1152/ajprenal.1999.277.1.F1. [DOI] [PubMed] [Google Scholar]
- YANG T., SINGH I., PHAM H., SUN D., SMART A., SCHNERMANN J.B., BRIGGS J.P. Regulation of cyclooxygenase expression by dietary salt intake. Am. J. Physiol. 1998;274:F481–F489. doi: 10.1152/ajprenal.1998.274.3.F481. [DOI] [PubMed] [Google Scholar]
- ZIPSER R., HOEFS J.C., SPECKART P.F., ZIA P.K., HORTON R. Prostaglandins: modulators of renal function and pressor resistance in chronic liver disease. J. Clin. Endocrinol. Metab. 1979;48:895–900. doi: 10.1210/jcem-48-6-895. [DOI] [PubMed] [Google Scholar]