

Abstract
This study has administered pirfenidone (5-methyl-1-phenyl-2-[1H]-pyridone) or amiloride to attenuate the remodelling and associated functional changes, especially an increased cardiac stiffness, in DOCA-salt hypertensive rats.
In control rats, the elimination half-life of pirfenidone following a single intravenous dose of 200 mg kg−1 was 37 min while oral bioavailability at this dose was 25.7%. Plasma pirfenidone concentrations in control rats averaged 1.9±0.1 μg ml−1 over 24 h after 14 days' administration as a 0.4% mixture in food.
Pirfenidone (approximately 250 – 300 mg kg−1 day−1 as 0.4% in food) and amiloride (1 mg kg−1 day−1 sc) were administered for 2 weeks starting 2 weeks post-surgery. Pirfenidone but not amiloride attenuated ventricular hypertrophy (2.69±0.09, UNX 2.01±0.05. DOCA-salt 3.11±0.09 mg kg−1 body wt) without lowering systolic blood pressure.
Collagen deposition was significantly increased in the interstitium after 2 weeks and further increased with scarring of the left ventricle after 4 weeks; pirfenidone and amiloride reversed the increases and prevented further increases. This accumulation of collagen was accompanied by an increase in diastolic stiffness constant; both amiloride and pirfenidone reversed this increase.
Noradrenaline potency (positive chronotropy) was decreased in right atria (neg log EC50: control 6.92±0.06; DOCA-salt 6.64±0.08); pirfenidone but not amiloride reversed this change. Noradrenaline was a more potent vasoconstrictor in thoracic aortic rings (neg log EC50: control 6.91±0.10; DOCA-salt 7.90±0.07); pirfenidone treatment did not change noradrenaline potency.
Thus, pirfenidone and amiloride reverse and prevent cardiac remodelling and the increased cardiac stiffness without reversing the increased vascular responses to noradrenaline.
Keywords: DOCA-salt hypertension, hypertrophy, fibrosis, pirfenidone, amiloride
Introduction
Cardiac remodelling as in chronic hypertension involves myocyte hypertrophy as well as fibrosis, an increased and non-uniform deposition of extracellular matrix proteins, especially collagens (Weber et al., 1993; 1994). Although hypertension-induced left ventricular hypertrophy has been implicated as the single most important prognostic indicator for adverse cardiovascular events in humans (Levy et al., 1990), changes in the quality of the interstitial space are also critical. The extracellular matrix connects myocytes, aligns contractile elements, prevents overextending and disruption of myocytes, transmits force and provides tensile strength to prevent rupture. Fibrosis occurs in many models of hypertension leading to an increased diastolic stiffness, a reduction in cardiac function (Weber et al., 1993; 1994) and an increased risk of arrhythmias (Assayag et al., 1997). If fibrosis rather than myocyte hypertrophy is the critical factor in impaired cardiovascular function, then reversal of cardiac fibrosis by itself may return cardiac function towards normal. Since collagen deposition is a dynamic process (Sun & Weber, 2000), appropriate pharmacological intervention could selectively reverse existing fibrosis and prevent further fibrosis and thereby improve function, even if the increased systolic blood pressure was unchanged.
Pirfenidone and amiloride are candidate drugs for the attenuation of cardiac fibrosis based on the relevant literature. Pirfenidone prevented and reversed lung fibrosis and attenuated pulmonary dysfunction following bleomycin treatment in hamsters (Iyer et al., 1995; Schelegle et al., 1997) by suppressing inflammatory events (Iyer et al., 2000) and down-regulating lung procollagen I over-expression (Iyer et al., 1999a). Pirfenidone decreased collagen production and deposition in a rat model of hepatic fibrosis (Tada et al., 2001). In addition, this compound reversed cardiac and renal fibrosis and attenuated the increase in diastolic stiffness of diabetic hearts from streptozotocin-treated rats without normalizing cardiac contractility or renal function (Miric et al., 2001). Amiloride selectively prevented cardiac collagen accumulation in aldosterone-salt hypertensive rats (Campbell et al., 1993). However, the more clinically relevant reversal of cardiac fibrosis by amiloride in hypertension has not been studied, although the ACE inhibitor, lisinopril, has been shown to reverse cardiac fibrosis in humans (Brilla et al., 2000).
The aim of this study was to determine the consequences of pirfenidone and amiloride treatment on collagen deposition and cardiac function in the deoxycorticosterone acetate (DOCA)-salt hypertensive rat heart. This model develops ventricular hypertrophy with cardiac fibrosis (Young et al., 1995) following increased expression of collagen I and III mRNA (Robert et al., 1994; Brown et al., 1999).
Methods
Pharmacokinetic studies
Blood sample collection after intravenous dosing
Wistar rats (150 – 200 g) were anaesthetized using Zoletil® i.p. (zolazepam 25 mg kg−1 and tiletamine 25 mg kg−1) with xylazine (10 mg kg−1) and catheterized via the femoral vein. Catheters were flushed with heparinized saline at the time of placement, four days later and on the day of experimentation (7th day). Pirfenidone (200 mg kg−1 suspension in 1 ml normal saline) was infused intravenously over 30 s. Blood samples of less than 0.2 ml were collected in heparinized tubes up to 4 h after pirfenidone was infused, and the same volume of saline was injected into the catheter to maintain blood volume throughout the trial.
Blood sample collection after oral dosing
Pirfenidone (200 mg kg−1 suspension in 1 ml normal saline) was administered through a stomach tube. Blood samples (0.2 ml) were collected from a small incision in the tail vein for up to 4 h following a single oral dosage and every four hours for 24 h following chronic dosage. Rats were killed as above following collection of the last blood sample, and the pH of the stomach contents was measured using pH paper. Additionally, rats were fed an unrestricted diet of pulverized rat chow containing 0.4% pirfenidone for 14 days. All blood samples were collected in heparinized microfuge tubes, and centrifuged at 11,000×g for 3 min before plasma was collected from each sample and stored at −20°C until HPLC analysis for pirfenidone concentration as described below.
HPLC analysis of plasma pirfenidone concentration
A Waters Novapak C18 (Picotag) reverse phase column was used with 50% acetonitrile in HPLC grade water as the mobile phase at 1 ml min−1; pirfenidone was measured using a Beckman DU690 UV spectrometer at 280 nm. Elution of pirfenidone occurred at approximately 3.1 min after injection onto the column; a standard curve was constructed and unknown concentrations estimated from this standard curve.
DOCA-salt hypertensive rats
Male Wistar rats (8 – 10 weeks old) were obtained from the Central Animal Breeding House of The University of Queensland. All experimental protocols were approved by the Animal Experimentation Ethics Committee of The University of Queensland under the guidelines of the National Medical and Health Research Council of Australia. Uninephrectomy was performed on all treated rats. The rats were anaesthetized as above; a lateral abdominal incision was used to access the kidneys, and the left renal vessels and ureter were ligated. The left kidney was removed and weighed and the skin wound sutured. Uninephrectomized rats were given either no further treatment (UNX rats) or 1% NaCl in the drinking water with subcutaneous injections of deoxycorticosterone acetate (DOCA; 25 mg in 0.4 ml dimethylformamide every fourth day) (DOCA-salt rats) (Dallemagne et al., 2000). Experiments were performed 2 or 4 weeks after surgery. After 2 weeks, rats were given either pirfenidone (0.4% in powdered rat food) or amiloride (1 mg kg−1 day−1 sc) for a further 2 weeks.
Food and water intake and body weights were measured daily for all rats. Systolic blood pressure was measured in unanaesthetized rats using a tail-cuff method. Rats were killed with pentobarbitone (200 mg kg−1 ip). Blood was taken from the abdominal vena cava, centrifuged and the plasma frozen. Plasma aldosterone concentrations were measured by a commercial radioimmunoassay kit.
Isolated Langendorff heart preparation
Rats were anaesthetized with sodium pentobarbitone (100 mg kg−1 ip) and heparin (2000 iu) was administered via the femoral vein. After allowing 2 min for the heparin to fully circulate, the heart was excised and placed in cooled (0°C) crystalloid perfusate (Krebs-Henseleit solution of the following composition in mM: NaCl 118, KCl 4.7, MgSO4 1.2, KH2PO4 1.2, CaCl2 2.3, NaHCO3 25.0, glucose 11.0). The heart was then attached to the cannula (with the tip of the cannula positioned immediately above the coronary ostia of the aortic stump) and perfused in a non-recirculating Langendorff fashion at 100 cm of hydrostatic pressure as previously described (Smolenski et al., 1998). The buffer temperature was maintained at 35°C. The apex of the heart was pierced to facilitate thebesian drainage and paced at 250 b.p.m.
A balloon catheter was inserted in the left ventricle via the mitral orifice for measurement of left ventricular developed pressure (Smolenski et al., 1998). The catheter was connected via a three-way tap to a micrometer syringe and to a Statham P23 pressure transducer. The outer diameter of the catheter was similar to the mitral annulus to prevent ejection of the balloon during the systolic phase. After a 10 min stabilization period, steady-state left ventricular pressure was recorded from isovolumetrically beating hearts. Increments in balloon volume were applied to the heart until left ventricular end-diastolic pressure reached approximately 30 mmHg. At the end of the experiment, the atria and right ventricle were dissected away and the weight of the left ventricle plus septum was recorded.
Myocardial diastolic stiffness was calculated as the diastolic stiffness constant (k, dimensionless), the slope of the linear relation between tangent elastic modulus (E, dyne cm−2) and stress (σ, dyne cm−2) (Brilla et al., 1991; Brown et al., 1999). To assess contractile function, maximal+dP/dt values were calculated at a diastolic pressure of 10 mmHg.
Isolated cardiac muscles and thoracic aortic rings
The heart was removed under anaesthesia. The right atria and papillary muscles from the left ventricle were removed and suspended in organ baths at a resting tension of 5 – 10 mN adjusted to give the maximal twitch response. Tissues were bathed in a modified Tyrodes solution (in mM): NaCl 136.9, KCl 5.4, MgCl2 1.05, CaCl2 1.8, NaHCO3 22.6, NaH2PO4 0.42, glucose 5.5, ascorbic acid 0.28, sodium edetate 0.05, bubbled with 95%O2/5%CO2 and stimulated at 1 Hz at 35°C as previously described (Brown et al., 1991b). Cumulative concentration-response curves were measured for noradrenaline and, following washout and re-equilibration, to calcium chloride. At the end of the experiment, papillary muscle dimensions were measured under the loading conditions of the experiment; all tissues were blotted and weighed.
Thoracic aortic rings (approximately 4 mm in length) were suspended with a resting tension of 10 mN (Brown et al., 1991a) and contracted twice with isotonic KCl (100 mM). The presence of endothelium was demonstrated by addition of acetylcholine (10 μM). Cumulative contraction responses to noradrenaline were measured.
Collagen distribution
Collagen distribution was determined by image analysis of picrosirius red-stained sections of the hearts (Miric et al., 2001). In brief, transverse sections stored in 4% paraformaldehyde were dehydrated and embedded in wax and then sliced into 5 μm sections. These were stained with picrosirius red (0.1% Sirius Red F3BA in picric acid). Slides were left in 0.2% phosphomolybdic acid for 5 min, washed, left in picrosirius red for 90 min, then in 1 mM HCl for 2 min and 70% ethanol for 45 s. The stained sections were analysed with an Image Pro plus analysis program using an Olympus BH2 microscope with results expressed as a percentage of red area in each screen. At least four areas were examined in each heart.
Data analysis
All results are given as mean±s.e.m. The negative log EC50 of the increase in either force of contraction in mN or rate of contraction in beats/min was determined from the concentration giving half-maximal responses in individual concentration-response curves. These results were analysed by two-way analysis of variance followed by the Duncan test to determine differences between treatment groups and by paired or unpaired t-tests as appropriate; P<0.05 was considered significant.
Drugs
Deoxycorticosterone acetate, amiloride and noradrenaline were purchased from Sigma Chemical Company, St Louis, MO, U.S.A. Pirfenidone was provided by Marnac Inc., Dallas, TX, U.S.A. Noradrenaline was dissolved in distilled water; deoxycorticosterone acetate was dissolved in dimethylformamide with mild heating.
Results
Following a single intravenous injection, pirfenidone showed an elimination half-life of 37 min (Figure 1A). Comparison of plasma concentrations following intravenous and oral administration indicated an oral bioavailability of 25.7% in the rat. Oral administration of pirfenidone as a 0.4% mixture in powdered food for 14 days gave constant plasma concentrations of 1.9±0.1 μg/ml over 24 h (Figure 1B). Following the same protocol, plasma pirfenidone concentrations in DOCA-salt rats were 1.3±0.3 μg/ml (n=5).
Figure 1.
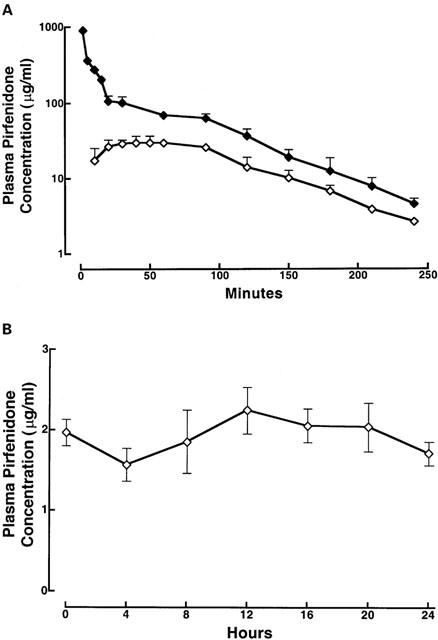
Plasma pirfenidone concentrations in control rats after a single dose (A) and throughout 24 h when pirfenidone added to food (B) for oral dosage (open diamonds) or intravenous dosage (filled diamonds); for (B), 0 h was 0900 h after 2 weeks' administration of pirfenidone in food; n=4 for all groups.
DOCA-salt treatment produced marked physiological changes including hypertension, hypertrophy of the left ventricle and remnant kidney (Table 1). Despite significant increases in water intake and decreases in food intake (Figure 2), no change in body weight was observed. Amiloride or pirfenidone administration did not modify these parameters in UNX or DOCA-salt hypertensive rats; pirfenidone, however, attenuated ventricular and renal hypertrophy (Figure 2; Table 1). Daily intake of pirfenidone averaged 287±12 mg kg−1 in UNX rats and 256±18 mg kg−1 in DOCA-salt rats (Figure 2).
Table 1.
Physiological parameters

Figure 2.
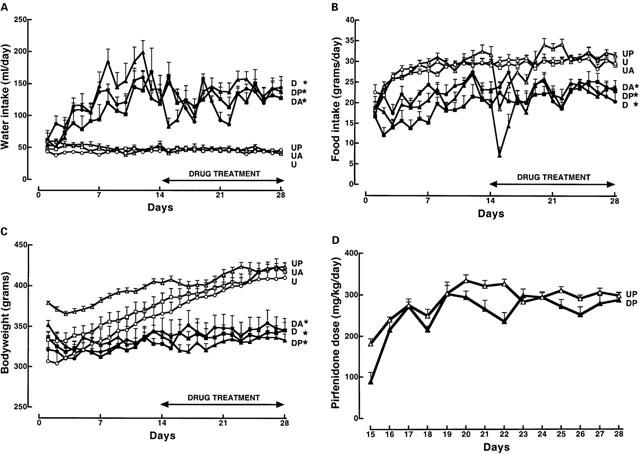
Water (A) and food (B) consumption, body weight (C) and pirfenidone consumption (D) in UNX (U, n=8; open circles), UNX+pirfenidone (UP, n=8; open triangles), UNX+amiloride (UA, n=8; open squares), DOCA-salt (D, n=8; filled circles), DOCA-salt+pirfenidone (DP, n=15; filled triangles), DOCA-salt+amiloride (DA, n=8; filled squares) rats; *P<0.05 vs control.
DOCA-salt hypertensive rats showed an increase in interstitial collagen deposition after 2 weeks which was further increased after 4 weeks (Figure 3A); perivascular collagen showed similar changes. Myocardial scarring increased only after 4 weeks (Figure 3B); administration of pirfenidone or amiloride normalised collagen deposition thus reversing the existing collagen deposition as well as preventing new collagen deposition. In the isolated Langendorff preparation, DOCA-salt hearts showed an increased diastolic stiffness constant; treatment with pirfenidone or amiloride normalized this constant (Figure 4, Table 1). Pirfenidone, but not amiloride, significantly decreased left ventricular contractility measured as+dP/dt in the DOCA-salt hearts (Figure 4, Table 1).
Figure 3.
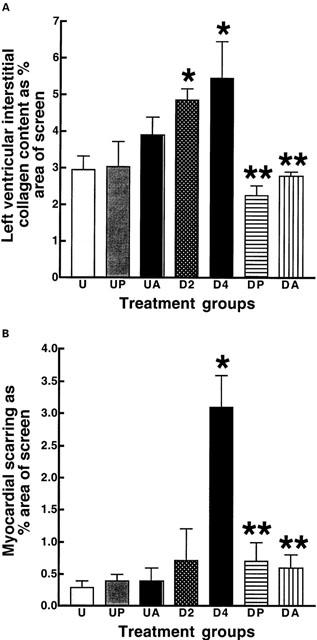
Interstitial collagen deposition (A) and scarring (B) in the ventricles (percent of total area): UNX (U, n=7), UNX+pirfenidone (UP, n=6), UNX+amiloride (UA, n=6), DOCA-salt (D2, after 2 weeks; D4, after 4 weeks; n=7), DOCA-salt+pirfenidone (DP, n=10), DOCA-salt+amiloride (DA, n=7); *P<0.05 compared to UNX, **P<0.05 compared to DOCA-salt.
Figure 4.
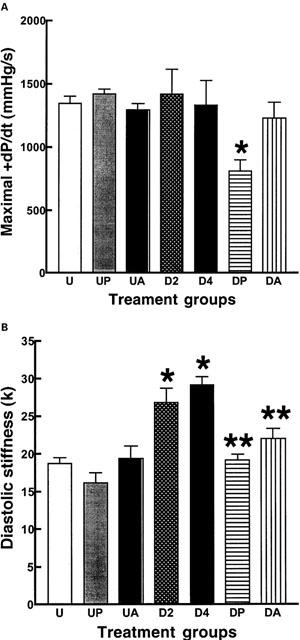
Contractility as maximal dP/dt (A) and diastolic stiffness (k) (B): UNX (U, n=7), UNX+pirfenidone (UP, n=6), UNX+amiloride (UA, n=6), DOCA-salt (D2, after 2 weeks; D4, after 4 weeks; n=7), DOCA-salt+pirfenidone (DP, n=10), DOCA-salt+amiloride (DA, n=7); *P<0.05 compared to UNX, **P<0.05 compared to DOCA-salt.
The potency of positive chronotropic responses to noradrenaline was increased in DOCA-salt rats (neg log EC50: control 6.92±0.06; DOCA-salt 6.64±0.08) (Figure 5A); pirfenidone but not amiloride reversed this change. Positive inotropic responses to noradrenaline and calcium chloride (Figure 5B) were unaltered in left ventricular papillary muscles taken from DOCA-salt hearts compared to UNX hearts. Pirfenidone treatment did not change noradrenaline responses but increased maximal responses to calcium chloride. No changes in responses were observed in responses in muscles from amiloride-treated DOCA-salt rat hearts. However, the responsiveness of isolated thoracic aortic rings to noradrenaline was increased in DOCA-salt rats compared to UNX controls (neg log EC50: control 6.91±0.10; DOCA-salt 7.90±0.07) (Figure 6). Neither pirfenidone nor amiloride treatment attenuated these increased vasoconstrictor responses to noradrenaline.
Figure 5.
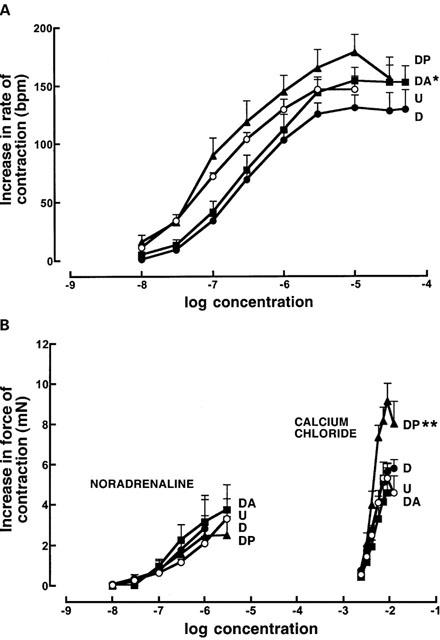
Cumulative concentration-response curves for noradrenaline in right atria (A) and noradrenaline and calcium chloride in left ventricular papillary muscles (B) from UNX (U, n=16; open circles), DOCA-salt (D, n=9; filled circles), DOCA-salt+pirfenidone (DP, n=5; filled triangles), and DOCA-salt+amiloride (DA, n=6; filled squares) treated rats; *P<0.05 compared to UNX, **P<0.05 compared to DOCA-salt values.
Figure 6.
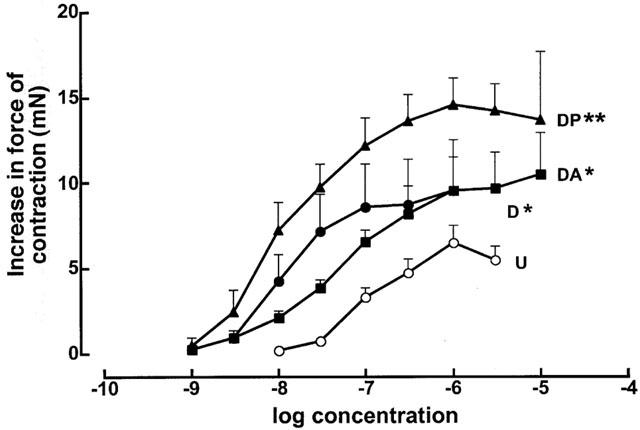
Cumulative concentration-response curves for noradrenaline in the thoracic aortic rings from UNX (U, n=16; open circles), DOCA-salt (D, n=7; filled circles), DOCA-salt+pirfenidone (DP, n=7; filled triangles) and DOCA-salt+amiloride (DA, n=5; filled squares) treated animals; *P<0.05 compared to UNX, **P<0.05 compared to DOCA-salt.
Discussion
The DOCA-salt hypertensive rat rapidly develops cardiac hypertrophy and fibrosis (Young et al., 1995; Doggrell & Brown, 1998). We have recently shown, in this model, that cardiac fibrosis can be reversed, without lowering blood pressure or decreasing the degree of cardiac hypertrophy, by inhibition of the renin-angiotensin system using the ACE inhibitor, captopril, the AT1 receptor antagonist, candesartan, or the aldosterone antagonist, spironolactone (Brown et al., 1999). This study extends the range of compounds that selectively reverse existing cardiac fibrosis and prevent further deposition to include pirfenidone and amiloride. Neither compound decreased the increased systolic blood pressure; pirfenidone attenuated the degree of cardiac and renal hypertrophy and also decreased ventricular contractility. Both compounds decreased the diastolic stiffness; neither reversed the increased vascular sensitivity to noradrenaline in these hypertensive rats. This study provides more evidence that cardiac fibrosis can be attenuated independently of blood pressure and ventricular hypertrophy and that this effect may improve cardiac function.
The antifibrotic actions of pirfenidone have been shown in different species and diseases, for example bleomycin-induced pulmonary fibrosis in hamsters (Iyer et al., 1995) with attenuation of pulmonary functional deficits (Schelegle et al., 1997), in sclerosing peritonitis in rats (Suga et al., 1995), in renal fibrosis following partial nephrectomy in rats (Shimizu et al., 1997), in human myometrial and leiomyoma cell cultures (Lee et al., 1998), in streptozotocin-diabetic rats (Miric et al., 2001) and in dimethylnitrosamine-induced hepatic fibrosis in rats (Tada et al., 2001). However, pirfenidone had no therapeutic activity in patients with myelofibrosis with myeloid metaplasia (Mesa et al., 2001). Reversal protocols were used by Miric et al. (2001) as well as by Shimizu et al. (1997) who started pirfenidone treatment two weeks post surgery, as in the current study. Daily pirfenidone doses ranged from 0.5% in food (250 – 375 mg kg−1 at food intakes of 20 – 30 g day−1 as in the current study), to 200 – 300 mg kg−1 (Miric et al., 2001 and current study), 350 mg kg−1 (Suga et al., 1995) or 500 mg kg−1 (Shimizu et al., 1997; Tada et al., 2001). Despite these differences in protocol, the results have shown that pirfenidone consistently prevents collagen accumulation. None of these literature studies have measured plasma pirfenidone concentrations. This study shows that plasma concentrations of 1.3 – 2 μg ml−1 are effective in attenuating fibrosis in the heart and that constant plasma levels are achieved in rats following administration in the powdered food.
The mechanisms of action of pirfenidone are unclear but this compound may increase collagen breakdown by reducing the TGFβ1-induced inhibition of the degrading enzymes, the matrix metalloproteinases (Suga et al., 1995). Pirfenidone also down-regulated bleomycin-induced over-expression of procollagen I and III genes in the lungs (Iyer et al., 1999a) and suppressed an increased TGFβ1 mRNA (Iyer et al., 1999b). Thus, suppression of the synthesis or activity of this cytokine is the most likely mechanism of action of pirfenidone. This is supported by results showing that suppression of an increased TGFβ1 expression by tranilast in hypertensive transgenic rats overexpressing human renin attenuated left ventricular hypertrophy and fibrosis without lowering blood pressure (Pinto et al., 2000). Pirfenidone may also show anti-inflammatory effects such as suppression of increased vascular permeability, neutrophil recruitment and influx of inflammatory cells, in addition to its effects on collagen turnover (Iyer et al., 2000; Corbel et al., 2001).
Amiloride prevented cardiac fibrosis in a model of mineralocorticoid excess (Campbell et al., 1993) similar to our model but the current study is the first demonstration of reversal of fibrosis with amiloride together with a normalization of cardiac stiffness. Amiloride is well-known as a sodium channel inhibitor with potassium-sparing properties. In electrolyte-steroid cardiopathy, amiloride protected the myocardium from the necrotizing effects of fludrocortisone (Selye, 1968). This prevention of cell death by maintenance of myocardial potassium concentrations is a possible mechanism for prevention of scarring by amiloride but does not explain the reversal of fibrosis.
Collagen synthesis, mostly in the fibroblasts, is activated by both cell surface receptors, especially for angiotensin II (Weber et al., 1993), and nuclear receptors such as the mineralocorticoid receptors activated by aldosterone (Young et al., 1995; Brilla et al., 1994) which may partially explain the responses to the renin-angiotensin system inhibitors. Mature collagens are degraded by matrix metalloproteinases (MMPs), especially MMP-1 and MMP-8 (collagenases); tissue inhibitors of metalloproteinases (TIMPs) control the activity of the MMPs (Dollery et al., 1995). Growth factors (for example TGFβ and bradykinin) modulate both the synthesis and degradation of collagens and the proliferation of fibroblasts (Weber et al., 1994). This level of complexity in the control of cardiac extracellular matrix proteins provides several points of attack for pharmacological therapy to reverse an existing increased collagen deposition and prevent further deposition in the heart. Thus, many compounds are likely to modulate cardiac fibrosis, including pirfenidone, amiloride and inhibitors of the renin-angiotensin system (Brown et al., 1999). Further, the cross-linking of myocardial collagen is another potential target as this parameter may determine the degree of cardiac dilatation in heart disease (Woodiwiss et al., 2001). Removal of collagen may carry some risks. Excessive degradation to below normal collagen concentrations may disrupt the collagen network and distort cardiac architecture leading to wall thinning, chamber dilatation and possibly wall rupture. The optimal extent of the removal of collagen and the extent of the decrease in cross-linking in chronic studies needs to be investigated.
Repair of the abnormal cardiac structure in hypertension by selective regression of the increased extracellular matrix provides an exciting possibility to return cardiac function towards normal. Previous studies have usually demonstrated prevention of fibrosis; however, both reversal of existing fibrosis and prevention of further collagen deposition are clinically relevant outcomes. Since both pirfenidone and amiloride appear to both reverse and prevent fibrosis, they may have considerable potential in attenuating cardiac fibrosis associated with chronic hypertension and also the functional impairment of the heart in hypertensive humans. These antifibrotic actions may help prevent the progression of hypertensive heart disease towards heart failure with its well-known poor prognosis.
Acknowledgments
This study was supported by The University of Queensland.
Abbreviations
- DOCA
deoxycorticosterone acetate
- UNX
uninephrectomized
References
- ASSAYAG P., CARRÉ F., CHEVALIER B., DELCAYRE C., MANSIER P., SWYNGHEDAUW B. Compensated cardiac hypertrophy, arrhythmogenicity and the new myocardial phenotype. I. Fibrosis. Cardiovasc. Res. 1997;34:439–444. doi: 10.1016/s0008-6363(97)00073-4. [DOI] [PubMed] [Google Scholar]
- BRILLA C.G., FUNCK R.C., RUPP H. Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation. 2000;102:1388–1393. doi: 10.1161/01.cir.102.12.1388. [DOI] [PubMed] [Google Scholar]
- BRILLA C.G., JANICKI J.S., WEBER K.T. Impaired diastolic function and coronary reserve in genetic hypertension. Circ. Res. 1991;69:107–115. doi: 10.1161/01.res.69.1.107. [DOI] [PubMed] [Google Scholar]
- BRILLA C.G., ZHOU G., MATSUBARA L., WEBER K.T. Collagen metabolism in cultured adult rat cardiac fibroblasts: response to angiotensin II and aldosterone. J. Mol. Cell. Cardiol. 1994;26:809–820. doi: 10.1006/jmcc.1994.1098. [DOI] [PubMed] [Google Scholar]
- BROWN L., CRAGOE E.J., JR, ABEL K.C., MANLEY S.W., BOURKE J.R. Amiloride analogues induce responses in isolated rat cardiovascular tissues by inhibition of Na+/Ca2+ exchange. Naunyn-Schmiedeberg's Arch. Pharmacol. 1991a;344:220–224. doi: 10.1007/BF00167222. [DOI] [PubMed] [Google Scholar]
- BROWN L., DUCE B., MIRIC G., SERNIA C. Reversal of cardiac fibrosis in deoxycorticosterone acetate-salt hypertensive rats by inhibition of the renin-angiotensin system. J. Am. Soc. Nephrol. 1999;10:S143–S148. [PubMed] [Google Scholar]
- BROWN L., SERNIA C., NEWLING R., FLETCHER P. Comparison of inotropic and chronotropic responses in rat isolated atria and ventricles. Clin. Exp. Pharmacol. Physiol. 1991b;18:753–760. doi: 10.1111/j.1440-1681.1991.tb01393.x. [DOI] [PubMed] [Google Scholar]
- CAMPBELL S.E., JANICKI J.S., MATSUBARA B.B., WEBER K.T. Myocardial fibrosis in the rat with mineralocorticoid excess - prevention of scarring by amiloride. Am. J. Hypertens. 1993;6:487–495. doi: 10.1093/ajh/6.6.487. [DOI] [PubMed] [Google Scholar]
- CORBEL M., LANCHOU J., GERMAIN N., MALLEDANT Y., BOICHOT E., LAGENTE V. Modulation of airway remodelling-associated mediators by the antibibrotic compound, pirfenidone, and the matrix metalloproteinase inhibitor, batimastat, during acute lung injury in mice. Eur. J. Pharmacol. 2001;426:113–121. doi: 10.1016/s0014-2999(01)01209-2. [DOI] [PubMed] [Google Scholar]
- DALLEMAGNE C., OOI S.-Y., BROWN L., GOBÉ G., ENDRE Z. Renal impairment in deoxycorticosterone acetate-salt hypertensive rats. Nephrology. 2000;5:255–284. [Google Scholar]
- DOGGRELL S.A., BROWN L. Rat models of hypertension, cardiac hypertrophy and failure. Cardiovasc. Res. 1998;39:89–105. doi: 10.1016/s0008-6363(98)00076-5. [DOI] [PubMed] [Google Scholar]
- DOLLERY C.M., MCEWAN J.R., HENNEY A.M. Matrix metalloproteinases and cardiovascular disease. Circ. Res. 1995;77:863–868. doi: 10.1161/01.res.77.5.863. [DOI] [PubMed] [Google Scholar]
- IYER S.N., WILD J.S., SCHIEDT M.J., HYDE D.M., MARGOLIN S.B., GIRI S. Dietary intake of pirfenidone ameliorates bleomycin induced lung fibrosis in hamsters. J. Lab. Clin. Med. 1995;125:779–785. [PubMed] [Google Scholar]
- IYER S.N., GURUJEYALAKSHMI G., GIRI S.N. Effects of pirfenidone on procollagen gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J. Pharmacol. Exp. Ther. 1999a;289:211–218. [PubMed] [Google Scholar]
- IYER S.N., GURUJEYALAKSHMI G., GIRI S.N. Effects of pirfenidone on transforming growth factor-β gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J. Pharmacol. Exp. Ther. 1999b;291:367–373. [PubMed] [Google Scholar]
- IYER S.N., HYDE D.M., GIRI S.N. Anti-inflammatory effect of pirfenidone in the bleomycin-hamster model of lung inflammation. Inflammation. 2000;24:477–491. doi: 10.1023/a:1007068313370. [DOI] [PubMed] [Google Scholar]
- LEE K.-S., MARGOLIN S.B., NOWAK R.A. Pirfenidone: a novel pharmacological agent that inhibits leiomyoma cell proliferation and collagen production. J. Clin. Endocrin. Metab. 1998;83:219–223. doi: 10.1210/jcem.83.1.4503. [DOI] [PubMed] [Google Scholar]
- LEVY D., GARRISON R.J., SAVAGE D.D., KANNEL W.B., CASTELLI W.P. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N. Engl. J. Med. 1990;322:1561–1566. doi: 10.1056/NEJM199005313222203. [DOI] [PubMed] [Google Scholar]
- MESA R.A., TEFFERI A., ELLIOTT M.A., HOAGLAND H.C., CALL T.G., SCHROEDER G.S., YOON S.Y., LI C.Y., GRAY L.A., MARGOLIN S., HOOK C.C. A phase II trial of pirfenidone (5-methyl-1-phenyl-2-[1H]-pyridone), a novel anti-fibrosing agent, in myelofibrosis with myeloid metaplasia. Br. J. Haematol. 2001;114:111–113. doi: 10.1046/j.1365-2141.2001.02883.x. [DOI] [PubMed] [Google Scholar]
- MIRIC G., DALLEMAGNE C., ENDRE Z., MARGOLIN S., TAYLOR S.M., BROWN L. Reversal of cardiac and renal fibrosis by pirfenidone and spironolactone in streptozotocin-diabetic rats. Br. J. Pharmacol. 2001;133:687–694. doi: 10.1038/sj.bjp.0704131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PINTO Y.M., PINTO-SIETSMA S.-J., PHILIPP T., ENGLER S., KOβMEHL P., HOCHER B., MARQUARDT H., SETHMANN S., LAUSTER R., MERKER H.-J., PAUL M. Reduction in left ventricular messenger RNA for transforming growth factor β1 attenuates left ventricular fibrosis and improves survival without lowering blood pressure in the hypertensive TGR(mRen2)27 rat. Hypertension. 2000;36:747–754. doi: 10.1161/01.hyp.36.5.747. [DOI] [PubMed] [Google Scholar]
- ROBERT V., THIEM N.V., CHEAV S.L., MOUAS C., SWYNGHEDAUW B., DELCAYRE C. Increased cardiac types I and III collagen mRNA's in aldosterone-salt hypertension. Hypertension. 1994;24:30–36. doi: 10.1161/01.hyp.24.1.30. [DOI] [PubMed] [Google Scholar]
- SCHELEGLE E.S., MANSOOR J.K., GIRI S. Pirfenidone attenuates bleomycin-induced changes in pulmonary functions in hamsters. PSEBM. 1997;218:392–397. doi: 10.3181/00379727-216-44187. [DOI] [PubMed] [Google Scholar]
- SELYE H. Prevention of cardiac necrosis by amiloride. J. Am. Med. Assoc. 1968;206:103–104. [PubMed] [Google Scholar]
- SHIMIZU T., FUKAGAWA M., KURODA T., HATA S., IWASAKI Y., NEMOTO M., SHIRAI K., YAMAUCHI S., MARGOLIN S.B., SHIMIZU F., KUROKAWA K. Pirfenidone prevents collagen accumulation in the remnant kidney in rats with partial nephrectomy. Kidney Int. 1997;52 suppl 63:S239–S243. [PubMed] [Google Scholar]
- SMOLENSKI R.T., JAYAKUMAR J., SEYMOUR A.-M.L., YACOUB M.H. Energy metabolism and mechanical recovery after cardioplegia in moderately hypertrophied rats. Molec. Cell Biochem. 1998;180:137–143. [PubMed] [Google Scholar]
- SUGA H., TERAOKA S., OTA K., KOMEMUSHI S., FURUTANI S., YAMAUCHI S., MARGOLIN S.B. Preventive effect of pirfenidone against sclerosing peritonitis in rats. Exp. Toxic. Pathol. 1995;47:287–291. doi: 10.1016/s0940-2993(11)80261-7. [DOI] [PubMed] [Google Scholar]
- SUN Y., WEBER K.T. Infarct scar: a dynamic tissue. Cardiovasc. Res. 2000;46:250–256. doi: 10.1016/s0008-6363(00)00032-8. [DOI] [PubMed] [Google Scholar]
- TADA S., NAKAMUTA M., ENJOJI M., SUGIMOTO R., IWAMOTO H., KATO M., NAKASHIMA Y., NAWATA H. Pirfenidone inhibits dimethylnitrosamine-induced hepatic fibrosis in rats. Clin. Exp. Pharmacol. Physiol. 2001;28:522–527. doi: 10.1046/j.1440-1681.2001.03481.x. [DOI] [PubMed] [Google Scholar]
- WEBER K.T., BRILLA C.G., JANICKI J.S. Myocardial fibrosis: functional significance and regulatory factors. Cardiovasc. Res. 1993;27:341–348. doi: 10.1093/cvr/27.3.341. [DOI] [PubMed] [Google Scholar]
- WEBER K.T., SUN Y., TYAGI S.C., CLEUTJENS J.P.M. Collagen network of the myocardium: function, structural remodeling and regulatory mechanisms. J. Mol. Cell. Cardiol. 1994;26:279–292. doi: 10.1006/jmcc.1994.1036. [DOI] [PubMed] [Google Scholar]
- WOODIWISS A.J., TSOTETSI O.J., SPROTT S., LANCASTER E.J., MELA T., CHUNG E.S., MEYER T.E., NORTON G.R. Reduction in myocardial collagen cross-linking parallels left ventricular dilatation in rat models of systolic chamber dysfunction. Circulation. 2001;103:155–160. doi: 10.1161/01.cir.103.1.155. [DOI] [PubMed] [Google Scholar]
- YOUNG M., HEAD G., FUNDER J. Determinants of cardiac fibrosis in experimental hyper-mineralocorticoid states. Am. J. Physiol. 1995;269:E657–E662. doi: 10.1152/ajpendo.1995.269.4.E657. [DOI] [PubMed] [Google Scholar]