

Abstract
The aim of the present study was to examine the effects of glucocorticoid dexamethasone on airway responsiveness to albuterol after intratracheal instillation of saline or IL-1β in Brown-Norway rats in vivo and to elucidate the molecular mechanism of this effect.
IL-1β caused a significant reduction in albuterol-mediated relaxation to protect against MCh-induced bronchoconstriction. Dexamethasone attenuated the IL-1β-induced impaired relaxation while alone had no effect when compared to rats treated identically with saline.
The density of β2-adrenoceptors was significantly reduced in lung membranes harvested from IL-1β-treated rats, which was associated with impaired isoproterenol- and forskolin-stimulated cyclic AMP accumulation and adenylyl cyclase (AC) activity ex vivo. Dexamethasone did not prevent IL-1β-induced down-regulation of β2-adrenoceptors but completely blocked IL-1β-induced impairment of cyclic AMP accumulation and AC activity stimulated by isoproterenol and forskolin.
The inhibitory G-protein subtypes, Giα1, Giα2 and Giα3, were detected in lung membranes prepared from all groups of rats but the intensity of Giα1 and Giα2 was markedly increased in IL-1β-treated rats, which were not prevented by dexamethasone.
The activity of cytosolic GRK and the expression of GRK2 and GRK5 were elevated in the lung of IL-1β-treated rats, which were completely abolished by dexamethasone.
These results indicate that treatment of rats with IL-1β results in desensitization of pulmonary β2-adrenoceptors. In light of data obtained in this study, we propose that both the decrease in AC activity and the increase in GRK activity, which are reversed by dexamethasone, may underlie β2-adrenoceptor desensitization.
Keywords: Cytokines, β2-adrenoceptor, dexamethasone, adenylyl cyclase, G-protein, gene expression
Introduction
Corticosteroids and β-adrenergic agonists are the mainstay of asthma therapy and positive interactions between these agents in the airways have been noted. For example, glucocorticoids increase β2-adrenoceptor numbers and prevent the homologous downregulation of β2-adrenoceptors in vivo (Mak et al., 1995a). Glucocorticoids directly influence many steps in the β-adrenoceptor signaling pathway, including supersensitization through increased β-agonist stimulation of adenylyl cyclase (AC) (Davies & Lefkowitz, 1984), potentiation of high-affinity β-agonist binding (Kalavantavanich & Schramm, 2000), and upregulation of β-adrenoceptor gene transcription and receptor expression in lung tissues (Mak et al., 1995b; Mcgraw et al., 1995). Exposure of the airway smooth muscle to proinflammatory cytokines such as IL-1β, or to allergic inflammation leads to a reduction in β2-agonist induced relaxation (Hakonarson et al., 1995; Koto et al., 1996; Shore et al., 1997; Wills-Karp et al., 1993). Some of the underlying mechanisms include uncoupling of receptor to AC with an upregulation of Giα (Hakonarson et al., 1995; Koto et al., 1996). Moore et al. (1999) have recently demonstrated that glucocorticoids ablate IL-1β-induced β-adrenoceptor-mediated hyporesponsiveness in human airway smooth muscle cells.
β2-Adrenoceptors belong to the G-protein-coupled receptor family, that are coupled to AC activation following activation of the intermediary heterotrimeric G protein, Gs. Adenylyl cyclase can also be inhibited by a range of receptors acting via inhibitory G protein, Gi. The specificity of distinct Giα proteins in receptor-AC coupling and the role of specific G protein-coupled receptor kinases on IL-1β-induced attenuation of β-adrenoceptor response remain incompletely understood. There are three known Giα subunits that relay inhibitory signals through the receptor-AC coupling, whose responsiveness is actively ‘turned off' by members of the G-protein-coupled receptor kinase (GRK) family. This family consists of seven known members that have been further classified into three subfamilies according to their sequence homology and functional similarity (Penn et al., 2000; Pitcher et al., 1998): (1) the rhodopsin kinase (GRK1) which is predominantly localized to the retina and phosphorylates light-bleached (agonist-activated) rhodopsin in rod outer segments; (2) the GRK2 subfamily including GRK2 and GRK3, which are more widely distributed; and (3) the GRK4 subfamily including GRK4 to GRK7. GRK5 and GRK6 are more ubiquitously expressed. GRKs phosphorylate serine/threonine residues in the carboxyl-tail and/or intracellular loops of receptors in an agonist-dependent manner. They preferentially phosphorylate agonist-occupied receptors, and on agonist-mediated β-adrenoceptor activation, are targeted to the plasma membrane (Debburman et al., 1996). The phosphorylated form of these receptors act as substrates for a class of inhibitory proteins called β-arrestins, which sterically inhibit further receptor/G-protein coupling (Hausdorff et al., 1990), hence leading to receptor down-regulation. Of interest, GRK2, GRK3, and GRK5 target β-adrenoceptors (Pitcher et al., 1998), and have been reported to be expressed in various tissues. GRK2 and GRK3 predominantly reside in the cytosol (Lefkowitz et al., 1998), whereas GRK5 is predominantly plasma-membrane-bound (Ishizaka et al., 1997).
There is growing evidence to support the hypothesis that GRKs are important modulators of β-adrenoceptor signaling in vivo. Indeed, myocardial overexpression of GRK2 or GRK5 in mice impairs β-adrenoceptor/G protein coupling (Koch et al., 1995; Rockman et al., 1996). Furthermore, the β-adrenoceptor desensitization observed in cardiac myocytes from human failing hearts is related to increased GRK activity (Ungerer et al., 1994). However, little is known about the modulation of GRK expression, particularly on the effect of glucocorticoids. We hypothesized that the impaired relaxation to β2-agonists induced by IL-1β may be due to an increase in the expression and activity of GRKs, and that this could be reversed by glucocorticoids. In order to address this issue, we first determined whether IL-1β-induced-attenuation of airway relaxation by a β-agonist could be reproduced in vivo and then examined whether dexamethasone was able to reverse this attenuation of β-adrenoceptor-mediated relaxation. We also measured AC activity and GRK activity and expression of GRK2, GRK3, and GRK5, as well as the expression of Giα isoforms in lung tissues in order to elucidate the mechanisms.
Methods
Intratracheal instillation of IL-1β
Inbred, pathogen-free male Brown-Norway rats (Harlan-Olac, Bicester, U.K.) weighing 200 – 300 g were used for all studies. Animals were anaesthetized with an i.p. injection of 2 mg kg−1 midazolam (Roche Products Ltd., Welwyn Garden City, U.K.) and s.c. injection of 0.4 mg kg−1 Hypnorm (Janssen Pharmaceuticals Ltd., Wantage, U.K.), which contains 0.315 mg ml−1 of fentanyl citrate and 10 mg ml−1 of fluanisone. After adequate anaesthesia was achieved, animals were intubated with a nylon cannula (1.02-mm OD), through which recombinant human IL-1β (500 U in 50 μl 0.9% NaCl solution) or 50 μl 0.9% NaCl solution (control group) were instilled intratracheally. Because recombinant human IL-1β has been reported to displace antibody binding to recombinant rat IL-1β (Derijk & Berkenbosch, 1992), in addition to the considerable homology of 78% between human and rat IL-1β (Nishida et al., 1987), we used human recombinant IL-1β.
Experimental protocol
Animals were kept in a special caging system with its own air circulation (Maximizer; Thorens Caging Systems Inc., Hazleton, PA, U.S.A.). They were divided into four groups according to the protocol. Dexamethasone (DEX; 3 mg kg−1) was dissolved in sterilized isotonic saline and injected intraperitoneally 2 h prior to the instillation of IL-1β. Group 1: rats instilled with 0.9% NaCl only; Group 2: rats instilled with IL-1β only; Group 3: rats pretreated with DEX and instilled with 0.9% NaCl; Group 4: rats pretreated with DEX and instilled with IL-1β. For studies of lung function, rats were anaesthetized 24 h after the instillation of IL-1β or saline. For studies involving cyclic AMP assay, rats were killed by a lethal dose of pentobarbitone (200 mg kg−1, i.p.) at 24 h after instillation of IL-1β or saline and the lungs were quickly removed.
Evaluation of relaxation effect of albuterol in vivo
Lung function was measured 24 h after the instillation of IL-1β or saline. Rats were anaesthetized with an i.p. injection of 2 mg kg−1 midazolam and an s.c. injection of 0.4 mg kg−1 Hypnorm, which contains 0.315 mg ml−1 of fentanyl citrate and 10 mg ml−1 of fluanisone. After adequate anaesthesia was achieved, a tracheal cannula (1.02-mm outer diameter) was inserted into the lumen of the cervical trachea through a tracheostomy and tied snugly with suture material. A polyethylene catheter was inserted into the left carotid artery to monitor blood pressure and heart rate with a pressure transducer. The right external jugular vein was cannulated for administration of intravenous drugs and fluids. The animals were then connected to a small-animal respirator (Harvard Apparatus, Edenbridge, U.K.) and ventilated with 10 ml kg−1 of air at a rate of 90 strokes min−1. Transpulmonary pressure was measured with a pressure transducer (model FCO 40±1000 mm H2O, Furness Controls, Bexhill, U.K.) with one side attached to an air-filled catheter inserted into the right pleural cavity and the other side attached to a catheter connected to a side port of the intratracheal cannula. The ventilatory circuit had a total volume of 20 ml.
Airflow was measured with a pneumotachograph (model F1L, Mercury Electronics, Glasgow, U.K.) connected to a transducer (model FCO 40±20 mm H2O, Furness Controls). The signals from the transducers were digitized with a 12-bit analog-to-digital board (NB-MIO-16, National Instruments, Austin, TX, U.S.A.) connected to a Macintosh II computer (Apple Computer, Cupertino, CA, U.S.A.) and analysed with software (Lab VIEW 2, National Instruments, Austin, TX, U.S.A.) that was programmed according to the method of von neergaard & Wirz (1927). Arterial blood pressure was also monitored throughout the experiments.
Aerosols were generated with an ultrasonic nebulizer (model 2511, PulmoSonic, DeVilbiss, Hazeltown, PA, U.S.A.) and were administered to the airways through a separate ventilator system that bypassed the pneumotachograph. The volume of this circuit was 50 ml. The mean mass diameter of the aerosol was 3.8 μm, with a geometric standard deviation of 1.3, measured with a laser droplet and particle analyzer (model 2600C, Malvern Instruments, Derbyshire, U.K.).
In order to evaluate the protective effect of albuterol, a group of rats were preconstricted with MCh, followed by inhalation of increasing doses of albuterol. Because IL-1β does not alter the airway responses to cholinergic agonists (Koto et al., 1997), the same concentration of MCh was used to induce bronchoconstriction in all groups. After obtaining a stable RL, MCh (4 mg ml−1) was administered by inhalation for 45 breaths to cause airway constriction and RL measurements were made every 10 min until a stable increased RL value was obtained (initial contraction). Subsequently, saline solution was administered by inhalation first as a control, followed by albuterol. Increasing concentrations of albuterol (0.0001, 0.001, 0.01 and 0.1%, w v−1) were administered by inhalation (45 breaths) at 5-min intervals, and RL values were recorded. The maximal relaxation effects were calculated as per cent changes from baseline RL (control saline) (i.e. baseline RL value – minimum RL value / baseline RL value).
Determination of cyclic AMP accumulation
In separate experiments, animals were killed 24 h after the instillation of IL-1β or saline by instillation of a lethal dose of pentobarbitone. The lungs were quickly removed and placed in oxygenated modified Krebs-Henseleit (KH) solution of the following composition (mM): NaCl 118, KCl 5.9, MgCl2 1.2, CaCl2 2.5, NaHCO3 25.5 and glucose 5.05. Tissue blocks (∼2×2×10 mm) were cut out from freshly excised lung tissues of each animals and placed in ice-cold KH solution containing 10 μM indomethacin. Tissue blocks were pre-incubated with phosphodiesterase inhibitor, 100 μM isobutylmethylxanthine (IBMX) for 5 min and then incubated in 1 ml KH solution with either no drugs (baseline), 10 μM (−)-isoproterenol or 10 μM forskolin for 10 min at 37°C. Tissues were removed, blotted on absorbent paper, frozen in liquid nitrogen and stored at −70°C until assay for cyclic AMP.
Frozen lung tissue was homogenized in ice-cold 1 M trichloroacetic acid and centrifuged at 2500×g to precipitate particulate material. The cyclic AMP content in the supernatant was measured by radioimmunoassay as described previously (Koto et al., 1996). Cyclic AMP concentrations were determined by interpolation from a standard curve and expressed as fmol mg−1 wet weght.
Adenylyl cyclase assay
Frozen lung tissue was homogenized in ice-cold 50 mM Tris-HCl buffer, pH 7.4, containing 0.25 mM EDTA. The homogenate was filtered through a single layer of nylon gauze and centrifuged at 1000×g to pellet the unbroken cell debris. Only freshly prepared homogenates maintained at 4°C were used for AC assay. Adenylyl cyclase activity was determined using a modification of the method described previously (Salomon et al., 1974). Briefly, the standard assay system contained 50 mM Tris-HCl buffer pH 7.4, 5 mM MgCl2, 20 mM creatine phosphate, 10 IU creatine kinase, 1 mM cyclic AMP, 0.25 mM Ro20-1724 (a phosphodiesterase inhibitor), 1 mM [α-32P]-ATP (i.e. 2 μCi tube−1), 4 μM GTP and 200 – 400 μg homogenate in the absence or presence of NaF (10 mM), (−)-isoproterenol (10 μM) and forskolin (10 μM). The reaction was started by the addition of 30 μl of homogenate and incubated at 37°C. After 15 min the reaction was terminated by the addition of 800 μl of 6.25% (w v−1) trichloroacetic acid. [8-3H]-cAMP (approximately 10,000 c.p.m. in 100 μl of water) was added to each tube, and the mixtures centrifuged at 1000×g for 20 min at 4°C. The [α-32P]-ATP and [32P]-cAMP were then separated using a two-step chromatographic procedure. The losses of [32P]-cAMP on the columns were corrected for by measurement of the recovery of [8-3H]-cAMP.
Sample preparations
For the preparation of cytosolic and membrane fractions, frozen lung was ground in liquid nitrogen and homogenized in ice-cold Tris-HCl (pH 7.4) 10 mM containing MgCl2 7.5 mM, EDTA 5 mM, PMSF 0.1 mM, aprotinin 25 μg ml−1, leupeptin 5 μg ml−1, pepstatin 10 μg ml−1, bacitracin 100 μg ml−1, soyabean trying inhibitor 10 μg ml−1 and benzamidine 2 mM. Unbroken debris was pelleted by centrifugation at 1000×g at 4°C for 10 min and discarded. The supernatant was then centrifuged at 100,000×g at 4°C for 60 min to separate plasma membrane from the cytosol. The resulting membrane pellet was resuspended in the homogenization buffer and the supernatant was collected, aliquoted and frozen in liquid nitrogen. Protein concentration was determined with a BioRad protein assay reagent, using bovine serum albumin as standard.
Radioligand binding assay
To determine β2-adrenoceptor binding, aliquots of membrane preparations from various groups of animals were incubated for 120 min at 37°C with different concentrations (3 – 100 pM) of [125I]-iodocyanopindolol (ICYP; 2000 Ci mmol−1) in the absence or presence of either 0.1 μM CGP 20712A (a highly selective β1-antagonist) or 200 μM (−)-isoproterenol. Incubations were stopped by rapid vacuum filtration through Whatman GF/C filters. Specific binding was calculated as the difference between ICYP binding values in the absence (total) or presence (non-specific) of (−)-isoproterenol. The values for maximal binding (Bmax) and dissociation constant (Kd) were calculated from the Scatchard plot analysis of the binding data according to the interactive LIGAND program of Munson & Rodbard (1980).
Measurement of GRK activity
GRK enzymatic activity was assessed using light-dependent phosphorylation of rhodopsin (Benovic et al., 1987). Rod outer segment (ROS) membranes were prepared from dark-adapted bovine retinas via stepwise sucrose gradient centrifugation, and then treated with 5 M urea to inactivate endogenous kinase activity. GRK-dependent phosphorylation was determined by incubating 60 μg of lung cytosolic protein with 0.5 μM ROS in a buffer containing Tris-HCl (pH 7.4) 20 mM, MgCl2 5 mM, EDTA 2 mM, ∼1 μCi [γ-32P]-ATP and 2 nmol ATP in a final reaction volume of 20 μl. The reactions were carried out at 30°C for 30 min in the presence or absence of light. The incubations were terminated by the addition of 10 μl of 3×sodium dodecyl sulphate (SDS) sample buffer (8% SDS, 50 mM Tris-HCl, pH 6.8, 20% glycerol, 5% β-mercaptoethanol, and 0.005% bromophenol blue). Samples were then electrophoresed on 10% SDS-polyacrylamide gel electrophoresis (PAGE). After electrophoresis, the gel was stained with Coomassie blue, dried, and phosphorylated rhodopsin was visualized by autoradiography. Bands corresponding to rhodopsin (∼38 kDa) were cut from the gel and quantitated by liquid scintillation counting. The extent of GRK-mediated phosphorylation was determined as the difference between light- and dark-dependent phosphorylation, each determined in triplicate. One unit of activity is defined as that amount of GRK that catalyzed the incorporation of 1 pmol phosphate from ATP into ROS in 1 min per mg protein.
Western blot analyses
An aliquot of the cytosolic or membrane fraction was mixed with Laemmli sample buffer (Laemmli, 1970), boiled for 5 min and loaded onto 10% SDS/Tris polyacrylamide gel. Proteins were size fractionated at 80 mA and then transferred to presoaked Hybond ECL nitrocellulose membranes (Amersham Inc., Amersham, U.K.) by electroblotting at 400 mA for 1 h in 50 mM Tris containing 200 mM glycine, 20% (v v−1) methanol and 0.03% SDS. Efficiency of transfer was verified by Ponceau red staining. Membranes were soaked in a blocking buffer solution of 5% non-fat dry milk in TBS-T Tris-HCl pH 7.5 10 mM, NaCl 154 mM, 0.05% Tween-20) overnight at 4°C, and then incubated with rabbit polyclonal antibody specific to either Giα1, Giα2, Giα3, GRK2, GRK3 or GRK5 (1 : 200 dilution in all cases) diluted in blocking buffer for 1 h at room temperature. After extensive washing in TBS-T, membranes were incubated for 1 h at room temperature with either a donkey, anti-rabbit horseradish peroxidase-conjugated antibody at 1 : 4000 dilution (for all antibodies). Immunoreactivity was detected with an enhanced chemiluminescence detection system (ECL, Amersham) and bands were visualized after exposing blots to X-ray film. The same membranes were used for subsequent reprobing with specific antibodies after stripping. All protein bands were quantified by laser-scanning densitometry. All protein bands were quantified by densitometric scanning (UVP's Gel Documentation and Analysis System – GDS8000, Cambridge, U.K.).
Northern blot analyses of GRK expression
Total RNA was isolated from frozen lung tissues of various treatment groups by phenol/chloroform extraction and isopropanol precipitation (Chomczynski & Sacchi, 1987). An mRNA isolation kit system (PolyATtract IV; Promega, Southampton, U.K.) was used to prepare poly(A)+ RNA according to the manufacturer's instructions. Samples of poly(A)+ RNA were size-fractionated on a 1% agarose/formaldehyde gel, blotted onto nylon membrane (Magna, MA, U.S.A.) by capillary action, and immobilized with a UV Stratalinker 2400 (Stratagene; Cambridge, U.K.).
The human GRK2 cDNA (284 bp EcoRI/SalI fragment), GRK3 cDNA (281 bp EcoRI/XhoI fragment), GRK5 cDNA (188 bp EcoRI/XhoI fragment) or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA (1272 bp PstI fragment) were radiolabelled by random primer labelling kit in the presence of [α-32P]-dCTP. The blot was prehybridized for 4 – 5 h in 50% formamide, 5×SSC, 5×Denhardt's solution, 0.1% SDS, 10 mM NaH2PO4 and 100 mg ml−1 sonicated denatured salmon sperm DNA, and then hybridized with 32P-labelled cDNA probes for 12 – 16 h at 42°C. After hybridization, the blot was washed at high stringency in 0.1×SSC/0.1% SDS at 55°C for 30 min. The blot was exposed to Kodak OMAT XS film at −70°C with an intensifying screen for up to 7 days. The blot was hybridized subsequently to each cDNA probe after stripping.
Materials
The radioisotopes [γ-32P]-ATP, [α-32P]-ATP, [8-3H]-cAMP and [125I]-ICYP (specific activities 20 – 40, 40 – 50, ∼24, and 2000 Ci mmol−1 respectively) were supplied by Amersham International (Amersham, U.K.). All primary and secondary antibodies were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, U.S.A.) and DAKO respectively. Unless otherwise stated, all drugs were purchased from Sigma Chemical Co. (Poole, Dorset, U.K.). Recombinant human IL-1β was a generous gift of Glaxo Laboratories Ltd. (Greenford, U.K.).
Statistical analysis
All values are expressed as means±s.e.mean. Statistical differences between two groups were determined by Mann-Whitney U-test. The effects of the combination of dexamethasone and IL-1β, dexamethasone alone, or IL-1β alone on baseline and agonist- or drug-induced cyclic AMP accumulation and AC activity as well as GRK activity and protein expressions of GRKs and Giα isoforms were examined by ANOVA. The Bonferroni correction was used to correct for multiple comparisons. A P value less than 0.05 was regarded as significant.
Results
In vivo bronchial responses
After exposure to 4 mg ml−1 MCh, the four groups of animals showed a similar time-course increase in RL with plateauing of RL at 10 min when RL was increased by 270% above baseline. RL remained stable for at least 35 min at that level (Figure 1). IL-1β caused a significant reduction of relaxation induced by 0.01% (P<0.05) and 0.1% (P<0.01) albuterol. Dexamethasone per se did not have significant effect on relaxation. However, dexamethasone reversed the impaired relaxation to 0.1% albuterol induced by IL-1β (44.8±3.0% vs 61.5±3.9%, P<0.05) (Figure 2).
Figure 1.

Time-course of per cent change in RL after inhalation of 4 mg ml−1 methacholine (MCh). The bronchoconstrictor response to the inhalation of MCh was rapid and was maximal by 5 min. The bronchoconstrictor response was stable for 45 min. All groups of animals showed a similar time-course and there were no significant differences between the groups. n=3 in each group.
Figure 2.
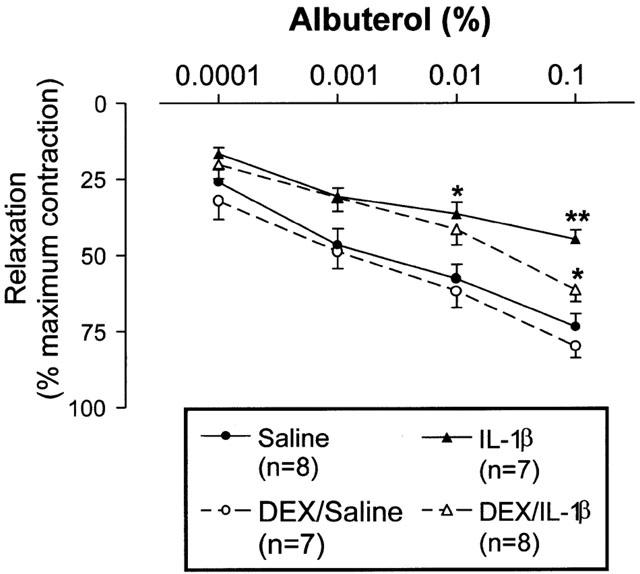
Effect of IL-1β instillation (500 U), dexamethasone (DEX; 3 mg kg−1, i.p.) and/or combined treatments on the protective actions of albuterol against methacholine-induced bronchoconstriction. IL-1β attenuated the relaxant effect of albuterol. DEX alone did not have any effects while DEX reversed the IL-1β-induced attenuation of albuterol effect. *P<0.05, **P<0.01 in comparison to saline-treated group.
Radioligand β-adrenoceptor binding assay
There was significant decrease in the density of β2-adrenoceptors in lung membranes from IL-1β-treated group compared to control (Bmax: 237.6±9.8 vs 147.9±6.3 fmol mg−1 protein, P<0.01) without any alteration in the affinity (Kd: 13.3±1.9 vs 11.3±1.2 pM). The presence of dexamethasone did not prevent the IL-1β-induced down-regulation of β2-adrenoceptors (Bmax: 147.9±6.3 vs 150.8±5.9 fmol mg−1 protein for IL-1β alone and dexamethasone plus IL-1β, respectively) (Figure 3).
Figure 3.

Effect of IL-1β and/or dexamethasone (DEX) on the density (as measured by the maximal binding, Bmax) and the affinity (as measured by the dissociation constant, Kd) of β2-adrenoceptors (β2ARs). Total [125I]-iodocyanopindolol ([125I]-ICYP) was displaced by the specific β1-receptor antagonist, CGP-20712A (100 nM), or isoproterenol (200 μM, to give nonspecific binding). The affinity of β2-adrenoceptors was not altered by either IL-1β or by dexamethasone. However, IL-1β caused a significant reduction in the density of receptors, an effect that was not prevented by dexamethasone. Data are means±s.e.mean of four animals in each group. **P<0.01.
Isoproterenol- and forskolin-stimulated cyclic AMP accumulation and AC activity
There were no significant differences between the groups in basal cyclic AMP accumulation level (Figure 4). In the presence of IBMX, cyclic AMP accumulation stimulated by isoproterenol (10 μM) was significantly less in IL-1β-treated group than in control (862.6±78.1 vs 613.6±35.7 fmol mg−1 wet weight, P<0.01) and pretreatment with dexamethasone reversed this attenuation (839.4±105.7 fmol mg−1 wet weight, P<0.05) (Figure 4). Similarly, cyclic AMP accumulation stimulated by forskolin (10 μM) was significantly less in IL-1β-treated group than in control (1729.4±97.9 vs 1347.4±111.5 fmol mg−1 wet weight, P<0.05) and pretreatment with dexamethasone reversed this attenuation (1750.1±123.9 fmol mg−1 wet weight, P<0.05) (Figure 4A). Dexamethasone alone per se did not have any significant effect on either isoproterenol- or forskolin-stimulated cyclic AMP accumulation and AC activity. Isoproterenol- and forskolin-stimulated AC activity was significantly lower in IL-1β-treated group compared to control, which was prevented by pretreatment with dexamethasone (Figure 4B), consistent with the cyclic AMP accumulation data. There was no difference on NaF-stimulated AC activity in all four groups.
Figure 4.
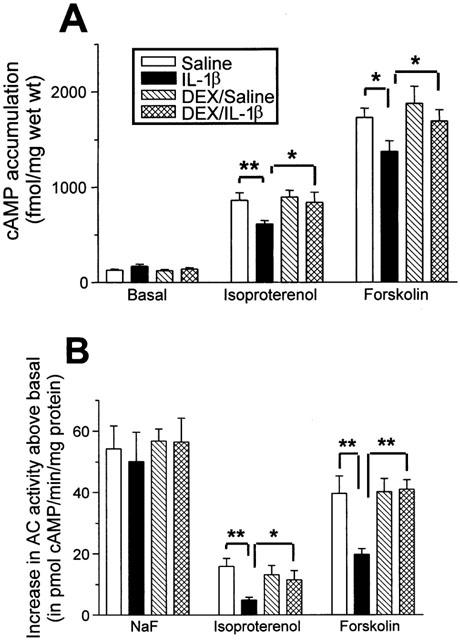
Effect of IL-1β and/or dexamethasone (DEX) on cyclic adenosine monophosphate (cyclic AMP) accumulation stimulated by isoproterenol (10 μM) and forskolin (10 μM) (A), and on adenylyl cyclase activity stimulated by sodium fluoride (10 mM), isoproterenol and forskolin (B). Intratracheal instillation of IL-1β caused significant attenuation in isoproterenol- and forskolin-stimulated cyclic AMP accumulation and adenylyl cyclase activity, which was reversed by DEX. Data are means±s.e.means of 4 – 6 animals in each group. *P<0.05, **P<0.01.
Giα isoform protein expression in lung
As shown in Figure 5, each antibody recognized a single protein of around 40 kDa referred to as Giα1, Giα2 and Giα3 respectively in lung membrane fractions from all groups. In comparison to saline-treated animals, IL-1β caused significant increases (about 75%) in Giα1 and Giα2 protein expression that was not blocked by dexamethasone. Dexamethasone alone did not cause significant increase in these protein expressions although there was a tendency for the increase in Giα1, which did not reach statistical significance. In contrast, Giα3 expression in lung membrane fractions was not significantly different between the groups.
Figure 5.

Comparison of Western blots for the expression of Giα isoforms in lung membranes prepared from each group of animals. The left-hand panel shows a representative example of Western blot analysis from a rat from each experimental group. Note enhanced expression of Giα1 and Giα2 in IL-1β-treated (lane 2), and dexamethasone (DEX) plus IL-1β-treated (lane 4) compared with saline (lane 1), and DEX plus saline (lane 3) groups. Right-hand panels (A,B) Densitometric measurements of Giα1 and Giα2 immunoreactivity were normalized to Giα3. There was a significant increase in Giα1 and Giα2 induced by IL-1β, but dexamethasone had no significant effect. Data are means±s.e.mean of 4 – 6 animals in each group. *P<0.05.
GRK activity, and GRK protein and gene expression in lung
IL-1β induced a significant (53%) increase in cytosolic GRK activity in lung tissue when compared with saline-treated animals (Figure 6), which was associated with a greater than 2 fold increase in the expression of GRK2 as assessed by Western blotting of the lung cytosolic fractions (Figure 7) as well as GRK5 (data not shown). Dexamethasone alone had no significant effect but prevented IL-1β-induced up-regulation of GRK activity (Figure 6). However, significant reduction in GRK2 protein expression was observed in lung tissues after dexamethasone treatment alone (Figure 7). No detectable level of GRK3 protein was observed in all lung tissues. Despite the increase in GRK activity and protein expression, the steady-state level of GRK2 and GRK5 mRNAs (∼3.8 and ∼2.8 kb transcripts respectively) was unchanged in lung harvested from different treatment groups (Figure 8) with no detectable GRK3 mRNA.
Figure 6.

Assessment of GRK activity in lung cytosolic fractions from different treatment groups. (A) Autoradiograph depicting light-dependent phosphorylation of rhodopsin (∼38 kD). Rat lungs from saline (lane 1), IL-1β-treated (lane 2), DEX plus saline (lane 3), and DEX plus IL-1β-treated (lane 4) were shown. (B) Quantification of GRK activity measurements in each group of animals. Data are means±s.e.mean of four animals in each group. *P<0.05.
Figure 7.
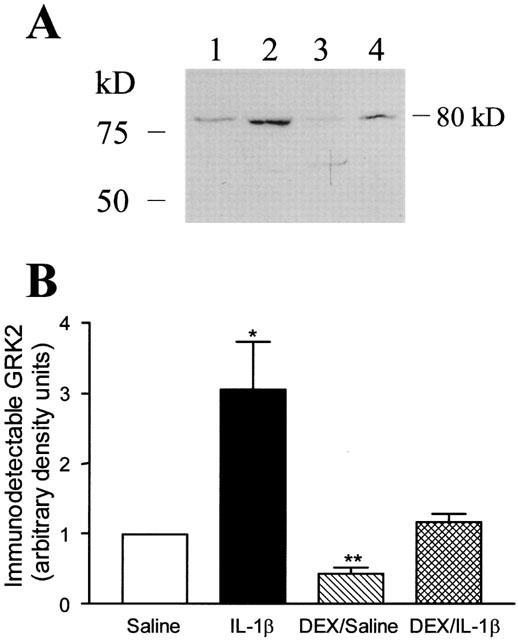
(A) Representative autoradiograph of Western blot depicting immuno-detectable GRK2 (∼80 kD) in lung cytosolic fractions from different treatment groups. Rat lungs from saline (lane 1), IL-1β-treated (lane 2), DEX plus saline (lane 3), and DEX plus IL-1β-treated (lane 4) were shown. (B) Densitometric measurements of GRK2 immunoreactivity. Data are means±s.e.mean of 4 – 6 animals in each group. *P<0.05, **P<0.01.
Figure 8.

Representative autoradiographs of Northern blot for steady-state level of GRK2, GRK5 and house-keeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA expression in lung from different treatment groups. Rat lungs from saline (lane 1), IL-1β-treated (lane 2), DEX plus saline (lane 3), and DEX plus IL-1β-treated (lane 4) were shown.
Discussion
The present manuscript describes the effects of IL-1β and/or dexamethasone on β-adrenoceptor-mediated relaxation in vivo and the different components of the β2-adrenoceptor-mediated signalling in lung tissues ex vivo. Most of the information to date on IL-1β-induced β2-adrenoceptor desensitization has been gathered from cultured cell systems with single cell type in particular airway smooth muscle cells (Moore et al., 1999; Shore et al., 1997) and the extent to which this applies to the in vivo situation has been little investigated. We have now demonstrated that IL-1β administered to the airways of rats reduced albuterol-induced relaxation of airways that were pre-constricted with methacholine in vivo and that dexamethasone partially reversed IL-1β-induced impairment of β2-adrenoceptor-mediated relaxation. Although dexamethasone reversed IL-1β-induced reduction of both isoproterenol- and forskolin-stimulated cyclic AMP accumulation and AC activity, the reduction in the density of β2-adrenoceptors by IL-1β was not prevented by dexamethasone in lung tissues ex vivo. However, NaF-stimulated AC activity was not altered by IL-1β. Interestingly, forskolin-stimulated cyclic AMP accumulation and AC activity was reduced, suggesting a direct IL-1β-induced defect on AC. These data are in contrast to either the increase or the lack of effect on forskolin-stimulated cyclic AMP accumulation previously reported in the literature albeit in other species (Billington et al., 2001; Shore et al., 1997).
We have established an in vivo model of pulmonary β2-adrenoceptor desensitization was established in rats by administering IL-1β intratracheally. In this study, we measured airway relaxation in vivo by first inducing a constant level of bronchoconstriction with a cholinergic bronchoconstrictor, methacholine, then followed by the adminstration of albuterol to induce relaxation. As shown previously in our ex vivo studies (Koto et al., 1996), there was attenuation of the relaxant response to albuterol after exposure to IL-1β. The inhibitory effect of dexamethasone was only observed on the highest concentration of albuterol (0.1%), and only about 50% of IL-1β-induced attenuation of albuterol-mediated relaxation was reversed by dexamethasone; it is to be noted that the down-regulation of β2-adrenoceptor was not reversed by dexamethasone. Our results are in contrast to those obtained in airway smooth muscle cells in vitro in which dexamethasone totally prevented the IL-1β-induced β-adrenoceptor hyporesponsiveness across a wide range of doses of β-agonists (Moore et al., 1999). The difference in effect of dexamethasone may reflect the in vivo and in vitro nature of the studies. Although the effects that we are studying (changes in lung resistance) are very much secondary to changes in airway smooth muscle cells, it is possible that effects on other cells in lung (i.e. inflammatory cells and resident cells) may influence the contractile responses. We also recognize that in our study, the lack of steroid effect may be related to the dose of dexamethasone administered to these animals in vivo, which may be insufficient to reach the target cells at the timepoint chosen to cause a greater reversal of IL-1β-induced attenuation of the relaxant effect of albuterol.
Our previous and current data confirm that there is an upregulation of Gi after IL-1β, as previously shown (Hakonarson et al., 1996; Koto et al., 1996). In our study, we found that Giα protein expression is associated with enhanced expression of the Giα1 and Giα2 subunits, but not of the Giα3 subunit in lung membranes isolated from IL-1β-treated rats. The present results are in accordance with elevated levels of Giα2 detected in lung membranes from antigen-challenged guinea-pigs (Lee et al., 1994), and induced increased Giα2 mRNA expression after IL-1β administration in guinea-pig tracheal smooth muscle and in cultured human endothelial cells (Hirata et al., 1994; Lee et al., 1989). In addition to an enhancement of Giα2 subunit, Giα1 was also increased in lung tissues from IL-1β-treated animals in our present findings. Dexamethasone alone did not cause any significant increase in these protein expressions although there was a tendency for the increase in Giα1, which did not reach statistical significance. The IL-1β-induced up-regulation of the Giα1 and Giα2 subunits was not reversed by dexamethasone in this study.
In subsequent experiments, cytosolic GRK activity was significantly increased in the lung of IL-1β-treated rats when compared with those animals that received saline. This effect was associated with increased protein expression of GRK2 and GRK5 but not steady-state levels of mRNA. It has previously shown that overexpression of GRK2 or GRK5 in transgenic mice led to a marked β-adrenoceptor desensitization (Koch et al., 1995; Korzick et al., 1997; Rockman et al., 1996). Mice overexpressing GRK2 showed attenuation of isoproterenol-stimulated left ventricular contraction in vivo, reduced sarcolemmal AC activity, and diminished functional coupling of β-adrenoceptors (Koch et al., 1995). Although the mechanism of action of IL-1β to induce an increase in GRK activity and protein expression is unclear. Upregulation of GRK2 expression in lymphocytes has been observed following chronic activation of protein kinase C (PKC) with phorbol ester (Chuang et al., 1995; de blasi et al., 1995). PKC is capable of phosphorylating β-adrenoceptors and the α-subunit of Gs that couples β-adrenoceptors to AC (Bouvier et al., 1987). Recent studies have found that IL-1β upregulates β2-adrenoceptor expression through the involvement of the activation of PKC in human airway epithelial cells (Bin et al., 2001). Although these data are in contrast to our present findings of IL-1β-induced down-regulation of β2-adrenoceptor in lung tissues ex vivo, this may reflect differences between in vivo and in vitro studies, and the specific cell types involved. Dexamethasone caused complete inhibition of the increased GRK activity as well as the increased GRK2 and GRK5 protein expression induced by IL-1β. However, significant reduction in GRK2 protein expression was observed in lung tissues ex vivo after dexamethasone treatment alone without any change in GRK activity. Clearly, therefore, this mechanism may be important for the action of dexamethasone in the regulation of GRK function.
We have studied lung tissues, rather than isolated airway smooth muscle cells, and therefore observed the responses of a number of different cell types that have varying degrees of β2-adrenoceptor expression. Pulmonary cells that express a high density of β2-adrenoceptor include alveolar and airway epithelial cells, airway smooth muscle cells, vascular endothelial cells and inflammatory cells such as neutrophils and eosinophils. In addition, it is likely that lung tissues from IL-1β-treated rats contain increased numbers of neutrophils (Tsukagoshi et al., 1994). Previous studies have shown that neutrophils expressed far less GRK2 compared with monocytes and peripheral blood lymphocytes (Loudon et al., 1996), and it is unlikely that the influx of neutrophils could account for the increased in GRK activity in this study. Using lung tissues populated by a large number of different cell types, it is difficult to say whether differences in the expression of Giα subunits, AC and GRK activity occur in a given cell type, and whether these correlate with differences in functional effect on β2-adrenoceptor signal transduction. It is in these cell types that IL-1β causes a significant reduction in β2-adrenoceptors, as we have previously shown by receptor autoradiography (Koto et al., 1996).
There may be alternative interpretations of our findings. The observed in vivo IL-1β-induced β2-adrenoceptor desensitization may be due to IL-1β-mediated induction of PGE2 release, resulting in cyclic AMP accumulation, protein kinase A activation, and phosphorylation of the β2-adrenoceptor by protein kinase A, and uncoupling of the β2-adrenoceptor from Gsα (Laporte et al., 1998). Furthermore, rats rendered tolerant to PGE2 have been shown to be insensitive to albuterol in vivo (Finney et al., 2000). Therefore, IL-1β-induced β2-adrenoceptor desensitization may involve multiple components of the β2-adrenoceptor-mediated signalling pathways. Dexamethasone only partially reversed the loss of IL-1β-induced relaxation activity at the highest concentration of albuterol but caused complete inhibition of IL-1β-induced increase in GRK activity and protein expression, and IL-1β-induced reduction in isoproterenol- and forskolin-stimulated cyclic AMP accumulation and AC activity despite no effect on IL-1β-induced down-regulation of β2-adrenoceptors. The absence of a direct relationship between the β2-adrenoceptor-mediated responsiveness and the β2-adrenoceptor number of lung tissues as demonstrated in the present study is supported by the findings of a previous study (Whicker et al., 1991). It is not known whether the effect of dexamethasone in IL-1β-induced desensitization of β2-adrenoceptors and of specific AC isoforms is reflected in any specific pulmonary cell type. This needs to be established in future in vitro experiments. Our findings indicate that IL-1β-induced defect in AC and up-regulation of GRK activity may not be solely responsible for IL-1β-induced pulmonary β2-adrenoceptor desensitization in vivo.
Acknowledgments
This study is partly supported by Imperial College Initiative Award and by GlaxoSmithKline.
Abbreviations
- AC
adenylyl cyclase
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GRK
G-protein-coupled receptor kinase
- IBMX
3-isobutyl-1-methylxanthine
- IL-1β
interleukin-1β
- KH
Krebs-Henseleit
- RL
lung resistance
- ROS
rod outer segment
- SSC
standard sodium citrate
References
- BENOVIC J.L., MAYOR F., STANISZEWSKI C., LEFKOWITZ R.J., CARON M.G. Purification and characterization of the β-adrenergic receptor kinase. J. Biol. Chem. 1987;262:9026–9032. [PubMed] [Google Scholar]
- BILLINGTON C.K., PASCUAL R.M., HAWKINS M.L., PENN R.B., HALL I.P. Interleukin-1β and rhinovirus sensitize adenylyl cyclase in human airway smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 2001;24:633–639. doi: 10.1165/ajrcmb.24.5.4215. [DOI] [PubMed] [Google Scholar]
- BIN W., AKSOY M.O., YANG Y., KELSEN S.G. IL-1β enhances β2-adrenergic receptor expression in human airway epithelial cells by activating PKC. Am. J. Physiol. Lung Cell Mol. Physiol. 2001;280:L675–L679. doi: 10.1152/ajplung.2001.280.4.L675. [DOI] [PubMed] [Google Scholar]
- BOUVIER M., LEEB-LUNDBERG L.M., BENOVIC J.L., CARON M.G., LEFKOWITZ R.J. Regulation of adrenergic receptor function by phosphorylation. II. Effects of agonist occupancy on phosphorylation of α1- and β2-adrenergic receptors by protein kinase C and the cyclic AMP-dependent protein kinase. J. Biol. Chem. 1987;262:3106–3113. [PubMed] [Google Scholar]
- CHOMCZYNSKI P., SACCHI N. Single step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987;162:156–160. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- CHUANG T.T., LEVINE H., DE BLASI A. Phosphorylation and activation of β-adrenergic receptor kinase by protein kinase C. J. Biol. Chem. 1995;270:18660–18665. doi: 10.1074/jbc.270.31.18660. [DOI] [PubMed] [Google Scholar]
- DAVIES A.O., LEFKOWITZ R.J. Regulation of β-adrenergic receptors by steroid hormones. Ann. Rev. Physiol. 1984;46:119–130. doi: 10.1146/annurev.ph.46.030184.001003. [DOI] [PubMed] [Google Scholar]
- DE BLASI A., PARRUTI G., SALLESE M. Regulation of G protein-coupled receptor kinase subtypes in activated T lymphocytes. Selective increase of β-adrenergic receptor kinase 1 and 2. J. Clin. Invest. 1995;95:203–210. doi: 10.1172/JCI117641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEBBURMAN S.K., PTASIENSKI J., BENOVIC J.L., HOSEY M.M. G protein-coupled receptor kinase GRK2 is a phospholipid-dependent enzyme that can be conditionally activated by G protein betagamma subunits. J. Biol. Chem. 1996;271:22552–22562. doi: 10.1074/jbc.271.37.22552. [DOI] [PubMed] [Google Scholar]
- DERIJK R., BERKENBOSCH F. Development and application of a radioimmunoassay to detect interleukin-1 in rat peripheral circulation. Am. J. Physiol. 1992;263:E1092–E1098. doi: 10.1152/ajpendo.2006.263.6.E1092. [DOI] [PubMed] [Google Scholar]
- FINNEY P.A., BELVISI M.G., DONNELLY L.E., CHUANG T.-T., MAK J.C.W., SCORER C., BARNES P.J., ADCOCK I.M., GIEMBYCZ M.A. Albuterol-induced downregulation of Gsα accounts for pulmonary β2-adrenoceptor desensitization in vivo. J. Clin. Invest. 2000;106:125–135. doi: 10.1172/JCI8374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAKONARSON H., HERRICK D.J., GRUNSTEIN M.M. Mechanism of impaired β-adrenoceptor responsiveness in atopic sensitized airway smooth muscle. Am. J. Physiol. 1995;269:L645–L652. doi: 10.1152/ajplung.1995.269.5.L645. [DOI] [PubMed] [Google Scholar]
- HAKONARSON H., HERRICK D.J., SERRANO P.G., GRUNSTEIN M.M. Mechanism of cytokine-induced modulation of β-adrenoceptor responsiveness in airway smooth muscle. J. Clin. Invest. 1996;97:2593–2600. doi: 10.1172/JCI118708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAUSDORFF W.P., CARON M.G., LEFKOWITZ R.J. Turning off the signal: desensitization of β-adrenergic receptor function. FASEB J. 1990;4:2881–2889. [PubMed] [Google Scholar]
- HIRATA F., LEE J.Y., SAKAMOTO T., NOMURA A., UCHIDA Y., HIRATA A., HASEGAWA S. IL-1β regulates the expression of the Gi2α gene via lipid mediators in guinea pig tracheal muscle. Biochem. Biophys. Res. Commun. 1994;203:1889–1896. doi: 10.1006/bbrc.1994.2408. [DOI] [PubMed] [Google Scholar]
- ISHIZAKA N., ALEXANDER R.W., LAURSEN J.B., KAI H., FUKUI T., OPPERMANN M., LEFKOWITZ R.J., LYONS P.R., GRIENDLING K.K. G protein-coupled receptor kinase 5 in cultured vascular smooth muscle cells and rat aorta. Regulation by angiotensin II and hypertension. J. Biol. Chem. 1997;272:32482–32488. doi: 10.1074/jbc.272.51.32482. [DOI] [PubMed] [Google Scholar]
- KALAVANTAVANICH K., SCHRAMM C.M. Dexamethasone potentiates high-affinity β-agonist binding and Gsα protein expression in airway smooth muscle. Am. J. Physiol. Lung Cell Mol. Physiol. 2000;278:L1101–L1106. doi: 10.1152/ajplung.2000.278.5.L1101. [DOI] [PubMed] [Google Scholar]
- KOCH W.J., ROCKMAN H.A., SAMAMA P., HAMILTON R.A., BOND R.A., MILANO C.A., LEFKOWITZ R.J. Cardiac function in mice overexpressing the β-adrenergic receptor kinase or a beta ARK inhibitor. Science. 1995;268:1350–1353. doi: 10.1126/science.7761854. [DOI] [PubMed] [Google Scholar]
- KORZICK D.H., XIAO R.P., ZIMAN B.D., KOCH W.J., LEFKOWITZ R.J., LAKATTA E.G. Transgenic manipulation of β-adrenergic receptor kinase modifies cardiac myocyte contraction to norepinephrine. Am. J. Physiol. 1997;272:H590–H596. doi: 10.1152/ajpheart.1997.272.1.H590. [DOI] [PubMed] [Google Scholar]
- KOTO H., MAK J.C.W., HADDAD E.B., XU W.B., SALMON M., BARNES P.J., CHUNG K.F. Mechanisms of impaired β-adrenoceptor-induced airway relaxation by interleukin-1β in vivo in the rat. J. Clin. Invest. 1996;98:1780–1787. doi: 10.1172/JCI118977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOTO H., SALMON M., HADDAD E.B., HUANG T.J., ZAGORSKI J., CHUNG K.F. Role of cytokine-induced neutrophil chemoattractant (CINC) in ozone-induced airway inflammation and hyperresponsiveness. Am. J. Respir. Crit. Care Med. 1997;156:234–239. doi: 10.1164/ajrccm.156.1.9606095. [DOI] [PubMed] [Google Scholar]
- LAEMMLI U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- LAPORTE J.D., MOORE P.E., PANETTIERI R.A., MOELLER W., HEYDER J., SHORE S.A. Prostanoids mediate IL-1β-induced β-adrenergic hyporesponsiveness in human airway smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 1998;275:L491–L501. doi: 10.1152/ajplung.1998.275.3.L491. [DOI] [PubMed] [Google Scholar]
- LEE J.Y., UCHIDA Y., SAKAMOTO T., HIRATA A., HASEGAWA S., HIRATA F. Alteration of G protein levels in antigen-challenged guinea pigs. J. Pharmacol. Exp. Ther. 1994;271:1713–1720. [PubMed] [Google Scholar]
- LEE R.T., BROCK T.A., TOLMAN C., BLOCOH K.D., SEIDMAN J.G., NEER E.J. Subtype-specific increase in G-protein α-subunit mRNA by interleukin 1β. FEBS Lett. 1989;249:139–142. doi: 10.1016/0014-5793(89)80610-6. [DOI] [PubMed] [Google Scholar]
- LEFKOWITZ R.J., PITCHER J., KRUEGER K., DAAKA Y. Mechanisms of β-adrenergic receptor desensitisation and resensitization. Adv. Pharmacol. 1998;42:416–420. doi: 10.1016/s1054-3589(08)60777-2. [DOI] [PubMed] [Google Scholar]
- LOUDON R.P., PERUSSIA B., BENOVIC J.L. Differentially regulated expression of the G-protein-coupled receptor kinases, βARK and GRK6, during myelomonocytic cell development in vitro. Blood. 1996;88:4547–4557. [PubMed] [Google Scholar]
- MAK J.C.W., NISHIKAWA M., BARNES P.J. Glucocorticosteroids increase β2-adrenergic receptor transcription in human lung. Am. J. Physiol. 1995a;268:L41–L46. doi: 10.1152/ajplung.1995.268.1.L41. [DOI] [PubMed] [Google Scholar]
- MAK J.C.W., NISHIKAWA M., SHIRASAKI H., MIYAYASU K., BARNES P.J. Protective effects of a glucocorticoid on downregulation of pulmonary β2-adrenergic receptors in vivo. J. Clin. Invest. 1995b;96:99–106. doi: 10.1172/JCI118084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCGRAW D.W., CHAI S.E., HILLER F.C., CORNETT L.E. Regulation of the β2-adrenergic receptor and its mRNA in the rat lung by dexamethasone. Exp. Lung Res. 1995;21:535–546. doi: 10.3109/01902149509031757. [DOI] [PubMed] [Google Scholar]
- MOORE P.E., LAPORTE J.D., GONZALEZ S., MOLLER W., HEYDER J., PANETTIERI R.A., SHORE S.A. Glucocorticoids ablate IL-1β-induced β-adrenergic hyporesponsiveness in human airway smooth muscle cells. Am. J. Physiol. 1999;277:L932–L942. doi: 10.1152/ajplung.1999.277.5.L932. [DOI] [PubMed] [Google Scholar]
- MUNSON P.J., RODBARD D. Ligand: a versatile computerized approach for characterization of ligand-binding systems. Anal. Biochem. 1980;107:220–239. doi: 10.1016/0003-2697(80)90515-1. [DOI] [PubMed] [Google Scholar]
- NISHIDA T., NISHINO N., TAKANO M., KAWAI K., BANDO K., MASUI Y., NAKAI S., HIRAI Y. cDNA cloning of IL-1α and IL-1β from mRNA of U937 cell line. Biochem. Biophys. Res. Commun. 1987;143:345–352. doi: 10.1016/0006-291x(87)90671-1. [DOI] [PubMed] [Google Scholar]
- PENN R.B., PRONIN A.N., BENOVIC J.L. Regulation of G protein-coupled receptor kinases. Trends Cardiovasc. Med. 2000;10:81–89. doi: 10.1016/s1050-1738(00)00053-0. [DOI] [PubMed] [Google Scholar]
- PITCHER J.A., FREEDMAN N.J., LEFKOWITZ R.J. G protein-coupled receptor kinases. Ann. Rev. Biochem. 1998;67:653–692. doi: 10.1146/annurev.biochem.67.1.653. [DOI] [PubMed] [Google Scholar]
- ROCKMAN H.A., CHOI D.J., RAHMAN N.U., AKHTER S.A., LEFKOWITZ R.J., KOCH W.J. Receptor-specific in vivo desensitization by the G protein-coupled receptor kinase-5 in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 1996;93:9954–9959. doi: 10.1073/pnas.93.18.9954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SALOMON Y., LONDOS C., RODBELL M. A highly sensitive adenylate cyclase assay. Anal. Biochem. 1974;58:541–548. doi: 10.1016/0003-2697(74)90222-x. [DOI] [PubMed] [Google Scholar]
- SHORE S.A., LAPORTE J., HALL I.P., HARDY E., PANETTIERI R.A.J. Effect of IL-1β on responses of cultured human airway smooth muscle cells to bronchodilator agonists. Am. J. Respir. Cell Mol. Biol. 1997;16:702–712. doi: 10.1165/ajrcmb.16.6.9191472. [DOI] [PubMed] [Google Scholar]
- TSUKAGOSHI H., SAKAMOTO T., XU W., BARNES P.J., CHUNG K.F. Effect of interleukin-1β on airway hyperresponsiveness and inflammation in sensitized and nonsensitized Brown-Norway rats. J. Allergy Clin. Immunol. 1994;93:464–469. doi: 10.1016/0091-6749(94)90355-7. [DOI] [PubMed] [Google Scholar]
- UNGERER M., PARRUTI G., BOHM M., PUZICHA M., DE BLASI A., ERDMANN E., LOHSE M.J. Expression of β-arrestins and β-adrenergic receptor kinases in the failing human heart. Circ. Res. 1994;74:206–213. doi: 10.1161/01.res.74.2.206. [DOI] [PubMed] [Google Scholar]
- VON NEERGAARD K., WIRZ K. Die messung der stromungswideerstande in den Atemwegen des Menschen, insbesondere bei Asthma und Emphysem. Z. Klin. Med. 1927;105:51–82. [Google Scholar]
- WHICKER S.D., LUMMIS S.C.R., BLACK J.L. β-Adrenoceptors in human airway tissue: relationship between functional responsiveness and receptor number. Life Sci. 1991;49:1021–1029. doi: 10.1016/0024-3205(91)90303-s. [DOI] [PubMed] [Google Scholar]
- WILLS-KARP M., UCHIDA Y., LEE J.Y., JINOT J., HIRATA A., HIRATA F. Organ culture with proinflammatory cytokines reproduces impairment of the β-adrenoceptor-mediated relaxation in tracheas of a guinea pig antigen model. Am. J. Respir. Cell Mol. Biol. 1993;8:153–159. doi: 10.1165/ajrcmb/8.2.153. [DOI] [PubMed] [Google Scholar]