

Abstract
This study deals with phosphorylation and activation of p38 mitogen-activated protein kinase (MAPK) via β3-adrenoceptor (AR) and the signal transduction pathway in 3T3-L1 adipocytes.
β3-AR agonist BRL37344A (10 nM) caused phosphorylation and activation of p38 MAPK in 3T3-L1 adipocytes but not in fibroblasts. BRL37344A and also the other β3-AR agonists, CGP12177A and SR58611A, caused p38 MAPK phosphorylation in dose-dependent manners.
The p38 MAPK phosphorylations by BRL37344A (10 nM), CGP12177A (100 nM), and SR58611A (10 nM) were not antagonized by β1- and β2-ARs antagonist 1-propranolol (100 nM) but blocked by β3-AR antagonist SR59230A (10 μM), suggesting the phosphorylation was caused via β3-AR.
The phosphorylations of p38 MAPK were completely abolished by treatment with cholera toxin (CTX) but not pertussis toxin (100 ng ml−1, 24 h). Activation of Gs by CTX (100 ng ml−1) and adenylyl cyclase by forskolin mimicked p38 MAPK phosphorylation.
p38 MAPK phosphorylation by BRL37344A was reduced to almost 50% by cyclic AMP-dependent protein kinase (PKA) inhibitors such as H89 (10 μM) and PKI (10 μM). A src-family tyrosine kinases inhibitor PP2 (1 μM) also halved the p38 MAPK phosphorylation. Combined use of H89 (10 μM) and PP2 (10 μM) did not bring about further inhibition.
These results suggest that β3-AR caused phosphorylation of p38 MAPK via Gs protein and partly through a pathway involving PKA and src-family kinase(s), although the contribution of the unidentified pathway remains to be clarified.
Keywords: β3-adrenoceptors, p38 MAPK, 3T3-L1 adipocytes, BRL37344A, CGP12177A, SR58611A, SR59230A
Introduction
β3-Adrenoceptors (ARs) are expressed predominantly, though not exclusively, in white and brown adipocytes (Emorine et al., 1989; Granneman et al., 1991; Muzzin et al., 1991; Nahmias et al., 1991). Treatment of animals with β3-AR selective agonists causes a number of diverse metabolic effects including lipolysis (van liefde et al., 1992), increase in energy expenditure (Weyer et al., 1998), reduction of food intake (Mantzoros et al., 1996), and dramatic increase of insulin level in blood (Susulic et al., 1995). These effects have been shown to disappear in β3-AR knock-out mice (Susulic et al., 1995). Re-expression of the receptors in both white and brown adipocytes of the knock-out mice recovered these effects, whereas re-expression of the receptors only in brown adipocytes did not (Grujic et al., 1997). This interesting phenomenon strongly suggests the significance of β3-AR-mediated responses in the functioning white adipocytes. In addition, it has been reported that amelioration of type 2 diabetes was caused by treatment with β3-AR agonists; thus, β3-AR agonists are expected to act as anti-obesity and/or anti-diabetic compounds in humans as well (Weyer et al., 1998; Sum et al., 1999; Kato et al., 2001). Despite its increasingly apparent significance, however, our understanding of the signalling pathway of β3-AR in adipocytes is insufficient, suggesting the need for further investigation of the signalling of β3-AR in order to realize an efficient treatment for obesity and/or insulin resistance (Klein et al., 1999).
Mitogen-activated protein kinases (MAPKs) such as extracellular signal-regulated kinase 1 and 2 (ERK1/2) and p38 MAPK play key roles in many physiological events, such as proliferation (Pages et al., 1993; Kanda et al., 2001), differentiation (Pang et al., 1995; Engelman et al., 1998), cell survival (Lindquist & Rehnmark, 1998, Kuroki et al., 2000) and gene expression (Vanhoute et al., 1998). Recently, it was found that stimulation of β3-AR caused phosphorylation and activation of ERK1/2 in brown adipocytes (Shimizu et al., 1997; Lindquist & Rehnmark, 1998; Lindquist et al., 2000), 3T3-L1 adipocytes (Mizuno et al., 1999; 2000), 3T3-F442A (Soeder et al., 1999), and exogenous β3-AR expression system with CHO/K1 cells (Gerhardt et al., 1999) or HEK293 cells (Soeder et al., 1999), although the physiological functions of this novel signalling pathway have been clarified only limitedly in brown adipocytes (Lindquist & Rehnmark, 1998). In addition, the pathway leading to ERK1/2 activation and phosphorylation, which includes coupling of G proteins to β3-AR, and the modulation mechanisms are controversial (Gerhardt et al., 1999; Lindquist et al., 2000; Mizuno et al., 2000; Soeder et al., 1999; Cao et al., 2000) and remain insufficiently elucidated. In the complexity of the β3-AR signalling pathways, very little is known about the activation of p38 MAPK via β3-AR. The β-AR agonist isoproterenol has been shown to cause activation of p38 MAPK in freshly isolated white adipocytes of rat (Moule & Denton, 1998), whereas a study with CGP12177A, a β3-AR agonist, failed to obtain clear phosphorylation of p38 MAPK in CHO/K1 cells which expressed exogenous β3-AR (Gerhardt et al., 1999). Very recently, it was confirmed by the study with C3H10T1/2 adipocytes and brown adipocytes that a β3-AR agonist CL316,243 caused p38 MAPK phosphorylation and that the phosphorylation was completely inhibited by treatment with a PKA inhibitor H89 (Cao et al., 2001).
In this study, we investigated whether selective β3-AR agonists had the potential to induce p38 MAPK phosphorylation using 3T3-L1 fibroblasts and 3T3-L1 adipocytes. We also examined the downstream pathway of β3-AR that leads to phosphorylation and activation of p38 MAPK including participations of G proteins, and found that PKA and src-family tyrosine kinase(s) were likely to participate in phosphorylation of p38 MAPK.
Methods
Materials
The 3T3-L1 fibroblast cells (Green & Kehinde, 1976), JCRB 9014, were obtained from The Health Science Research Resources Bank of the Japan Health Sciences Foundation. The β3-AR antagonist SR59230A and agonist SR58611A were the kind gifts of Dr. Luciano Manara (Research Centre Sanofi Midy) and Dr. Martine Combes (SANOFI RECHERCHE). The following materials were purchased from the sources indicated: Dulbecco's modified Eagle medium (DMEM) from Nissui Pharmaceutical Co., Ltd. (Tokyo, Japan); foetal bovine serum (FBS) from JRH Biosciences (Lenexa, KS, U.S.A.); penicillin and streptomycin from Life technologies (Grand Island, NY, U.S.A.); BRL37344A and CGP12177A from Research Biochemical International (Natick, MA, U.S.A.); cholera toxin (CTX) of Vibrio cholerae from List Biological Laboratories, Inc. (Campbell, CA, U.S.A.); pertussis toxin (PTX) of Bordetella pertussis from Seikagaku Corporation (Tokyo, Japan). H89 (N-[2-(p-bromocinnamylamino) ethyl]-5-isoquinolinesulfonamide dihydrochloride), PP2 (4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine) and cell-permeable cyclic AMP-dependent protein kinase inhibitor peptide (PKI-(14 – 22)-amide) were from Calbiochem-Novabiochem Corporation (La Jolla, CA, U.S.A.). Other reagents used were of the highest grade commercially available.
Cell culture and differentiation
3T3-L1 fibroblast cells were maintained in high-glucose (25 mM) DMEM supplemented with 10% FBS at 37°C (95% air/5% CO2) and treated with 0.5 mM 3-isobutyl-1-methylxanthine, 1 mM dexamethasone and 10 mg ml−1 insulin to initiate adipogenesis as described previously (Mizuno et al., 1999). After appropriate cultivation, the adipocytes were washed and cultured in the absence of FBS for 4 h and then treated with various reagents. To confirm effect of SR59230A, 3T3-L1 adipocytes were cultured in glass dishes to avoid binding of SR59230A to the walls, as it does to plastic culture dishes (Nisoli et al., 1996). In some cases, the adipocytes were pretreated and serum-starved in the presence of either PTX or CTX prior to the experiments. The conditions are described in detail in the results for each trial.
Immunoblot analysis
Whole-cell extracts of 3T3-L1 cells were prepared as described previously (Mizuno et al., 1999). Briefly, the cells were washed with ice-cold phosphate-buffered saline containing 1 mM sodium vanadate and 1 mM phenylmethylsulfonyl fluoride (PMSF), and then gently scraped into cell lysis buffer consisting of in mM: Tris-HCl (pH 7.5) 20, EDTA-2Na 10, β-glycerophosphate 60, MgCl2 10, Triton X-100 1%, dithiothreitol 1, sodium vanadate 1, PMSF 1, leupeptin 2 μg ml−1 and aprotinin 2 μg ml−1. After sonication (1 s×5 times, on ice) and centrifugation (12,000 × g, 20 min, 4°C), the obtained supernatants of fibroblasts or infranatants of adipocytes were used as cell extracts.
To prepare membrane fractions, the washed cells were scraped and homogenized in ice-cold homogenizing buffer consisting of in mM Tris-HCl (pH 7.5) 25, EDTA-2Na 1, 5 μg ml−1 leupeptin, 1 μg ml−1 aprotinin, and PMSF 1. The homogenates were centrifuged (500×g, 10 min, 4°C) and the recovered supernatant was centrifuged again (40,000 × g, 10 min, 4°C). The resultant pellets were suspended and used as membrane fractions.
The protein contents of these fractions were determined using a BCA Protein Assay Reagent Kit from Pierce (Rockford, IL, U.S.A.).
The cell extracts or membrane fractions containing 10 μg of crude proteins were subjected to sodium dodecyl sulphate-polyacrylamide gel electrophoresis. The separated proteins were transferred to a polyvinilidene difluoride membrane (IPVH00010; Millipore, Bedford, MA, U.S.A.), and immunoblotted with anti-phospho p38 MAPK (Thr180/Tyr182) specific antibody (number 9210; New England Biolabs, Beverly, MA, U.S.A.), anti-p38 MAPK antibody (number 9212; New England Biolabs), anti-Gs protein antibody (sc-383; Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.), or anti-β3-AR antibody (sc-1473; Santa Cruz Biotechnology), and then with horseradish peroxidase-conjugated anti-rabbit IgG antibody (Calbiochem-Novabiochem, La Jolla, CA, U.S.A.) or anti-goat IgG (ICN Biomedicals, Inc. Aurora, OH, U.S.A.) as a secondary antibody. Bound antibodies were detected by enhanced chemiluminescence using ECL Western blotting detection reagents (Amersham Pharmacia Biotech, Buckinghamshire, U.K.) and photographed using Fuji X-ray film (Fuji Film Co., Tokyo, Japan). The intensity of each band was quantitatively analysed using NIH image software ver. 1.61 (National Institute of Health).
Activation of p38 MAPK was assayed with a p38 MAP Kinase Assay Kit (number 9820; New England Biolabs).
Statistical analysis
Each experiment was performed independently at least three times in triplicate. The data within groups were compared using an analysis of variance (one way-ANOVA) with Dunnett's or Tukey's post-hoc correction for multiple comparisons. Detailed condition was shown in each result.
Results
Stimulation with β3-AR agonists induced p38 MAPK phosphorylation in 3T3-L1 adipocytes, but not in fibroblasts
Stimulation with the β3-AR agonist BRL37344A did not cause phosphorylation of p38 MAPK in either 3T3-L1 fibroblasts or the cells, when given immediately after the initiation of adipogenesis (Figure 1a,b). On the other hand, when administrated 5 days or more after the initiation of adipogenesis, the stimulation induced clear and statistically significant increases in the phosphorylation levels of threonine (180) and tyrosine (182) residues of p38 MAPK (Figure 1a,b). The phosphorylated p38 MAPK showed the ability to phosphorylate ATF-2 (Figure 1b).
Figure 1.
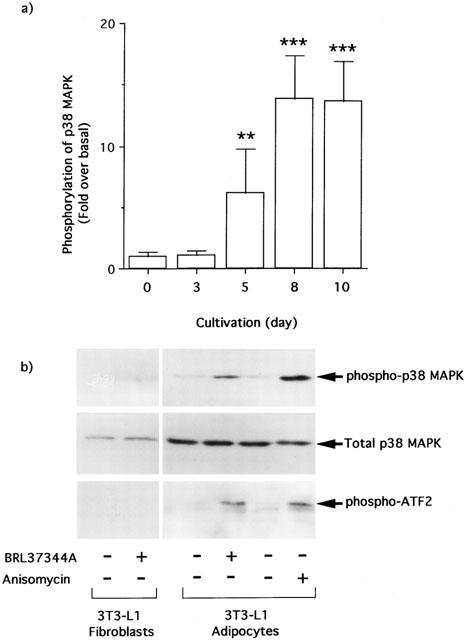
Cultivation-dependent occurrence of p38 MAPK phosphorylation and activation by the stimulation with BRL37344A in 3T3-L1 cells. The 3T3-L1 fibroblast cells were grown and treated with differentiation reagents for initiation of adipogenesis. After appropriate cultivation, the cells were serum-starved and stimulated with 10 nM BRL37344A for 30 min at 37°C. Open bars represent the degree of p38 MAPK phosphorylation at each period, expressed as the fold increase in phosphorylation level over respective basal level (a). Values represent the means±s.d. (four independent experiments). The values are significantly different from that obtained at day 0 by one-way ANOVA and Dunnett's multiple comparison (**:P<0.01, ***:P<0.001). The activation of p38 MAPK was confirmed at 8-day cultivation after the initiation by immunoprecipitation kinase assay with ATF-2 as described in the Methods, and representative results are shown (b). A positive control was established using 10 μM anisomycin under the same condition.
The increase in phosphorylation fold of p38 MAPK by the stimulation with BRL37344A occurred in a time-dependent manner. The stimulation increased the phosphorylation level of p38 MAPK, reached a maximal level at 15 min, and was sustained for at least 120 min after the stimulation (Figure 2a). The protein contents of p38 MAPK were not changed over this time period (see Figure 1b). As summarized in Figure 2b, BRL37344A and the other β3-AR agonists, CGP12177A and SR58611A, increased the degree of p38 MAPK phosphorylation in a dose-dependent manner.
Figure 2.
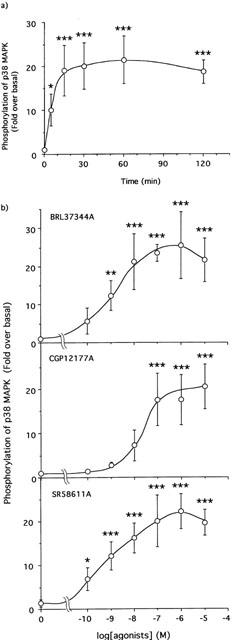
Time- (a) and dose-dependency (b) of p38 MAPK phosphorylation by BRL37344A in 3T3-L1 adipocytes. 3T3-L1 adipocytes (35 mm dish, 8-day culture) were treated for the indicated periods with 10 nM BRL37344A (a) or for 30 min at the indicated concentrations with either BRL37344A, CGP12177, or SR58611A (b) at 37°C. The increases of phosphorylation were measured as described in Figure 1. Open circles represent the results obtained at each period or with each agonist (means±s.d. of three independent experiments). The data were compared with the values obtained without agonists as controls by one-way ANOVA and Dunnett's multiple comparison (*:P<0.05, **:P<0.01, ***:P<0.001).
Effect of β-AR antagonists on the phosphorylation of p38 MAPK in 3T3-L1 adipocytes
As shown in Figure 3a, the β1- and β2-ARs specific antagonist l-propranolol (100 nM) did not affect the degree of p38 MAPK phosphorylation caused by BRL37344A (10 nM), CGP12177A (100 nM) or SR58611A (10 nM). On the other hand, the β3-AR specific antagonist SR59230A (Manara et al., 1996) decreased the phosphorylation of p38 MAPK caused by BRL37344A in a dose-dependent manner and almost completely blocked it at a concentration of 10 μM (Figure 3b). In addition, the phosphorylations of p38 MAPK by CGP12177A and SR58611A were also decreased to the unstimulated level by the β3-AR antagonist at this concentration (Figure 3c), although a slight increase in p38 MAPK phosphorylation was seen even in the absence of agonist(s) (Figure 3b,c).
Figure 3.
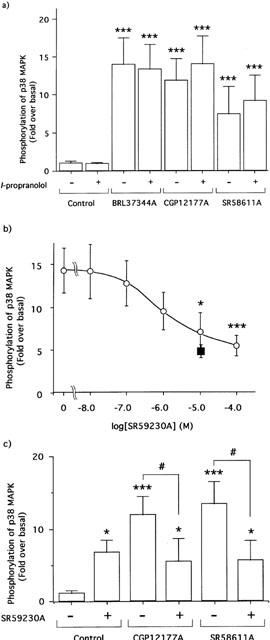
Effects of β-AR antagonists on p38 MAPK phosphorylations by the β3-AR agonists. 3T3-L1 adipocytes (35 mm dish, 8-day cultivation) were treated with 10 nM BRL37344A, 100 nM CGP12177A and 10 nM SR58611A in the absence or presence of 100 nM l-propranolol (a). The adipocytes were cultured in glass dishes and stimulated by 10 nM BRL37344A for 30 min in the presence of SR59230A at the indicated concentrations (b) or by 100 nM CGP12177A and 10 nM SR58611A for 30 min in the presence of 10 μM SR59230A (c). The increases of p38 MAPK phosphorylations were measured and expressed as described in Figure 1 (means±s.d. of three independent experiments). Open bars in (a,c) represent the results obtained with/without each antagonist. The open circles and closed square in (b) represent the results obtained with 10 nM BRL37344A and SR59230A at the indicated concentrations or that with 10 μM SR59230A alone, respectively. The data in (a) and (c) were compared with the controls (*:P<0.05, ***:P<0.001) or each other (#:P<0.001) by one-way ANOVA and Tukey's multiple comparison. The data in (b) were compared with the values obtained with BRL37344A as the control by one-way ANOVA and Dunnett's multiple comparison (*:P<0.05, ***:P<0.001).
Effects of bacterial toxins on the phosphorylations of p38 MAPK by β3-AR agonists
Long-term treatment with CTX dramatically decreased immuno-reactive Gs protein contents in 3T3-L1 adipocytes (Figure 4a,b) without effects on the contents of p38 MAPK and β3-AR on membrane of the adipocytes (Figure 4a). At the same time, the increases in phosphorylations of p38 MAPK by BRL37344A (10 nM), CGP12177A (100 nM), and SR58611A (10 nM) were completely abolished by the treatment (Figure 4c), whereas lysophosphatidic acid (LPA) has been shown to remain effective at increasing ERK1/2 phosphorylations after the same treatment (Mizuno et al., 2000). On the other hand, as shown in Figure 4d, treatment with PTX failed to block the p38 MAPK phosphorylations caused by BRL37344A (10 nM), CGP12177A (100 nM), and SR58611A (10 nM), whereas ERK1/2 phosphorylations by LPA (10 μM) were completely abolished (Mizuno et al., 2000).
Figure 4.
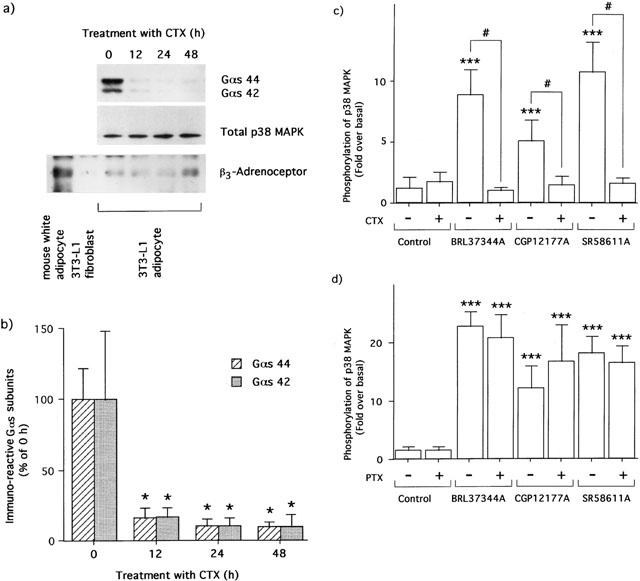
Effects of CTX and PTX on p38 MAPK phosphorylations caused by β3-AR agonists in 3T3-L1 adipocytes. 3T3-L1 adipocytes were treated with 100 ng ml−1 CTX for the indicated periods at 37°C. The contents of immuno-reactive Gs, p38 MAPK and β3-AR proteins are shown in upper, middle, and lower panels of (a), respectively. The hatched and shaded bars express the relative contents of two isoforms of Gs proteins in the membrane of 3T3-L1 adipocytes at indicated periods (b). After treatment with 100 ng ml−1 CTX (c) or PTX (d) for 24 h, 3T3-L1 adipocytes were serum-starved and then stimulated by the β3-AR agonists as described in the legend for Figure 3. The increases of p38 MAPK phosphorylations were measured as described in Figure 1 and expressed as open bars (means±s.d. of three independent experiments). The data in (b) were compared with values obtained at 0 h as the controls by one-way ANOVA and Dunnett's multiple comparison (*:P<0.001). The data in (c) and (d) were compared with the values obtained without agonists (***:P<0.001) or each other (#:P<0.001) by one-way ANOVA with Tukey's multiple comparison.
Phosphorylation of p38 MAPK by activation of the Gs-dependent pathway
Treatment of 3T3-L1 adipocytes for several hours with CTX, which is known as an activator of Gs protein (Cassel & Selinger, 1997), increased p38 MAPK phosphorylation in a time- and dose-dependent manner (Figure 5a,b).
Figure 5.
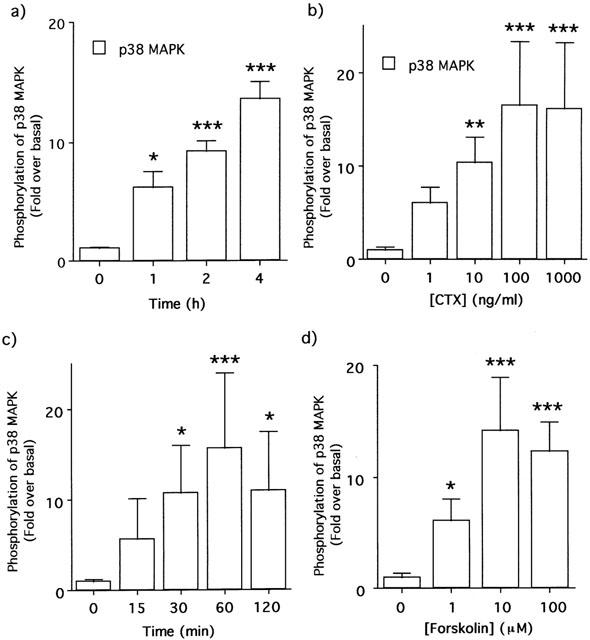
p38 MAPK phosphorylations in 3T3-L1 adipocytes by CTX (a and b) and forskolin (c and d). 3T3-L1 adipocytes were treated with 100 ng ml−1 CTX (a) or 1 μM forskolin (c) for the indicated periods and at the indicated concentrations for 4 h with CTX (b) or for 30 min with forskolin (d). The increases of p38 MAPK phosphorylations were measured as described in Figure 1 and expressed as open bars (means±s.d. of three independent experiments). The data were compared with the values obtained at 0 min or 0 h without reagents as the control by one-way ANOVA with Dunnett's multiple comparison (*:P<0.05, **:P<0.01, ***:P<0.001).
Forskolin, by which adenylyl cyclase (AC) is directly activated (Seamon & Daly, 1981), also caused p38 MAPK phosphorylation in 3T3-L1 adipocytes (Figure 5c,d).
Phosphorylation of p38 MAPK by the β3-AR agonists is mediated via a pathway involving PKA and src-family tyrosine kinase(s)
As shown in Figure 6a, treatment of the adipocytes with H89, the highly selective inhibitor for cyclic AMP-dependent protein kinase (PKA), decreased the phosphorylation of p38 MAPK in a dose-dependent manner, achieving a maximal reduction of approximately 50% at a concentration of 10 μM. In addition, another PKA inhibitor, PKI-(14 – 22)-amide also decreased the phosphorylation of p38 MAPK in a dose-dependent manner and almost halved the p38 MAPK phosphorylation at 10 μM (Figure 6b). Treatment with a src-family tyrosine kinases inhibitor, PP2, also decreased the phosphorylation of p38 MAPK by BRL37344A in a dose-dependent manner, and also reached a maximal reduction of about 50% (Figure 6c). Combined use of 10 μM H89 and 10 μM PP2 did not enhance the decrease in phosphorylation of p38 MAPK by 10 nM of BRL37344A (Figure 6d).
Figure 6.
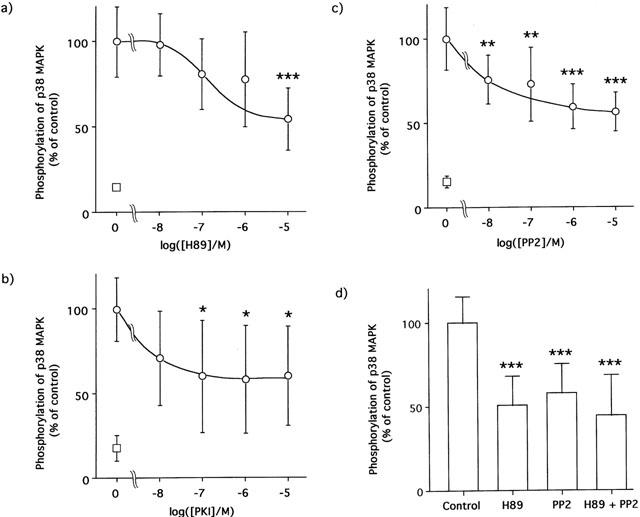
Effects of PKA and a src-family kinases inhibitors on p38 MAPK phosphorylation by BRL37344A in 3T3-L1 adipocytes. The adipocytes were treated with H89, PKI-(14 – 22)-amide and/or PP2 at the indicated concentrations for 30 min, and then stimulated with 10 nM BRL37344A for 30 min at 37°C. The degree of p38 MAPK phosphorylation was expressed as open circle and bars as a percentage of control that obtained without inhibitors (means±s.d. of four independent experiments). The open square expressed the basal value obtained without BRL37344A and inhibitors. The data in (a, b and c) were compared with the values obtained without inhibitors as controls by one-way ANOVA with Dunnett's multiple comparison (*:P<0.05, **:P<0.01, ***:P<0.001). The values in (d) are significantly different from that obtained without inhibitors by one-way ANOVA and Tukey's multiple comparison (***:P<0.001).
Discussion
We here showed that stimulation of β3-AR caused phosphorylation of p38 MAPK in 3T3-L1 adipocytes via Gs but not Gi protein, and that the downstream pathway of this phosphorylation may have involved AC, PKA and src-family tyrosine kinase(s).
As shown in Figure 1a,b, the β3-AR agonist BRL37344A was effective at inducing p38 MAPK phosphorylation and activation in 3T3-L1 adipocytes, whereas it was not effective in 3T3-L1 fibroblasts that did not express β3-AR (Mizuno et al., 1999). The activation of p38 MAPK by the phosphorylation was confirmed by immunoprecipitation kinase assay (Figure 1b). The result was consistent with the previous report that p38 MAPK was activated by the phosphorylation of these residues (Derijard et al., 1995).
The kinetics of p38 MAPK phosphorylation by BRL37344A in 3T3-L1 adipocytes and that of activation by isoproterenol in rat white adipocytes (Moule & Denton, 1998) were well consistent with each other. Both results showed sustained activation with a peak at 15 – 30 min after stimulation.
As shown in Figure 3a, the β1- and β2-ARs-specific antagonist l-propranolol did not affect the degree of p38 MAPK phosphorylation caused by BRL37344A (10 nM), CGP12177A (100 nM) or SR58611A (10 nM) despite the fact that this concentration was previously reported to be sufficient to completely block β1- and β2-ARs (Arch et al., 1984). On the other hand, the β3-AR-specific antagonist SR59230A decreased the BRL37344A-induced phosphorylation of p38 MAPK in a dose-dependent manner and nearly reached the unstimulated level at a concentration of 10 μM (Figure 3b). SR59230A also completely blocked the p38 MAPK phosphorylations by CGP12177A and SR58611A (Figure 3c), whereas SR59230A only slightly blocked the stimulation of β1- and β2-ARs by specific agonists at this concentration (Nisoli et al., 1996). These results strongly suggest that the phosphorylations of p38 MAPK in 3T3-L1 adipocytes by BRL37344A, CGP12177A and SR58611A were caused via β3-AR, rather than by β1- or β2-ARs.
It has previously been shown that β3-ARs are coupled to Gs protein (Guan et al., 1995; Soeder et al., 1999). Thus, stimulation of the receptors rapidly activates the Gs protein and Gs-coupled AC. The activated AC generates cyclic AMP (Emorine et al., 1989; Nahmias et al., 1991), leading to activation of cyclic AMP-dependent protein kinase (PKA). The lipolysis, one of the most characteristic responses mediated through β3-AR in white adipocytes, is induced via activation of hormone-sensitive lipase by PKA (Langin et al., 1992; Anthonsen et al., 1998). However, some studies have provided evidence that β3-AR receptors also constitutively couple to Gi protein in adipocytes (Chaudhry et al., 1994; Soeder et al., 1999) and the heart (for review see Gauthier et al., 2000). Recently, it has been confirmed that stimulation of β3-AR causes phosphorylation and activation of ERK1/2 in brown adipocytes (Lindquist et al., 2000) and 3T3-L1 adipocytes (Mizuno et al., 1999; 2000) through a Gs-protein-dependent pathway. On the other hand, it has been reported that ERK1/2 activation via β3-AR was caused through pertussis toxin (PTX)-sensitive Gi protein in 3T3-F442A adipocytes (Soeder et al., 1999) as well as CHO/K1 cells (Gerhardt et al., 1999) and HEK293 cells (Soeder et al., 1999) expressing exogenous β3-AR. As mentioned above, the complicated signal-transduction pathways of β3-AR, including the coupling of β3-AR to G protein(s), are insufficiently understood, prompting us to examine what kinds of G protein(s) were involved in the p38 MAPK phosphorylation.
To elucidate the role of Gs and Gi proteins in the p38 MAPK phosphorylations via β3-AR, we examined the effects of bacterial toxins that selectively impair the individual signalling of each G protein. It has been reported that long-term treatment with CTX caused selective down regulation of Gs proteins and blocked signalling of Gs protein-coupled receptors in various cells (Chang & Bourne, 1989; Milligan et al., 1989; Carr et al., 1990). These phenomena were also found in 3T3-L1 adipocytes. Treatment of the adipocytes with CTX (100 ng ml−1) for 24 h was sufficient to bring almost complete reduction of Gs proteins (Figure 4a,b). The total content of p38 MAPK in the cells and that of β3-AR in the membrane fractions remained almost unchanged even after the treatments (Figure 4a). As shown in Figure 4c, the stimulation of 3T3-L1 adipocytes by BRL37344A (10 nM), CGP12177A (100 nM) and SR58611A (10 nM) did not increase phosphorylation of p38 MAPK after long-term treatment with CTX. These results suggest that Gs protein plays an important role in the phosphorylation of p38 MAPK by the three β3-AR specific agonists in 3T3-L1 adipocytes. On the other hand, long-term treatment of the cells with PTX (100 ng ml−1), which is known to diminish Gi protein-dependent signalling (van corven et al., 1993), did not alter the phosphorylation of p38 MAPK caused by the β3-AR agonists (Figure 4d), whereas phosphorylations of ERK1/2 by LPA that occurred through Gi protein was completely abolished by the same treatment even in 3T3-L1 adipocytes (Mizuno et al., 2000). These results suggest that the Gs protein rather than the Gi protein plays an indispensable role in p38 MAPK phosphorylation caused by the stimulation of β3-AR in 3T3-L1 adipocytes.
Although, in a previous report, stimulation of β3-ARs expressed in CHO/K1 cells failed to provide clear demonstration of p38 MAPK phosphorylation (Gerhardt et al., 1999), such phosphorylation was evident in the present report using 3T3-L1 adipocytes. The most prominent difference between the cells used here and those used previous is suggested to be the receptor-G protein coupling. The β3-AR coupled to Gi protein in the CHO/K1 cells (Gerhardt et al., 1999), whereas it was suggested that β3-AR coupled functionally to Gs protein in 3T3-L1 adipocytes (Figure 4c,d). This difference in receptor-G protein coupling might bring about the difference in the ability to activate p38 MAPK. Very recently, during revising this manuscript, it was confirmed that a β3-AR agonist CL316,243 caused p38 MAPK phosphorylation in C3H10T1/2 and brown adipocytes in a cyclic AMP/PKA dependent pathway (Cao et al., 2001). This report might strengthen the possibility that phosphorylation of p38 MAPK is determined by G protein(s) functionally coupling to β3-AR, although it was not confirmed which G protein was involved in the phosphorylation of p38 MAPK.
CTX causes activation of Gs protein by the ADP-ribosylation of Gs protein at specific amino acid residues, which leads to inhibition of GTP-hydrolyzing activity (Cassel & Selinger, 1997). As shown in Figure 5a,b, short-term treatment of 3T3-L1 adipocytes with CTX caused phosphorylation of p38 MAPK in a dose- and time-dependent manner. Forskolin is known as a direct activator of AC (Seamon & Daly, 1981) although it has the ability to inhibit AC under certain circumstances (Watanabe & Jakobs, 1986). Treatment of the 3T3-L1 adipocytes with forskolin also increased p38 MAPK phosphorylation (Figure 5c,d). These results indicate that activation of Gs protein and AC is sufficient to cause phosphorylation of p38 MAPK in 3T3-L1 adipocytes, and that PKA is likely to participate in the phosphorylation.
As shown in Figure 6a, the phosphorylation of p38 MAPK by BRL37344A (10 nM) was inhibited by a highly selective PKA inhibitor H89 in a dose-dependent manner and reached a maximal reduction of 50% at a concentration of 10 μM, whereas ERK1/2 phosphorylations via β3-ARs were completely inhibited at the same concentration (Mizuno et al., 1999). Although H89 was reported to inhibit ligand binding to β1 and β2-ARs (Penn et al., 1999), such an antagonism was not found in the case with β3-AR even at 50 μM (Fredriksson et al., 2001), suggesting that the effect of H89 to inhibit the p38 MAPK phosphorylation was brought about by the inhibition of PKA but not by the inhibition of binding of BRL37344A to β3-AR. To support this hypothesis, the effect of cell-permeable PKI-(14 – 22)-amide was examined, which was also known as an inhibitor for PKA (Glass et al., 1989). PKI-(14 – 22)-amide decreased the phosphorylation of p38 MAPK by BRL37344A in a dose-dependent manner and the maximal effect was obtained at 10 μM although the inhibition was about 50%. The dose-dependency was well consistent with the previous report showing that PKI-(14 – 22)-amide almost completely inhibited PKA activity at 10 μM or higher concentrations (Glass et al., 1989). These result might show that the complete inhibition of PKA brought about almost 50% reduction of the p38 MAPK phosphorylation, in other words, PKA-dependent pathway(s) participate, at least to some degree, in β3-AR mediated p38 MAPK phosphorylations.
The participation of cAMP and PKA in p38 MAPK phosphorylation by a β3-AR agonist CL316,243 have already confirmed using C3H10T1/2 and brown adipocytes (Cao et al., 2001). Treatment of the cells with the PKA inhibitor H89 completely abrogated the p38 MAPK phosphorylation in C3H10T1/2 and brown adipocytes, suggesting that the PKA-dependent pathway was the sole pathway leading to the p38 MAPK phosphorylation at least in C3H10T1/2 and brown adipocytes (Cao et al., 2001). On the other hand, our result with 3T3-L1 adipocytes and two PKA inhibitors H89 and PKI-(14 – 22)-amide failed to show the obligatory role of PKA in p38 MAPK phosphorylation. These results suggested the possibility that other pathway(s) independent from PKA also play(s) a role in the phosphorylation of p38 MAPK via β3-AR.
Recent studies have shown that src-family tyrosine kinase(s) play(s) an important role not only in MAPK activation via various receptors but also in β3-AR signalling as a downstream element of PKA (Lindquist et al., 2000) or by directly binding to β3-AR in a Gi-protein dependent manner (Cao et al., 2000). To elucidate the role of src-family tyrosine kinase(s) in the p38 MAPK phosphorylation by BRL37344A, the effect of PP2, which selectively inhibits src-family tyrosine kinases (Hanke et al., 1996), was examined. Treatment with PP2 markedly decreased p38 MAPK phosphorylation by BRL37344A in 3T3-L1 adipocytes in a dose-dependent manner and achieved almost a maximal reduction of 50% at a concentration of 1.0 μM as shown in Figure 6c. At this concentration, PP2 have reported to selectively inhibit src-family kinases (Hanke et al., 1996). In addition, further inhibitions were not observed even at 10 times higher concentration (Figure 6c). The dose-dependency was well consistent with the previous report that examined the effect of PP2 to inhibit activity of a src-family tyrosine kinase lck (Hanke et al., 1996). Taken together, the result strongly suggested that the reduction of p38 MAPK phosphorylation by the treatment with PP2 was due to inhibition of src-family kinase(s) although the inhibition by PP2 was 50% even at the maximal. Combined use of H89 and PP2 even at 10 μM did not enhance the inhibition (Figure 6d), suggesting that PKA and src-family kinase(s) share the same pathway.
In conclusion, it was suggested that stimulation of β3-AR causes phosphorylation and activation of p38 MAPK and that these effects occur, at least in part, via a Gs-protein coupled pathway involving PKA and src-family tyrosine kinase(s), although the physiological significance of p38 MAPK activated via β3-ARs and the contribution of the unidentified pathway in the phosphorylation remain to be clarified.
Acknowledgments
We would like to acknowledge Dr Luciano Manara and Dr Martine Combes for kindly supplying us with the β3-AR antagonist SR59230A and β3-AR agonist SR58611A. This work was supported in part by a grant from The Smoking Research Foundation to Y.Watanabe.
Abbreviations
- β-AR
β-adrenoceptor
- BSA
bovine serum albumin
- BRL37344A
dl-4-[2′-{2-hydroxy-2-(3-cholorophenyl)ethylamino}propyl phenoxyacetic acid sodium salt sesquihydrate
- CGP12177A
dl-4-3[(1,1-dimethylethyl)amino]-(2-hydroxylpropoxy)1,3-dihydro-2H-benzimidazol-2-one hydrochloride
- CTX
cholera toxin
- DMEM
Dulbecco's modified Eagle medium
- ERK
extra cellular signal-regulated kinase
- MAPK
mitogen-activated protein kinase
- PTX
pertussis toxin
- SR58611A
[(7S)7-{(2R)2-(3-chlorophenyl)-2-hydroxyethyl-amino}-5,6,7,8-tetrahydronapht-2-yl]ethyl oxyacetate, hydrochloride
- SR59230A
SS-enantiomer 3-(2-ethylphenoxy)-1-[(1S)-1,2,3,4-tetrahydronapth-1-ylaminol]-(2S)-2-propanol oxalate
References
- ANTHONSEN M.W., RONNSTRAND L., WERNSTEDT C., DEGERMAN E., HOLM C. Identification of novel phosphorylation sites in hormone-sensitive lipase that are phosphorylated in response to isoporterenol and govern activation properties in vitro. J. Biol. Chem. 1998;273:215–221. doi: 10.1074/jbc.273.1.215. [DOI] [PubMed] [Google Scholar]
- ARCH J.R.S., ANISWORTH A.T., CAWTHORNE M.A., PIERCY V., SENNITT M.V., THODY V.E., WILSON C., WILSON S. Atypical β-adrenoceptor on brown adipocytes as target for anti-obesity drugs. Nature. 1984;309:163–165. doi: 10.1038/309163a0. [DOI] [PubMed] [Google Scholar]
- CAO W., MEDVEDEV A.V., DANIEL K.W., COLLINS S. β-Adrenergic activation of p38 MAP kinase in adipocytes. J. Biol. Chem. 2001;276:27077–27082. doi: 10.1074/jbc.M101049200. [DOI] [PubMed] [Google Scholar]
- CAO W., LUTTEREL L.M., MEDVEDEV A.V., PIERCE K.L., DANIEL K.W., DIXON T.M., LEFKOWITZ R.J., COLLINS S. Direct binding of activated c-src to the β3-adrenergic receptor is required for MAP kinase activation. J. Biol. Chem. 2000;275:38131–38134. doi: 10.1074/jbc.C000592200. [DOI] [PubMed] [Google Scholar]
- CARR C., LONEY C., UNSON C., KNOWLER J., MILLIGAN G. Chronic exposure of rat glioma C6 cells to cholera toxin induces loss of the α-subunit of the stimulatory guanine nucleotide-binding protein (Gs) Eur. J. Pharmacol. 1990;188:203–209. doi: 10.1016/0922-4106(90)90003-g. [DOI] [PubMed] [Google Scholar]
- CASSEL D., SELINGER Z. Mechanism of adenylate cyclase activation by cholera toxin: Inhibition of GTP hydrolysis at the regulatory site. Proc. Natl. Acad. Sci. U.S.A. 1997;74:3307–3311. doi: 10.1073/pnas.74.8.3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHANG F.-H., BOURNE H.R. Cholera toxin induces cAMP-independent degradation of Gs. J. Biol. Chem. 1989;264:5352–5357. [PubMed] [Google Scholar]
- CHAUDHRY A., MACKENZIE R.G., GEORGIC L.M., GRANNEMAN J.G. Differential interaction of β1- and β3-adrenergic receptors with Gi in rat adipocytes. Cell. Signal. 1994;6:457–465. doi: 10.1016/0898-6568(94)90093-0. [DOI] [PubMed] [Google Scholar]
- DERIJARD B., RAINGEAUD J., BARRETT T., WU I.H., HAN J., ULEVITCH R.J., DAVIS R.J. Independent human MAP-kinase signal transduction pathway defined by MEK and MKK isoforms. Science. 1995;267:682–685. doi: 10.1126/science.7839144. [DOI] [PubMed] [Google Scholar]
- EMORINE L.J., MARULLO S., BRIEND-SUTREN M.-M., PATEY G., TATE K., DELAVIER-KLUTCHKO C., STROSBERG A.D. Molecular characterization of the human β3-adrenergic receptor. Science. 1989;245:1118–1121. doi: 10.1126/science.2570461. [DOI] [PubMed] [Google Scholar]
- ENGELMAN J.A., LISANTI M.P., SCHERER P.E. Specific inhibitors of p38 mitogen-activated protein kinase block 3T3-L1 adipogenesis. J. Biol. Chem. 1998;273:32111–32120. doi: 10.1074/jbc.273.48.32111. [DOI] [PubMed] [Google Scholar]
- FREDRIKSSON J.M., THONBERG H., OHLSON K.B.E., OHBA K., CANNON B., NEDERGAARD J. Analysis of inhibition by H89 of UCP1 gene expression and thermogenesis indicates protein kinase a mediation of β3-adrenergic signaling rather than β3-adrenoceptor antagonism by H89. Biochim. Biophys. Acta. 2001;1538:206–217. doi: 10.1016/s0167-4889(01)00070-2. [DOI] [PubMed] [Google Scholar]
- GAUTHIER C., LNGIN D., BALLIGAND J.-L. β3-Adrenoceptors in the cardiovascular system. TiPS. 2000;21:426–431. doi: 10.1016/s0165-6147(00)01562-5. [DOI] [PubMed] [Google Scholar]
- GERHARDT C.C., GROS J., STROSBERG A.D., ISSAD T. Stimulation of the extracellular signal-regulated kinase 1/2 pathway by human Beta-adrenergic receptor: New Pharmacological profile and mechanism of activation. Mol. Pharmacol. 1999;55:255–262. doi: 10.1124/mol.55.2.255. [DOI] [PubMed] [Google Scholar]
- GLASS D.B., CHENG H.-C., MENDE-MUELLER L., REED J., WALSH D.A. Primary structural determinants essential for potent inhibition of cAMP-dependent protein kinase by inhibitory peptide corresponding to the active protein of the heat-stable inhibitor protein. J. Biol. Chem. 1989;264:8802–8810. [PubMed] [Google Scholar]
- GRANNEMAN J.G., LAHNERS K.N., CHAUDHRY A. Molecular cloning and expression of the rat β3-adrenergic receptor. Mol. Pharmacol. 1991;40:895–899. [PubMed] [Google Scholar]
- GREEN H., KEHINDE O. Spontaneous heritable changes leading to increased adipose conversion in 3T3 cells. Cell. 1976;7:105–113. doi: 10.1016/0092-8674(76)90260-9. [DOI] [PubMed] [Google Scholar]
- GRUJIC D., SUSULIC V.S., HARPER M.-E., HIMMS-HAGEN J., CUNNINGHAM B.A., CORKEY B.E., LOWELL B.B. β3-adrenergic receptors on white and brown adipocytes mediate β3-selective agonist-induced effects on energy expenditure, insulin secration, and food intake. J. Biol. Chem. 1997;272:17686–17693. doi: 10.1074/jbc.272.28.17686. [DOI] [PubMed] [Google Scholar]
- GUAN X.-M., AMEND A., STRADER C.D. Determination of structural domains for G protein coupling and ligand binding in β3-adrenergic receptor. Mol. Pharmacol. 1995;48:492–498. [PubMed] [Google Scholar]
- HANKE J.H., GARDNER J.P., DOW R.L., CHANGELIAN P.S., BRISSETTE W.H., WERINGER E.J., POLLOK B.A., CONNELLY P.A. Discovery of a novel, potent, and src family-selective tyrosine kinase inhibitor. J. Biol. Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- KANDA Y., MIZUNO K., KUROKI Y., WATANABE Y. Thrombin-induced p38 mitogen-activated protein kinase activation is mediated by epidermal growth factor receptor transactivation pathway. Br. J. Pharmacol. 2001;132:1657–1664. doi: 10.1038/sj.bjp.0703952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KATO H., OHUE M., KATO K., NOMURA A., TOYOSAWA K., FURUTANI Y., KIMURA S., KADOWAKI T. Mechanism of amelioration of insulin resistance by β3-adrenoceptor agonist AJ-9677 in the KK-Ay/Ta diabetic obese mouse model. Diabetes. 2001;50:113–122. doi: 10.2337/diabetes.50.1.113. [DOI] [PubMed] [Google Scholar]
- KLEIN J., FASSHAUER M., ITO M., LOWELL B.B., BENITO M., KAHN C.R. β3-Adrenergic stimulation differentially inhibits insulin signaling and decreases insulin-induced glucose uptake in brown adipocytes. J. Biol. Chem. 1999;274:34795–34802. doi: 10.1074/jbc.274.49.34795. [DOI] [PubMed] [Google Scholar]
- KUROKI Y., FUKUSHIMA K., KANDA Y., MIZUNO K., WATANABE Y. Neuroprotection by estrogen via extracellular signal-regulated kinase against quinolinic acid-induced cell death in the rat hippocampus. Eur. J. Neurosci. 2000;13:472–476. doi: 10.1046/j.0953-816x.2000.01409.x. [DOI] [PubMed] [Google Scholar]
- LANGIN D., EKHOLM D., RIDDERSTRALE M., LAFONTAN M., BELFRAGE P. cAMP-dependent protein kinase activation mediated by β3-adrenergic receptors parallels lipolysis in rat adipocytes. Biochim. Biophys. Acta. 1992;1135:349–352. doi: 10.1016/0167-4889(92)90242-4. [DOI] [PubMed] [Google Scholar]
- LINDQUIST J.M., FREDRIKSSON J.M., REHNMARK S., CANNON B., NEDERGAARD J. β3- and α1-Adrenegic Erk1/2 activation is src- but not Gi-mediated in brown adipocytes. J. Biol. Chem. 2000;275:22670–22677. doi: 10.1074/jbc.M909093199. [DOI] [PubMed] [Google Scholar]
- LINDQUIST J.M., REHNMARK S. Ambient temperature regulation of apoptosis in brown adipose tissue. J. Biol. Chem. 1998;273:30147–30156. doi: 10.1074/jbc.273.46.30147. [DOI] [PubMed] [Google Scholar]
- MANARA L., BADONE D., BARONI M., BOCCARDI G., CECCHI R., CROCI T., GIUDICE A., GUZZI U., LANDI M., LE FUR G. Functional identification of rat atypical beta-adrenoceptors by the first beta3-selective antagonists, aryloxypropanolaminotetralins. Br. J. Pharmacol. 1996;117:435–442. doi: 10.1111/j.1476-5381.1996.tb15209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MANTZOROS C.S., QU D., FREDERICH R.C., SUSLIC V.S., LOWELL B.B., MARATOS-FLIER E., FLIER J.S. Activation of β3-adrenergic receptors suppresses leptin expression and mediates a letpin-independent inhibition of food intake in mice. Diabetes. 1996;45:909–914. doi: 10.2337/diab.45.7.909. [DOI] [PubMed] [Google Scholar]
- MILLIGAN G., UNSON C.G., WAKELAM M.J.O. Cholera toxin treatment produces down-regulation of the α-subunit of the stimulatory guanine-nucleotide-binding protein (Gs) Biochem. J. 1989;262:643–649. doi: 10.1042/bj2620643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIZUNO K., KANDA Y., KUROKI Y., TOMIYAMA K., WATANABE Y. Phosphorylation of extracellular signal-regulated kinases 1 and 2 in 3T3-L1 adipocytes by stimulation of β3-adrenoceptor. Eur. J. Pharmacol. 1999;385:63–69. doi: 10.1016/s0014-2999(99)00733-5. [DOI] [PubMed] [Google Scholar]
- MIZUNO K., KANDA Y., KUROKI K., WATANABE Y. The stimulation of β3-adrenoceptor causes phosphorylation of extracellular signal-regulated kinases 1 and 2 through a Gs- but not Gi-dependent pathway in 3T3-L1 adipocytes. Eur. J. Pharmacol. 2000;404:63–68. doi: 10.1016/s0014-2999(00)00601-4. [DOI] [PubMed] [Google Scholar]
- MOULE S.K., DENTON R.M. The activation of p38 MAPK by the β-adrenergic agonist isoproterenol in rat epididymal fat cells. FEBS Lett. 1998;439:287–290. doi: 10.1016/s0014-5793(98)01392-1. [DOI] [PubMed] [Google Scholar]
- MUZZIN P., REVELLI J.-P., KUHNE F., GOCAYNE J.D., MCCOMBIE W.R., VENTER J.C., GIACOBINO J.-P., FRASER C.M. An adipose tissue-specific β3-adrenergic receptor. J. Biol. Chem. 1991;266:24053–24058. [PubMed] [Google Scholar]
- NAHMIAS C., BLIN N., ELALOUF J.-M., MATTEI M.G., STROSBERG A.D., EMORINE L.J. Molecular characterization of the mouse β3-adrenergic receptor: relationship with the atypical receptor of adipocytes. EMBO. J. 1991;10:3721–3727. doi: 10.1002/j.1460-2075.1991.tb04940.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NISOLI E., TONELLO C., LANDI M., CARRUBA M.O. Functional studies of the first selective β3-adrenergic receptor antagonist SR59230A in rat brown adipocytes. Mol. Pharmacol. 1996;49:7–14. [PubMed] [Google Scholar]
- PAGES G., LENORMAND P., L'ALLEMAIN G., CHAMBARD J.-C., MELOCHE S., POUYSSEGUR J. Mitogen-activated protein kinase p42mapk and p44mapk are required for fibroblast proliferation. Proc. Natl. Acad. Sci. U.S.A. 1993;90:8319–8323. doi: 10.1073/pnas.90.18.8319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PANG L., SAWADA T., DECKER S.J., SALTIEL A.R. Inhibition of MAP kinase blocks the differentiation of PC-12 cells induced by nerve growth factor. J. Biol. Chem. 1995;270:13585–13588. doi: 10.1074/jbc.270.23.13585. [DOI] [PubMed] [Google Scholar]
- PENN R.B., PARENT J.L., PRONIN A.N., PANETTIERI R.A., JR, BENOVIC J.L. Pharmacological inhibition of protein kinases in intact cells: antagonism of beta adrenergic receptor ligand binding by H-89 reveals limitations of usefulness. J. Pharmacol. Exp. Ther. 1999;288:428–437. [PubMed] [Google Scholar]
- SEAMON K.B., DALY J.W. Forskolin: a unique diterpene activator of cyclic AMP-generating systems. J. Cyclic. Nucleotide. Res. 1981;7:201–224. [PubMed] [Google Scholar]
- SHIMIZU Y., TANISHITA T., MINOKOSHI Y., SHIMAZY T. Activation of mitogen-activated protein kinase by norepinephrine in brown adipocytes from rats. Endocrinology. 1997;138:248–253. doi: 10.1210/endo.138.1.4832. [DOI] [PubMed] [Google Scholar]
- SOEDER K.J., SNEDDEN S.K., CAO W., ROCCA G.J.D., DANIEL K.W., LUTTRELL L.M., COLLINS S. The β3-adrenergic receptor activates mitogen-activated protein kinase in adipocytes through a Gi -dependent mechanism. J. Biol. Chem. 1999;274:12017–12022. doi: 10.1074/jbc.274.17.12017. [DOI] [PubMed] [Google Scholar]
- SUM F.W., GILBERT A., VENKATESAN A.M., LIM K., WONG V., O'DELL M., FRANCISCO G., CHEN Z., GROSU G., BAKER J., ELLINGBOE J., MALAMAS M., GUNAWAN I., PRIMEAU J., LARGIS E., STEINER K. Prodrugs of CL316243: a selective β3-adrenergic receptor agonist for treating obesity and diabetes. Bioorgan. Med. Chem. Letter. 1999;9:1921–1926. doi: 10.1016/s0960-894x(99)00316-9. [DOI] [PubMed] [Google Scholar]
- SUSULIC V.S., FREDERICH R.C., LAWITTS J., TOZZO E., KAHN B.B., HARPER M.-E., HIMMS-HAGEN J., FLIER J.S., LOWELL B.B. Target disruption of the β3-adrenergic receptor gene. J. Biol. Chem. 1995;270:29483–29492. doi: 10.1074/jbc.270.49.29483. [DOI] [PubMed] [Google Scholar]
- VAN CORVEN E.J., HORDIJIK P.L., MEDEMA R.H., BOS J.L., MOOLENAAR W.H. Pertussis toxin-sensitive activation of p21 ras by G protein-coupled receptor agonists in fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 1993;90:1257–1261. doi: 10.1073/pnas.90.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN LIEFDE I., VAN WITENBURG A., VAQUELIN G. Multiple beta-adrenergic receptor subclasses mediate the L-isoproterenol-induced lipolytic response in rat adipocytes. J. Pharmacol. Exp. Ther. 1992;262:552–558. [PubMed] [Google Scholar]
- VANHOUTE P., BARNIER J.-V., GUIBERT B., PAGES C., BESSON M.-J., HIPSKIND R.A., CABOCHE J. Glutamate induces phosphorylation of Elk-1 and CREB, along with c-fos activation, via an extracellular signal-regulated kinase-dependent pathway in brain slices. Mol. Cell. Biol. 1998;19:136–146. doi: 10.1128/mcb.19.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WATANABE Y., JAKOBS K.H. Inhibition of Ns-stimulated human platelet adenylate cyclase by forskolin. Mol. Pharmacol. 1986;29:258–263. [PubMed] [Google Scholar]
- WEYER C., TATARANNI P.A., SNITKER S., DANFORTH E., JR, RAVUSSIN E. Increase in insulin action and fat oxidation after treatment with CL316, 243, a highly selective β3-adrenoceptor agonists in Humans. Diabetes. 1998;47:1555–1561. doi: 10.2337/diabetes.47.10.1555. [DOI] [PubMed] [Google Scholar]